
Genetic Basis of Dilated Cardiomyopathy in Dogs and Its Potential as a Bidirectional Model

 , ,
, ,  ,
,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Clinical Aspects of Canine and Human DCM
3. DCM Histopathology

4. Breed-Specific Canine DCM Genetics
4.1. Dobermanns
4.2. Boxers
4.3. Irish Wolfhound
4.4. Welsh Springer Spaniels
4.5. Portuguese Water Dogs
5. Genetics of Human DCM
6. The Onset of DCM
6.1. Juvenile-Onset DCM
6.2. Adult-Onset DCM
7. Sex Differences in DCM
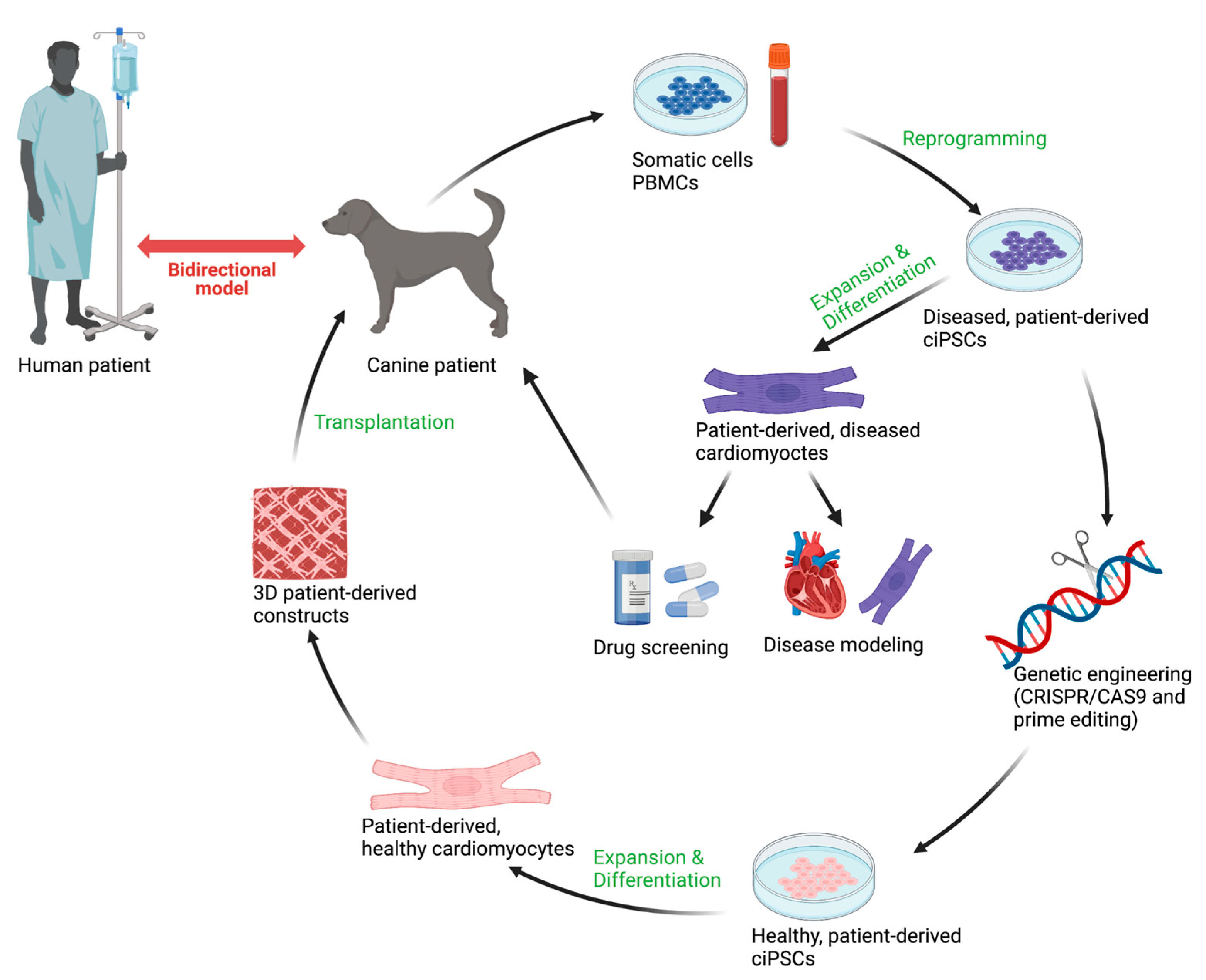
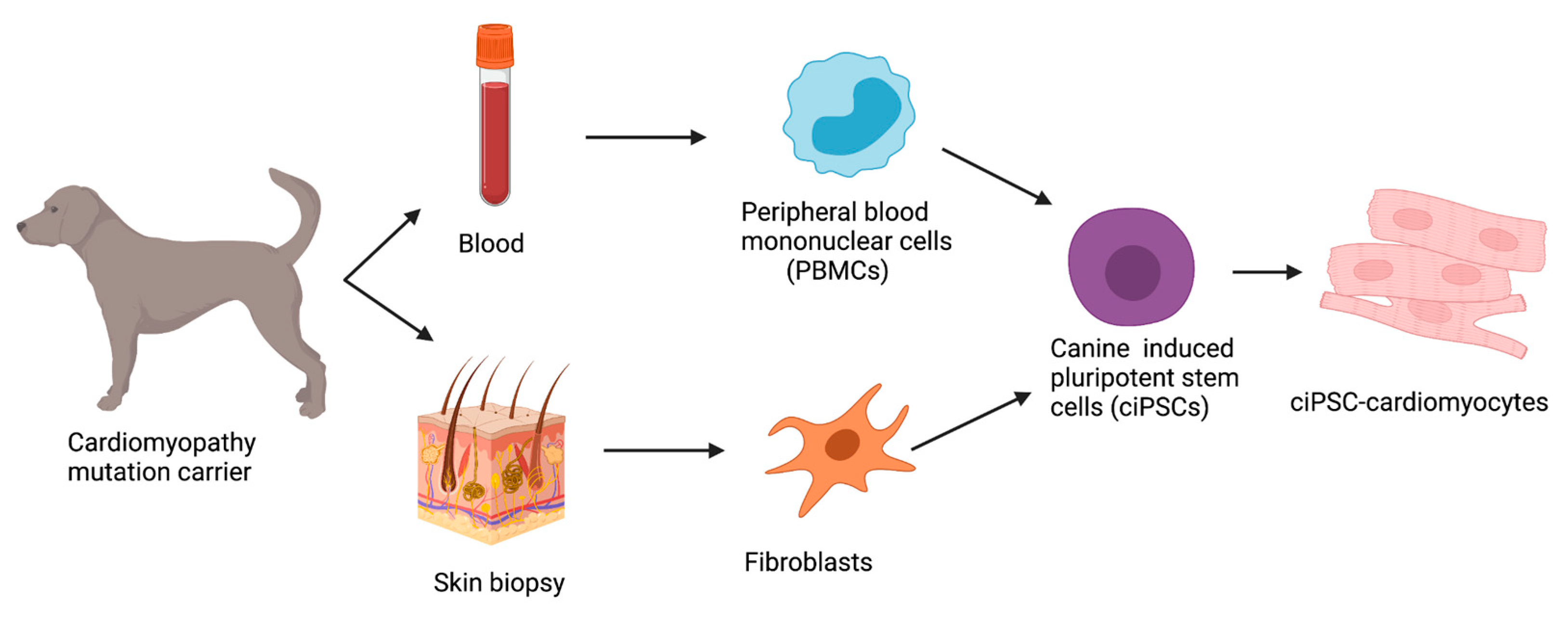
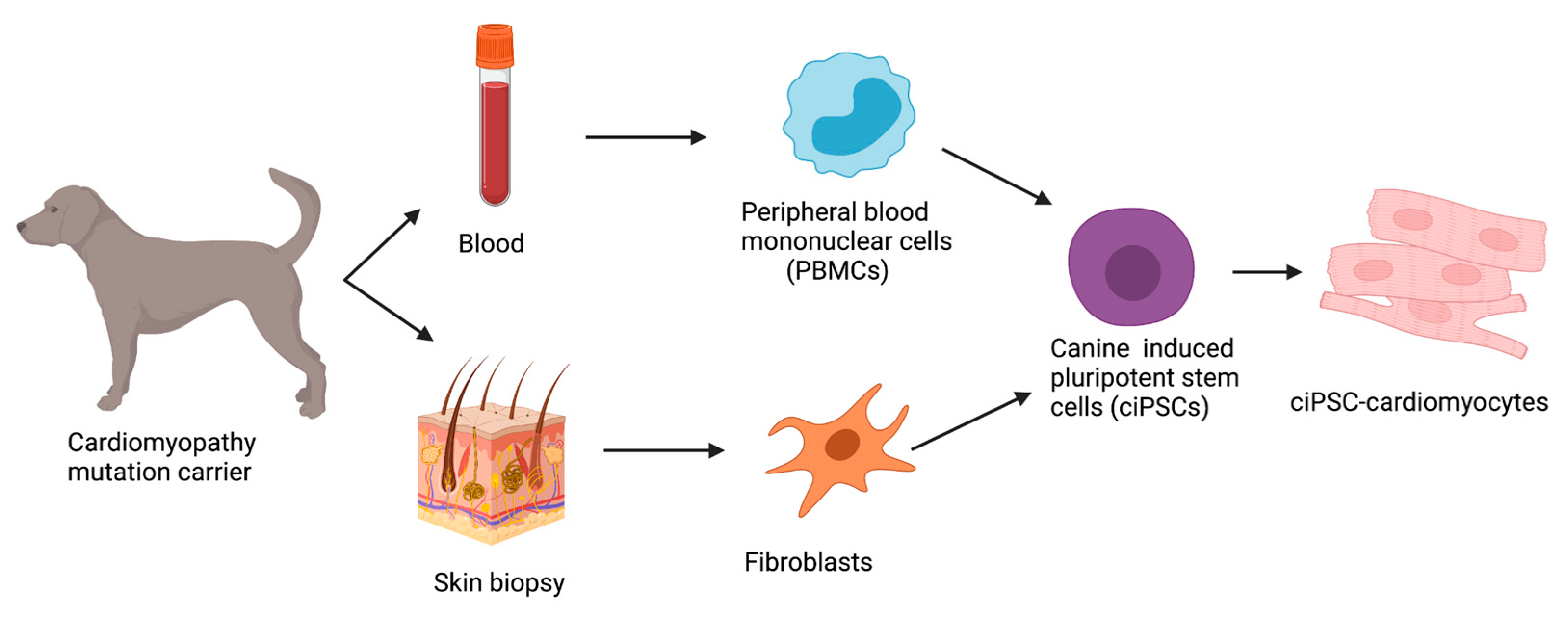
8. Disease-in-a-Dish
8.1. Reprogramming Strategies in ciPSCs
8.2. Challenges in ciPSCs
9. Discussion
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Simpson, S.; Edwards, J.; Mignan, T.; Cobb, M.; Mongan, N.P.; Rutland, C.S. Genetics of Human and Canine Dilated Cardiomyopathy. Int. J. Genom. 2015, 2015, 204823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.-D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Egenvall, A.; Bonnett, B.N.; Häggström, J. Heart Disease as a Cause of Death in Insured Swedish Dogs Younger Than 10 Years of Age. J. Veter. Intern. Med. 2006, 20, 894–903. [Google Scholar] [CrossRef]
- Raju, H.; Alberg, C.; Sagoo, G.S.; Burton, H.; Behr, E.R. Inherited cardiomyopathies. BMJ 2011, 343, d6966. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Mausberg, T.-B.; Wess, G.; Simak, J.; Keller, L.; Drögemüller, M.; Drögemüller, C.; Webster, M.; Stephenson, H.; Dukes-McEwan, J.; Leeb, T. A Locus on Chromosome 5 Is Associated with Dilated Cardiomyopathy in Doberman Pinschers. PLoS ONE 2011, 6, e20042. [Google Scholar] [CrossRef] [Green Version]
- Recchia, F.A.; Lionetti, V. Animal Models of Dilated Cardiomyopathy for Translational Research. Veter- Res. Commun. 2007, 31 (Suppl. S1), 35–41. [Google Scholar] [CrossRef]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Hasegawa, A.; Hosoi, Y.; Sugiura, K. Association between breed, gender and age in relation to cardiovascular disorders in insured dogs in Japan. J. Veter- Med Sci. 2016, 78, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chen, Y.; Liu, X.; Ye, L.; Yu, M.; Shen, Z.; Lei, W.; Hu, S. Uncovering Inherited Cardiomyopathy With Human Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2021, 9, 672039. [Google Scholar] [CrossRef]
- Deacon, D.C.; Happe, C.L.; Chen, C.; Tedeschi, N.; Manso, A.M.; Li, T.; Dalton, N.D.; Peng, Q.; Farah, E.N.; Gu, Y.; et al. Combinatorial interactions of genetic variants in human cardiomyopathy. Nat. Biomed. Eng. 2019, 3, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, S.; Fonfara, S.; Kitz, S.; Hetzel, U.; Kipar, A. Canine Dilated Cardiomyopathy: Diffuse Remodeling, Focal Lesions, and the Involvement of Macrophages and New Vessel Formation. Veter. Pathol. 2020, 57, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänselmann, A.; Veltmann, C.; Bauersachs, J.; Berliner, D. Dilated cardiomyopathies and non-compaction cardiomyopathy. Herz 2020, 45, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Elliott, P.M. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Cardiomyopathies. Circ. Res. 2017, 121, 711–721. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W. Precision Medicine and Dilated Cardiomyopathy. Precis. Med. 2020, 2204, 161–171. [Google Scholar] [CrossRef]
- McEwan, J.D. Canine dilated cardiomyopathy 2. Pathophysiology and treatment. Practice 2000, 22, 620–628. [Google Scholar] [CrossRef]
- Borgarelli, M.; Tarducci, A.; Tidholm, A.; Häggström, J. Canine Idiopathic Dilated Cardiomyopathy. Part II: Pathophysiology and therapy. Vet. J. 2001, 162, 182–195. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Kazzam, E.; Caidahl, K.; Hãllgren, R.; Gustafsson, R.; Landelius, J.; Waldenström, A. Non-invasive assessmenty of systolic left ventricular function in systemic sclerosis. Eur. Heart J. 1991, 12, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.; Ferraz, M.B.; Pessoa, A.P.; Fonseca, A.S.; Carvalho, A.C.; Hilario, M.O.; Atra, E. Symptomatic cardiac involvement in juvenile rheumatoid arthritis. Int. J. Cardiol. 1992, 34, 57–62. [Google Scholar] [CrossRef]
- Paradiso, M.; Gabrielli, F.; Masala, C.; Coppotelli, L.; Di Franco, M.; Paoletti, V.; Musca, A.; Mammarella, A. Evaluation of myocarial involvement in systemic lupus erythematosus by signal-averaged electrocardiography and echocardiography. Acta Cardiol. 2001, 56, 381–386. [Google Scholar] [CrossRef] [PubMed]
- McCauley, S.R.; Clark, S.D.; Quest, B.W.; Streeter, R.M.; Oxford, E.M. Review of canine dilated cardiomyopathy in the wake of diet-associated concerns. J. Anim. Sci. 2020, 98, skaa155. [Google Scholar] [CrossRef] [PubMed]
- Hantson, P. Mechanisms of toxic cardiomyopathy. Clin. Toxicol. 2018, 57, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.E.; Parnell, L.D.; Lai, C.-Q.; Rush, J.E.; Freeman, L.M. Investigation of diets associated with dilated cardiomyopathy in dogs using foodomics analysis. Sci. Rep. 2021, 11, 15881. [Google Scholar] [CrossRef]
- Van Vleet, J.F.; Ferrans, V.J. Myocardial diseases of animals. Am. J. Pathol. 1986, 124, 98–178. [Google Scholar]
- Reeves, W.C.; Marcuard, S.P.; Willis, S.E.; Movahed, A. Reversible Cardiomyopathy Due to Selenium Deficiency. J. Parenter. Enter. Nutr. 1989, 13, 663–665. [Google Scholar] [CrossRef]
- Stergiopoulos, K.; Lima, F.V. Peripartum cardiomyopathy-diagnosis, management, and long term implications. Trends Cardiovasc. Med. 2018, 29, 164–173. [Google Scholar] [CrossRef]
- Elliott, P. CARDIOMYOPATHY: Diagnosis and management of dilated cardiomyopathy. Heart 2000, 84, 106. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, H.; Fonfara, S.; López-Alvarez, J.; Cripps, P.; Dukes-McEwan, J. Screening for Dilated Cardiomyopathy in Great Danes in the United Kingdom. J. Veter. Intern. Med. 2012, 26, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Dukes-McEwan, J.; Borgarelli, M.; Tidholm, A.; Vollmar, A.C.; Häggström, J. Proposed Guidelines for the Diagnosis of Canine Idiopathic Dilated Cardiomyopathy. J. Veter. Cardiol. 2003, 5, 7–19. [Google Scholar] [CrossRef]
- Brownlie, S.E.; Cobb, M.A. Observations on the development of congestive heart failure in Irish wolfhounds with dilated cardiomyopathy. J. Small Anim. Pr. 1999, 40, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Tidholm, A.; Jönsson, L. Histologic Characterization of Canine Dilated Cardiomyopathy. Veter. Pathol. 2005, 42, 1–8. [Google Scholar] [CrossRef]
- Meurs, K.M.; Miller, M.W.; Wright, N.A. Clinical features of dilated cardiomyopathy in Great Danes and results of a pedigree analysis: 17 cases (1990-2000). J. Am. Veter. Med Assoc. 2001, 218, 729–732. [Google Scholar] [CrossRef]
- Calvert, C.A.; Pickus, C.W.; Jacobs, G.J.; Brown, J. Signalment, Survival, and Prognostic Factors in Doberman Pinschers with End-Stage Cardiomyopathy. J. Veter. Intern. Med. 1997, 11, 323–326. [Google Scholar] [CrossRef]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and Adipose Tissue Dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Lobo, L.; Carvalheira, J.; Canada, N.; Bussadori, C.; Gomes, J.L.; Faustino, A.M.R. Histologic Characterization of Dilated Cardiomyopathy in Estrela Mountain Dogs. Vet. Pathol. 2010, 47, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Davies, M. The cardiomyopathies: A review of terminology, pathology and pathogenesis. Histopathology 1984, 8, 363–393. [Google Scholar] [CrossRef]
- Sepehrkhouy, S.; Gho, J.M.; van Es, R.; Harakalova, M.; de Jonge, N.; Dooijes, D.; Vink, A. Distinct fibrosis pattern in desmosomal and phospholamban mutation carriers in hereditary cardiomyopathies. Heart Rhythm. 2017, 14, 1024–1032. [Google Scholar] [CrossRef]
- Dutton, E.; López-Alvarez, J. An update on canine cardiomyopathies—Is it all in the genes? J. Small Anim. Pr. 2018, 59, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Estrada, A.; Meurs, K.; Sleeper, M.; Vulpe, C.; Martyniuk, C.; Pacak, C. A review of the underlying genetics and emerging therapies for canine cardiomyopathies. J. Veter. Cardiol. 2021, 40, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Meurs, K.M.; Lahmers, S.; Keene, B.W.; White, S.N.; Oyama, M.A.; Mauceli, E.; Lindblad-Toh, K. A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Qual. Life Res. 2012, 131, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Bolfer, L.; Estrada, A.H.; Larkin, C.; Conlon, T.J.; Lourenco, F.; Taggart, K.; Suzuki-Hatano, S.; Pacak, C.A. Functional Consequences of PDK4 Deficiency in Doberman Pinscher Fibroblasts. Sci. Rep. 2020, 10, 3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taggart, K.; Estrada, A.; Thompson, P.; Lourenco, F.; Kirmani, S.; Suzuki-Hatano, S.; Pacak, C.A. PDK4 Deficiency Induces Intrinsic Apoptosis in Response to Starvation in Fibroblasts from Doberman Pinschers with Dilated Cardiomyopathy. BioResearch Open Access 2017, 6, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meurs, K.M.; Friedenberg, S.G.; Kolb, J.; Saripalli, C.; Tonino, P.; Woodruff, K.; Olby, N.J.; Keene, B.W.; Adin, D.B.; Yost, O.L.; et al. A missense variant in the titin gene in Doberman pinscher dogs with familial dilated cardiomyopathy and sudden cardiac death. Qual. Life Res. 2019, 138, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Wess, G.; Schulze, A.; Butz, V.; Simak, J.; Killich, M.; Keller, L.; Maeurer, J.; Hartmann, K. Prevalence of Dilated Cardiomyopathy in Doberman Pinschers in Various Age Groups. J. Veter. Intern. Med. 2010, 24, 533–538. [Google Scholar] [CrossRef]
- Meurs, K.M.; Stern, J.A.; Adin, D.; Keene, B.W.; De Francesco, T.C.; Tou, S.P. Assessment of PDK4 and TTN gene variants in 48 Doberman Pinschers with dilated cardiomyopathy. J. Am. Veter. Med Assoc. 2020, 257, 1041–1044. [Google Scholar] [CrossRef]
- Vollmar, A.C. The prevalence of cardiomyopathy in the Irish wolfhound: A clinical study of 500 dogs. J. Am. Anim. Hosp. Assoc. 2000, 36, 125–132. [Google Scholar] [CrossRef]
- Distl, O.; Vollmar, A.C.; Broschk, C.; Hamann, H.; Fox, P.R. Complex segregation analysis of dilated cardiomyopathy (DCM) in Irish wolfhounds. Heredity 2007, 99, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Vollmar, A.; Fox, P. Long-term Outcome of Irish Wolfhound Dogs with Preclinical Cardiomyopathy, Atrial Fibrillation, or Both Treated with Pimobendan, Benazepril Hydrochloride, or Methyldigoxin Monotherapy. J. Veter. Intern. Med. 2016, 30, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, S.; Dunning, M.; Brownlie, S.; Patel, J.; Godden, M.; Cobb, M.; Mongan, N.P.; Rutland, C.S. Multiple Genetic Associations with Irish Wolfhound Dilated Cardiomyopathy. BioMed Res. Int. 2016, 2016, 6374082. [Google Scholar] [CrossRef] [PubMed]
- Philipp, U.; Vollmar, A.; Häggström, J.; Thomas, A.; Distl, O. Multiple Loci Are Associated with Dilated Cardiomyopathy in Irish Wolfhounds. PLoS ONE 2012, 7, e36691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, O.; Friedenberg, S.G.; Jesty, S.A.; Olby, N.J.; Meurs, K.M. The R9H phospholamban mutation is associated with highly penetrant dilated cardiomyopathy and sudden death in a spontaneous canine model. Gene 2019, 697, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Young, H.S.; Ceholski, D.K.; Trieber, C.A. Deception in simplicity: Hereditary phospholamban mutations in dilated cardiomyopathy. Biochem. Cell Biol. 2015, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Werner, P.; Raducha, M.G.; Prociuk, U.; Sleeper, M.M.; Van Winkle, T.J.; Henthorn, P.S. A novel locus for dilated cardiomyopathy maps to canine chromosome 8. Genomics 2008, 91, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Yancy, C. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Fu, Y.; Eisen, H.J. Genetics of Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2018, 20, 121. [Google Scholar] [CrossRef]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: A Cardiomyopathy Clinic Doctor’s point of view. Hell. J. Cardiol. 2018, 59, 254–261. [Google Scholar] [CrossRef]
- Mestroni, L.; Rocco, C.; Gregori, D.; Sinagra, G.; Di Lenarda, A.; Miocic, S.; Vatta, M.; Pinamonti, B.; Muntoni, F.; Caforio, A.L.; et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. J. Am. Coll. Cardiol. 1999, 34, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Paldino, A.; De Angelis, G.; Merlo, M.; Gigli, M.; Ferro, M.D.; Severini, G.M.; Mestroni, L.; Sinagra, G. Genetics of Dilated Cardiomyopathy: Clinical Implications. Curr. Cardiol. Rep. 2018, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Jakobs, P.; Nauman, D.; Burgess, D.; Partain, J.; Litt, M. Coding Sequence Mutations Identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 Patients with Familial or Idiopathic Dilated Cardiomyopathy. Clin. Transl. Sci. 2008, 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Leach, S.B.; Briggs, M.; Hansen, L.; Johnson, G.S. Prevalence, geographic distribution, and impact on lifespan of a dilated cardiomyopathy-associated RNA-binding motif protein 20 variant in genotyped dogs. J. Veter. Cardiol. 2021, 40, 119–125. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Wilde, A.A.M.; Jongbloed, J.D.H. Recurrent and founder mutations in inherited cardiac diseases in the Netherlands. Neth. Hear. J. 2009, 17, 407–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Zwaag, P.A.; Van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; Van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; Van Lochem, L.T.; De Boer, R.A.; Hofstra, R.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Hear. Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Simpson, S.; Rutland, P.; Rutland, C.S. Genomic Insights into Cardiomyopathies: A Comparative Cross-Species Review. Veter-Sci. 2017, 4, 19. [Google Scholar] [CrossRef]
- Vollmar, A.; Fox, P.R.; Meurs, K.M.; Liu, S.-K. Dilated Cardiomyopathy in Juvenile Doberman Pinschers. J. Veter. Cardiol. 2003, 5, 23–27. [Google Scholar] [CrossRef]
- Yordy, J.; Kraus, C.; Hayward, J.J.; White, M.E.; Shannon, L.M.; Creevy, K.E.; Promislow, D.E.L.; Boyko, A.R. Body size, inbreeding, and lifespan in domestic dogs. Conserv. Genet. 2019, 21, 137–148. [Google Scholar] [CrossRef]
- Pelliccia, F.; Limongelli, G.; Autore, C.; Gimeno-Blanes, J.R.; Basso, C.; Elliott, P. Sex-related differences in cardiomyopathies. Int. J. Cardiol. 2019, 286, 239–243. [Google Scholar] [CrossRef]
- Halliday, B.P.; Gulati, A.; Ali, A.; Newsome, S.; Lota, A.; Tayal, U.; Prasad, S.K. Sex- and age-based differences in the natural history and outcome of dilated cardiomyopathy: Sex- and age-based differences in DCM. Eur. J. Heart Fail. 2018, 20, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Seidman, C.E. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amario, D.; Camilli, M.; Migliaro, S.; Canonico, F.; Galli, M.; Arcudi, A.; Montone, R.A.; Borovac, J.A.; Crea, F.; Savarese, G. Sex-Related Differences in Dilated Cardiomyopathy with a Focus on Cardiac Dysfunction in Oncology. Curr. Cardiol. Rep. 2020, 22, 102. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Ast, J.F.H.-V.; van der Kooi, A.J.; van Tintelen, J.P.; Berg, M.P.V.D.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Hear. Fail. 2013, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes—A systematic review. Pflüg Arch. Eur. J. Physiol. 2018, 471, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Micheu, M.M.; Rosca, A.-M. Patient-specific induced pluripotent stem cells as “disease-in-a-dish” models for inherited cardiomyopathies and channelopathies—15 years of research. World J. Stem Cells 2021, 13, 281–303. [Google Scholar] [CrossRef]
- Sampaio-Pinto, V.; Janssen, J.; Chirico, N.; Serra, M.; Alves, P.M.; Doevendans, P.A.; Voets, I.K.; Sluijter, J.P.G.; van Laake, L.W.; van Mil, A. A Roadmap to Cardiac Tissue-Engineered Construct Preservation: Insights from Cells, Tissues, and Organs. Adv. Mater. 2021, 33, 2008517. [Google Scholar] [CrossRef]
- Montero, P.; Flandes-Iparraguirre, M.; Musquiz, S.; Araluce, M.P.; Plano, D.; Sanmartín, C.; Orive, G.; Gavira, J.J.; Prosper, F.; Mazo, M.M. Cells, Materials, and Fabrication Processes for Cardiac Tissue Engineering. Front. Bioeng. Biotechnol. 2020, 8, 955. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Lu, A.T.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Lim, A.R.; Brooke, R.T.; Raj, K.; Serres-Armero, A.; Dreger, D.L.; et al. DNA methylation clocks for dogs and humans. Proc. Natl. Acad. Sci. USA 2022, 119, e2120887119. [Google Scholar] [CrossRef]
- Koh, S.; Piedrahita, J.A. Generation of Induced Pluripotent Stem Cells (iPSCs) from Adult Canine Fibroblasts. In Cell Reprogramming; Verma, P.J., Sumer, H., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1330, pp. 69–78. [Google Scholar] [CrossRef]
- Betts, D.H.; Tobias, I.C. Canine Pluripotent Stem Cells: Are They Ready for Clinical Applications? Front. Veter. Sci. 2015, 2, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, A.; Barsby, T.; Guest, D.J. Derivation of Canine Induced Pluripotent Stem Cells. Reprod. Domest. Anim. 2015, 50, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Nakada, A.; Hashimoto, Y.; Shigeno, K.; Shionoya, Y.; Nakamura, T. Generation of canine induced pluripotent stem cells by retroviral transduction and chemical inhibitors. Mol. Reprod. Dev. 2009, 77, 2. [Google Scholar] [CrossRef]
- Kim, S.; Kim, B.; Kim, J.; Kim, D.-H.; Lee, S.; Lee, D.-S.; Lee, H. L-myc Gene Expression in Canine Fetal Fibroblasts Promotes Self-Renewal Capacity but Not Tumor Formation. Cells 2021, 10, 1980. [Google Scholar] [CrossRef]
- Koh, S.; Thomas, R.; Tsai, S.; Bischoff, S.; Lim, J.-H.; Breen, M.; Olby, N.; Piedrahita, J.A. Growth Requirements and Chromosomal Instability of Induced Pluripotent Stem Cells Generated from Adult Canine Fibroblasts. Stem Cells Dev. 2013, 22, 951–963. [Google Scholar] [CrossRef]
- Lee, A.S.; Xu, D.; Plews, J.R.; Nguyen, P.K.; Nag, D.; Lyons, J.K.; Han, L.; Hu, S.; Lan, F.; Liu, J.; et al. Preclinical Derivation and Imaging of Autologously Transplanted Canine Induced Pluripotent Stem Cells. J. Biol. Chem. 2011, 286, 32697–32704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, D.V.; Bhaskar, S.; Sheshadri, P.; Joshi, C.G.; Patel, D.; Kumar, A. Positioning canine induced pluripotent stem cells (iPSCs) in the reprogramming landscape of naïve or primed state in comparison to mouse and human iPSCs. Life Sci. 2020, 264, 118701. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, N.; Bressan, F.; Roballo, K.; Meirelles, F.; Xavier, P.; Fukumasu, H.; Williams, C.; Breen, M.; Koh, S.; Sper, R.; et al. Generation of LIF-independent induced pluripotent stem cells from canine fetal fibroblasts. Theriogenology 2017, 92, 75–82. [Google Scholar] [CrossRef]
- Luo, J.; Suhr, S.T.; Chang, E.A.; Wang, K.; Ross, P.J.; Nelson, L.L.; Venta, P.J.; Knott, J.G.; Cibelli, J.B. Generation of Leukemia Inhibitory Factor and Basic Fibroblast Growth Factor-Dependent Induced Pluripotent Stem Cells from Canine Adult Somatic Cells. Stem Cells Dev. 2011, 20, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, D.J.; Ovchinnikov, D.; Wolvetang, E. Generation and Characterization of LIF-dependent Canine Induced Pluripotent Stem Cells from Adult Dermal Fibroblasts. Stem Cells Dev. 2012, 21, 2288–2297. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Nishimura, T.; Yodoe, K.; Kanegi, R.; Tsujimoto, Y.; Alam, E.; Kuramochi, M.; Kuwamura, M.; Ohtaka, M.; Nishimura, K.; et al. Generation of Footprint-Free Canine Induced Pluripotent Stem Cells Using Auto-Erasable Sendai Virus Vector. Stem Cells Dev. 2018, 27, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Tsukamoto, M.; Tanaka, M.; Kuwamura, M.; Ohtaka, M.; Nishimura, K.; Nakanishi, M.; Sugiura, K.; Hatoya, S. Efficient Reprogramming of Canine Peripheral Blood Mononuclear Cells into Induced Pluripotent Stem Cells. Stem Cells Dev. 2021, 30, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Johnson, V.; Regan, D.; Wheat, W.; Webb, S.; Koch, P.; Dow, S. Safety and immune regulatory properties of canine induced pluripotent stem cell-derived mesenchymal stem cells. Stem Cell Res. 2017, 25, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, M.; Kimura, K.; Tanaka, M.; Kuwamura, M.; Ohtaka, M.; Nakanishi, M.; Sugiura, K.; Hatoya, S. Generation of Footprint-Free Canine Induced Pluripotent Stem Cells from Peripheral Blood Mononuclear Cells Using Sendai Virus Vector. Mol. Reprod. Dev. 2020, 87, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Hwang, S.-U.; Yoon, J.D.; Jeong, Y.W.; Kim, E.; Hyun, S.-H. Optimized Approaches for the Induction of Putative Canine Induced Pluripotent Stem Cells from Old Fibroblasts Using Synthetic RNAs. Animals 2020, 10, 1848. [Google Scholar] [CrossRef]
- Yoshimatsu, S.; Edamura, K.; Yoshii, Y.; Iguchi, A.; Kondo, H.; Shibuya, H.; Sato, T.; Shiozawa, S.; Okano, H. Non-viral derivation of a transgene-free induced pluripotent stem cell line from a male beagle dog. Stem Cell Res. 2021, 53, 102375. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2014, 33, 58–63. [Google Scholar] [CrossRef]
- Debowski, K.; Warthemann, R.; Lentes, J.; Salinas-Riester, G.; Dressel, R.; Langenstroth, D.; Behr, R. Non-Viral Generation of Marmoset Monkey iPS Cells by a Six-Factor-in-One-Vector Approach. PLoS ONE 2015, 10, e0118424. [Google Scholar] [CrossRef]
- Yoshimatsu, S.; Nakajima, M.; Iguchi, A.; Sanosaka, T.; Sato, T.; Nakamura, M.; Nakajima, R.; Arai, E.; Ishikawa, M.; Imaizumi, K.; et al. Non-viral Induction of Transgene-free iPSCs from Somatic Fibroblasts of Multiple Mammalian Species. Stem Cell Rep. 2021, 16, 754–770. [Google Scholar] [CrossRef]
- Steinle, H.; Weber, M.; Behring, A.; Mau-Holzmann, U.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Generation of iPSCs by Nonintegrative RNA-Based Reprogramming Techniques: Benefits of Self-Replicating RNA versus Synthetic mRNA. Stem Cells Int. 2019, 2019, 7641767. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, B.; Amanullah, A.; Shafarenko, M.; Liebermann, D.A. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene 2002, 21, 3414–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, B.; Liebermann, D.A. The proto-oncogene c-myc and apoptosis. Oncogene 1998, 17, 3351–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Tobias, I.C.; Kao, M.-M.C.; Parmentier, T.; Hunter, H.; LaMarre, J.; Betts, D.H. Targeted expression profiling reveals distinct stages of early canine fibroblast reprogramming are regulated by 2-oxoglutarate hydroxylases. Stem Cell Res. Ther. 2020, 11, 528. [Google Scholar] [CrossRef]
- Van Steenbeek, F.G.; Hytönen, M.; Leegwater, P.A.J.; Lohi, H. The canine era: The rise of a biomedical model. Anim. Genet. 2016, 47, 519–527. [Google Scholar] [CrossRef]
- Lennermann, D.; Backs, J.; Hoogenhof, M.M.G.V.D. New Insights in RBM20 Cardiomyopathy. Curr. Hear. Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Sacchetto, C.; Vitiello, L.; De Windt, L.J.; Rampazzo, A.; Calore, M. Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures. Int. J. Mol. Sci. 2020, 21, 3404. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Canine | Human [60,61] | ||
|---|---|---|---|
| Sarcomeric | TTN (Dobbermann [45]) | TTN MYH6 MYBPC3 TNNT1 MYL3 MYL2 | ACTC1 MYH7 TNNC1 TPM1 TNNI3 TNNT2 |
| Z-disc | MYPN ANKRD1 CSRP3 ACTN2 | TCAP NEBL | |
| Cytoskeleton | DMD (German short-haired pointer [1]) ARHGAP8 (Irish wolfhound [52]) | DMD VCL DES ILK LDB3 LAM4 CRYAB | FLNC SGCD SGCB SGCA SGCG PDLIM3 |
| Nuclear envelope | LMNA TMPO EMD | SYNE1 SYNE2 | |
| Sarcoplasmic reticulum | PLN (Welsh springer spaniel [53]) PDE3B (Irish wolfhound [52]) | PLN | |
| RNA binding | RBM20 (Schnauzer [63]) | RBM20 | |
| Other | PDK4 (Dobbermann [42]) STRN (Boxer [40]) FSTL5 (Irish wolfhound [52]) | SCN5A BAG3 DSC2 DSP PSEN1 | ABCC9 DSG2 EYA4 TAZ PSEN2 |
| Reprogramming Strategy | Method |
|---|---|
| Retroviral transduction | Viral, genomic integrating [80,81,88,89,90,91,92] |
| Lentiviral transduction | Viral, genomic integrating [86,93,94] |
| Sendai viral transduction | Viral, non-integrating [84,85,95,96] |
| Synthetic RNA transfection | Nonviral, non-integrating [97] |
| Episomal plasmids | Nonviral, non-integrating [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaar-Humphreys, K.R.; Spanjersberg, T.C.F.; Santarelli, G.; Grinwis, G.C.M.; Szatmári, V.; Roelen, B.A.J.; Vink, A.; van Tintelen, J.P.; Asselbergs, F.W.; Fieten, H.; et al. Genetic Basis of Dilated Cardiomyopathy in Dogs and Its Potential as a Bidirectional Model. Animals 2022, 12, 1679. https://doi.org/10.3390/ani12131679
Gaar-Humphreys KR, Spanjersberg TCF, Santarelli G, Grinwis GCM, Szatmári V, Roelen BAJ, Vink A, van Tintelen JP, Asselbergs FW, Fieten H, et al. Genetic Basis of Dilated Cardiomyopathy in Dogs and Its Potential as a Bidirectional Model. Animals. 2022; 12(13):1679. https://doi.org/10.3390/ani12131679
Chicago/Turabian StyleGaar-Humphreys, Karen R., Talitha C. F. Spanjersberg, Giorgia Santarelli, Guy C. M. Grinwis, Viktor Szatmári, Bernard A. J. Roelen, Aryan Vink, J. Peter van Tintelen, Folkert W. Asselbergs, Hille Fieten, and et al. 2022. "Genetic Basis of Dilated Cardiomyopathy in Dogs and Its Potential as a Bidirectional Model" Animals 12, no. 13: 1679. https://doi.org/10.3390/ani12131679
APA StyleGaar-Humphreys, K. R., Spanjersberg, T. C. F., Santarelli, G., Grinwis, G. C. M., Szatmári, V., Roelen, B. A. J., Vink, A., van Tintelen, J. P., Asselbergs, F. W., Fieten, H., Harakalova, M., & van Steenbeek, F. G. (2022). Genetic Basis of Dilated Cardiomyopathy in Dogs and Its Potential as a Bidirectional Model. Animals, 12(13), 1679. https://doi.org/10.3390/ani12131679