
Machine Learning-Based Co-Expression Network Analysis Unravels Potential Fertility-Related Genes in Beef Cows


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Retrieval and Quality Control
2.2. Gene Expression Normalization and Supervised Machine Learning
2.3. Gene Co-Expression Network Analysis
2.4. Functional Over-Representation Analysis
3. Results
3.1. Identification of Potential Biomarker Genes through ML
3.2. Gene Network Analysis
3.3. Functional Over-Representation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, Y.; Peñagaricano, F. Unravelling the genomic architecture of bull fertility in Holstein cattle. BMC Genet. 2016, 17, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.F.; Schnabel, R.D.; Sutovsky, P. Review: Genomics of bull fertility. Animal 2018, 12, s172–s183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercadante, V.R.G.; Dias, N.W.; Timlin, C.L.; Pancini, S. 375 Economic consequences of pregnancy loss in beef cattle. J. Anim. Sci. 2020, 98, 124. [Google Scholar] [CrossRef]
- Bach, À.; Bach, À. Effects of nutrition and genetics on fertility in dairy cows. Reprod. Fertil. Dev. 2019, 31, 40–54. [Google Scholar] [CrossRef]
- Berry, D.P.; Wall, E.; Pryce, J.E. Genetics and genomics of reproductive performance in dairy and beef cattle. Animal 2014, 8, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorey, S.E.; Biase, F.H. Beef heifer fertility: Importance of management practices and technological advancements. J. Anim. Sci. Biotechnol. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.S. Identification of genes associated with reproductive function in dairy cattle. Anim. Reprod. 2018, 15, 923–932. [Google Scholar] [CrossRef]
- Olasege, B.S.; Tahir, M.S.; Gouveia, G.C.; Kour, J.; Porto-Neto, L.R.; Hayes, B.J.; Fortes, M.R.S. Genetic parameter estimates for male and female fertility traits using genomic data to improve fertility in Australian beef cattle. Anim. Prod. Sci. 2021, 61, 1863. [Google Scholar] [CrossRef]
- Ponsart, C.; Le Bourhis, D.; Knijn, H.; Fritz, S.; Guyader-Joly, C.; Otter, T.; Lacaze, S.; Charreaux, F.; Schibler, L.; Dupassieux, D.; et al. Reproductive technologies and genomic selection in dairy cattle. Reprod. Fertil. Dev. 2013, 26, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Moorey, S.E.; Walker, B.N.; Elmore, M.F.; Elmore, J.B.; Rodning, S.P.; Biase, F.H. Rewiring of gene expression in circulating white blood cells is associated with pregnancy outcome in heifers (Bos taurus). Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Phillips, K.M.; Read, C.C.; Kriese-Anderson, L.A.; Rodning, S.P.; Brandebourg, T.D.; Biase, F.H.; Marks, M.L.; Elmore, J.B.; Stanford, M.K.; Dyce, P.W. Plasma metabolomic profiles differ at the time of artificial insemination based on pregnancy outcome, in Bos taurus beef heifers. Sci. Rep. 2018, 8, 13196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cánovas, A.; Reverter, A.; DeAtley, K.L.; Ashley, R.L.; Colgrave, M.L.; Fortes, M.R.S.; Islas-Trejo, A.; Lehnert, S.; Porto-Neto, L.; Rincón, G.; et al. Multi-tissue omics analyses reveal molecular regulatory networks for puberty in composite beef cattle. PLoS ONE 2014, 9, e102551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Reverter, A.; Cánovas, A.; Venus, B.; Islas-Trejo, A.; Porto-Neto, L.R.; Lehnert, S.A.; Medrano, J.F.; Moore, S.S.; Fortes, M.R.S. Global differential gene expression in the pituitary gland and the ovaries of pre- and postpubertal Brahman heifers. J. Anim. Sci. 2017, 95, 599–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, T.W.; Burns, G.W.; Moraes, J.G.N.; Moss, J.I.; Denicol, A.C.; Dobbs, K.B.; Ortega, M.S.; Hansen, P.J.; Wehrman, M.E.; Neibergs, H.; et al. Identification of beef heifers with superior uterine capacity for pregnancy. Biol. Reprod. 2016, 95, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gómez, E.; Salvetti, P.; Gatien, J.; Carrocera, S.; Martín-González, D.; Muñoz, M. Blood plasma metabolomics predicts pregnancy in Holstein cattle transferred with fresh and vitrified/warmed embryos produced in vitro. J. Proteome Res. 2020, 19, 1169–1182. [Google Scholar] [CrossRef]
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.C.; Sibille, E. Beyond modules and hubs: The potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 2014, 13, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Hudson, N.J.; Dalrymple, B.P.; Reverter, A. Beyond differential expression: The quest for causal mutations and effector molecules. BMC Genom. 2012, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Jackson, S.A. Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef]
- Rabaglino, M.B.; Kadarmideen, H.N. Machine learning approach to integrated endometrial transcriptomic datasets reveals biomarkers predicting uterine receptivity in cattle at seven days after estrous. Sci. Rep. 2020, 10, 16981. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Suárez-Vega, A.; Cánovas, A. Weighted gene correlation network meta-analysis reveals functional candidate genes associated with high- and sub-fertile reproductive performance in beef cattle. Genes 2020, 11, 543. [Google Scholar] [CrossRef]
- Martins, T.; Sponchiado, M.; Silva, F.A.C.C.; Estrada-Cortés, E.; Hansen, P.J.; Peñagaricano, F.; Binelli, M. Progesterone-dependent and progesterone-independent modulation of luminal epithelial transcription to support pregnancy in cattle. Physiol. Genom. 2022, 54, 71–85. [Google Scholar] [CrossRef]
- Ewels, P. SRA-Explorer. Available online: https://sra-explorer.info/ (accessed on 13 May 2022).
- Andrews, S. FASTQC. A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 January 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. 2018. Available online: https://www.R-project.org (accessed on 6 January 2022).
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Di Pietro, A.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, M.; Vittrant, B.; Martin-Magniette, M.L.; Scott Boyer, M.P.; Perin, O.; Bergeron, A.; Fradet, Y.; Droit, A. Large-scale automatic feature selection for biomarker discovery in high-dimensional OMICS data. Front. Genet. 2019, 10, 452. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Reverter, A.; Chan, E.K.F. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Diniz, W.J.S.; Crouse, M.S.; Cushman, R.A.; McLean, K.J.; Caton, J.S.; Dahlen, C.R.; Reynolds, L.P.; Ward, A.K. Cerebrum, liver, and muscle regulatory networks uncover maternal nutrition effects in developmental programming of beef cattle during early pregnancy. Sci. Rep. 2021, 11, 2771. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Goenawan, I.H.; Bryan, K.; Lynn, D.J. DyNet: Visualization and analysis of dynamic molecular interaction networks. Bioinformatics 2016, 32, 2713–2715. [Google Scholar] [CrossRef] [Green Version]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Pryce, J.E.; Veerkamp, R.F. The incorporation of fertility indices in genetic improvement programmes. BSAP Occas. Publ. 2001, 26, 237–249. [Google Scholar] [CrossRef]
- Spencer, T.E. Early pregnancy: Concepts, challenges, and potential solutions. Anim. Front. 2013, 3, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Binelli, M.; Scolari, S.C.; Pugliesi, G.; Van Hoeck, V.; Gonella-Diaza, A.M.; Andrade, S.C.S.; Gasparin, G.R.; Coutinho, L.L. The transcriptome signature of the receptive bovine uterus determined at early gestation. PLoS ONE 2015, 10, e0122874. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, G.; Pedersen, H.S.; Rabaglino, M.B.; Hyttel, P.; Callesen, H.; Kadarmideen, H.N. Characterization of the endometrial transcriptome in early diestrus influencing pregnancy status in dairy cattle after transfer of in vitro-produced embryos. Physiol. Genom. 2020, 52, 269–279. [Google Scholar] [CrossRef]
- Estrada-Cortés, E.; Ortiz, W.G.; Chebel, R.C.; Jannaman, E.A.; Moss, J.I.; De Castro, F.C.; Zolini, A.M.; Staples, C.R.; Hansen, P.J. Embryo and cow factors affecting pregnancy per embryo transfer for multiple-service, lactating Holstein recipients. Transl. Anim. Sci. 2019, 3, 60–65. [Google Scholar] [CrossRef] [PubMed]
- França, M.R.; da Silva, M.I.S.; Pugliesi, G.; Van Hoeck, V.; Binelli, M. Evidence of endometrial amino acid metabolism and transport modulation by peri-ovulatory endocrine profiles driving uterine receptivity. J. Anim. Sci. Biotechnol. 2017, 8, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, B.; Oliveira, M.L.; Pugliesi, G.; Batista, E.O.S.; Binelli, M. Cytobrush: A tool for sequential evaluation of gene expression in bovine endometrium. Reprod. Domest. Anim. 2017, 52, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.P.; Zhang, Q.; McGowan, S.; Buckle, A.M.; Silverman, G.A.; Wong, W.; Rosado, C.J.; Langendorf, C.G.; Pike, R.N.; Bird, P.I.; et al. An overview of the serpin superfamily. Genome Biol. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanrattana, W.; Maas, C.; de Maat, S. SERPINs—From trap to treatment. Front. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Bédard, J.; Brûlé, S.; Price, C.A.; Silversides, D.W.; Lussier, J.G. Serine protease inhibitor-E2 (SERPINE2) is differentially expressed in granulosa cells of dominant follicle in cattle. Mol. Reprod. Dev. 2003, 64, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Li, H.; Cao, D.; Chen, Y. The development of endometrial hyperplasia in aged PD-1-deficient female mice. Diagn. Pathol. 2014, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Taglauer, E.S.; Trikhacheva, A.S.; Slusser, J.G.; Petroff, M.G. Expression and function of PDCD1 at the human maternal-fetal interface. Biol. Reprod. 2008, 79, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, S.E.; Griffin, B.A.; Elmore, M.F.; Kriese-Anderson, L.; Elmore, J.B.; Dyce, P.W.; Rodning, S.P.; Biase, F.H. Transcriptome profiles in peripheral white blood cells at the time of artificial insemination discriminate beef heifers with different fertility potential. BMC Genom. 2018, 19, 129. [Google Scholar] [CrossRef] [Green Version]
- Kishi, T.; Mayanagi, T.; Iwabuchi, S.; Akasaka, T.; Sobue, K. Myocardin-related transcription factor A (MRTF-A) activity-dependent cell adhesion is correlated to focal adhesion kinase (FAK) activity. Oncotarget 2016, 7, 72113–72130. [Google Scholar] [CrossRef]
- Di-Luoffo, M.; Daems, C.; Bergeron, F.; Tremblay, J.J. Novel targets for the transcription factors MEF2 in MA-10 Leydig cells. Biol. Reprod. 2015, 93, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Rubin, L.P.; Gong, X. Translational Physiology: MEF2 transcription factors in human placenta and involvement in cytotrophoblast invasion and differentiation. Physiol. Genom. 2018, 50, 10. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, L.S.; Sutherland, L.B.; Liu, Z.; Grinnell, F.; Kamm, K.E.; Schneider, J.W.; Olson, E.N.; Small, E.M. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl. Acad. Sci. USA 2013, 42, 16850–16855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtz, M.L.; Misra, R.P. Serum response factor is required for cell contact maintenance but dispensable for proliferation in visceral yolk sac endothelium. BMC Dev. Biol. 2011, 11, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scolari, S.C.; Pugliesi, G.; Strefezzi, R.D.F.; Andrade, S.C.D.S.; Coutinho, L.L.; Binelli, M. Dynamic remodeling of endometrial extracellular matrix regulates embryo receptivity in cattle. Reproduction 2017, 153, 49–61. [Google Scholar] [CrossRef]
- Banerjee, P.; Rodning, S.P.; Diniz, W.J.S.; Dyce, P.W. Co-expression network and integrative analysis of metabolome and transcriptome uncovers biological pathways for fertility in beef heifers. Metabolites 2022, 12, 708. [Google Scholar] [CrossRef]
- Calamita, P.; Gatti, G.; Miluzio, A.; Scagliola, A.; Biffo, S. Translating the game: Ribosomes as active players. Front. Genet. 2018, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Gershon, E.; Zeisel, A.; Jacob-Hirsch, J.; Neeman, M.; Winterhager, E.; Rechavi, G.; Domany, E.; Dekel, N. Blastocyst implantation failure relates to impaired translational machinery gene expression. Reproduction 2014, 148, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Xin, L.; Xu, B.; Ma, L.; Hou, Q.; Ye, M.; Meng, S.; Ding, X.; Ge, W. Proteomics study reveals that the dysregulation of focal adhesion and ribosome contribute to early pregnancy loss. PROTEOMICS—Clin. Appl. 2016, 10, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef]
- Klohonatz, K.M.; Nulton, L.C.; Hess, A.M.; Bouma, G.J.; Bruemmer, J.E. The role of embryo contact and focal adhesions during maternal recognition of pregnancy. PLoS ONE 2019, 14, e0213322. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, R.C.; Burghardt, J.R.; Taylor, J.D.; Reeder, A.T.; Nguen, B.T.; Spencer, T.E.; Bayless, K.J.; Johnson, G.A. Enhanced focal adhesion assembly reflects increased mechanosensation and mechanotransduction at maternal–conceptus interface and uterine wall during ovine pregnancy. Reproduction 2009, 137, 567–582. [Google Scholar] [CrossRef] [PubMed]

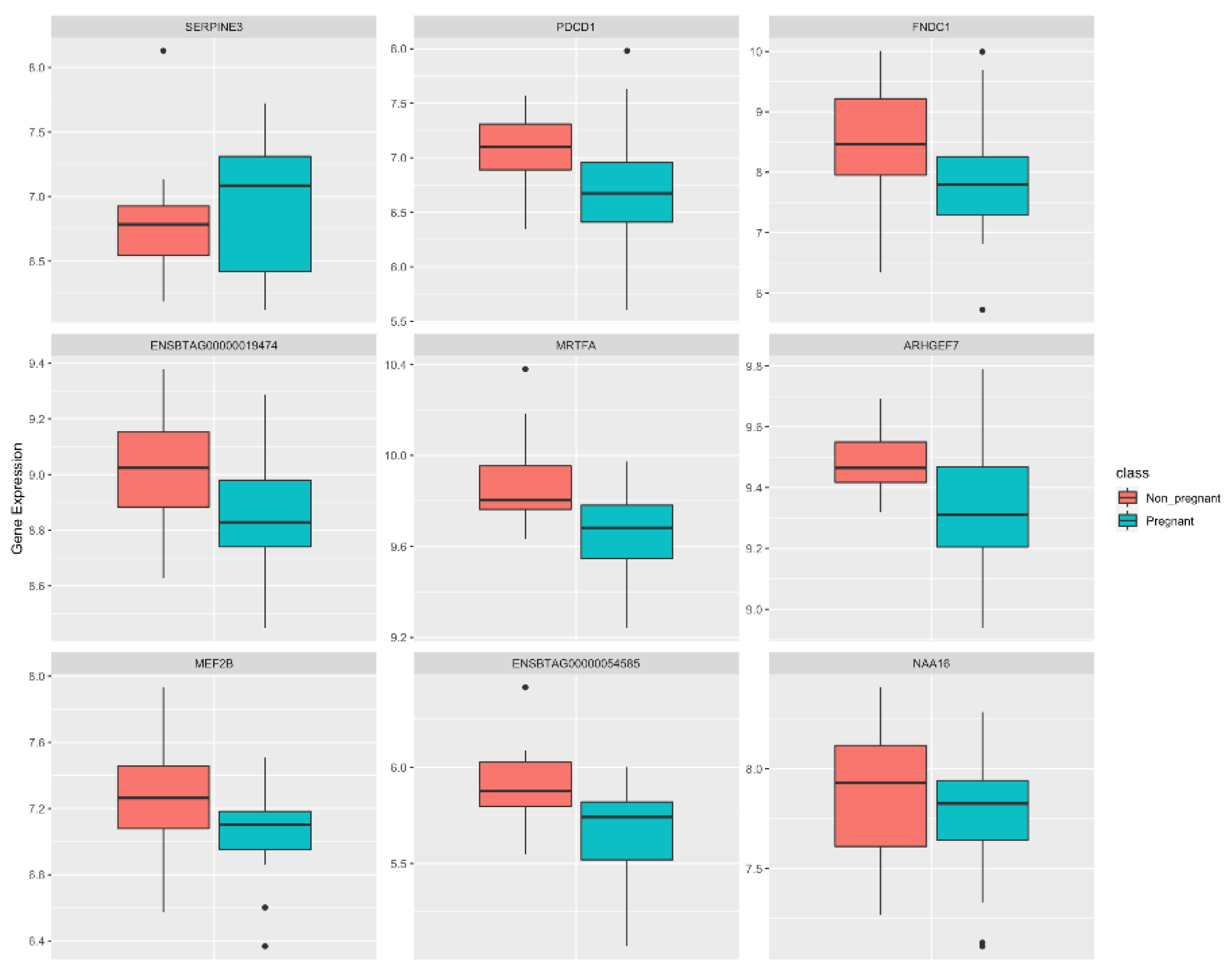



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl Gene ID | Gene Symbol | Nodes in NP | Nodes in P | DIFFK | z-Score * |
|---|---|---|---|---|---|
| ENSBTAG00000001818 | MEF2B | 794 | 169 | 0.88983 | 45.428 |
| ENSBTAG00000003938 | FNDC1 | 670 | 342 | 0.62088 | 31.6887 |
| ENSBTAG00000005284 | SERPINE3 | 646 | 401 | 0.55219 | 28.1798 |
| ENSBTAG00000019474 | ENSBTAG00000019474 | 577 | 507 | 0.39619 | 20.2104 |
| ENSBTAG00000002630 | MRTFA | 373 | 127 | 0.38698 | 19.74 |
| ENSBTAG00000038251 | NAA16 | 384 | 1488 | −0.4864 | −24.876 |
| ENSBTAG00000020726 | ARHGEF7 | 331 | 1534 | −0.5831 | −29.818 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diniz, W.J.S.; Banerjee, P.; Rodning, S.P.; Dyce, P.W. Machine Learning-Based Co-Expression Network Analysis Unravels Potential Fertility-Related Genes in Beef Cows. Animals 2022, 12, 2715. https://doi.org/10.3390/ani12192715
Diniz WJS, Banerjee P, Rodning SP, Dyce PW. Machine Learning-Based Co-Expression Network Analysis Unravels Potential Fertility-Related Genes in Beef Cows. Animals. 2022; 12(19):2715. https://doi.org/10.3390/ani12192715
Chicago/Turabian StyleDiniz, Wellison J. S., Priyanka Banerjee, Soren P. Rodning, and Paul W. Dyce. 2022. "Machine Learning-Based Co-Expression Network Analysis Unravels Potential Fertility-Related Genes in Beef Cows" Animals 12, no. 19: 2715. https://doi.org/10.3390/ani12192715