

Research on the Gut Microbiota of Hainan Black Goat
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Grouping Status
2.2. DNA Extraction
2.3. PCR Amplification and Miseq Sequencing
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Analysis of DNA Sequences
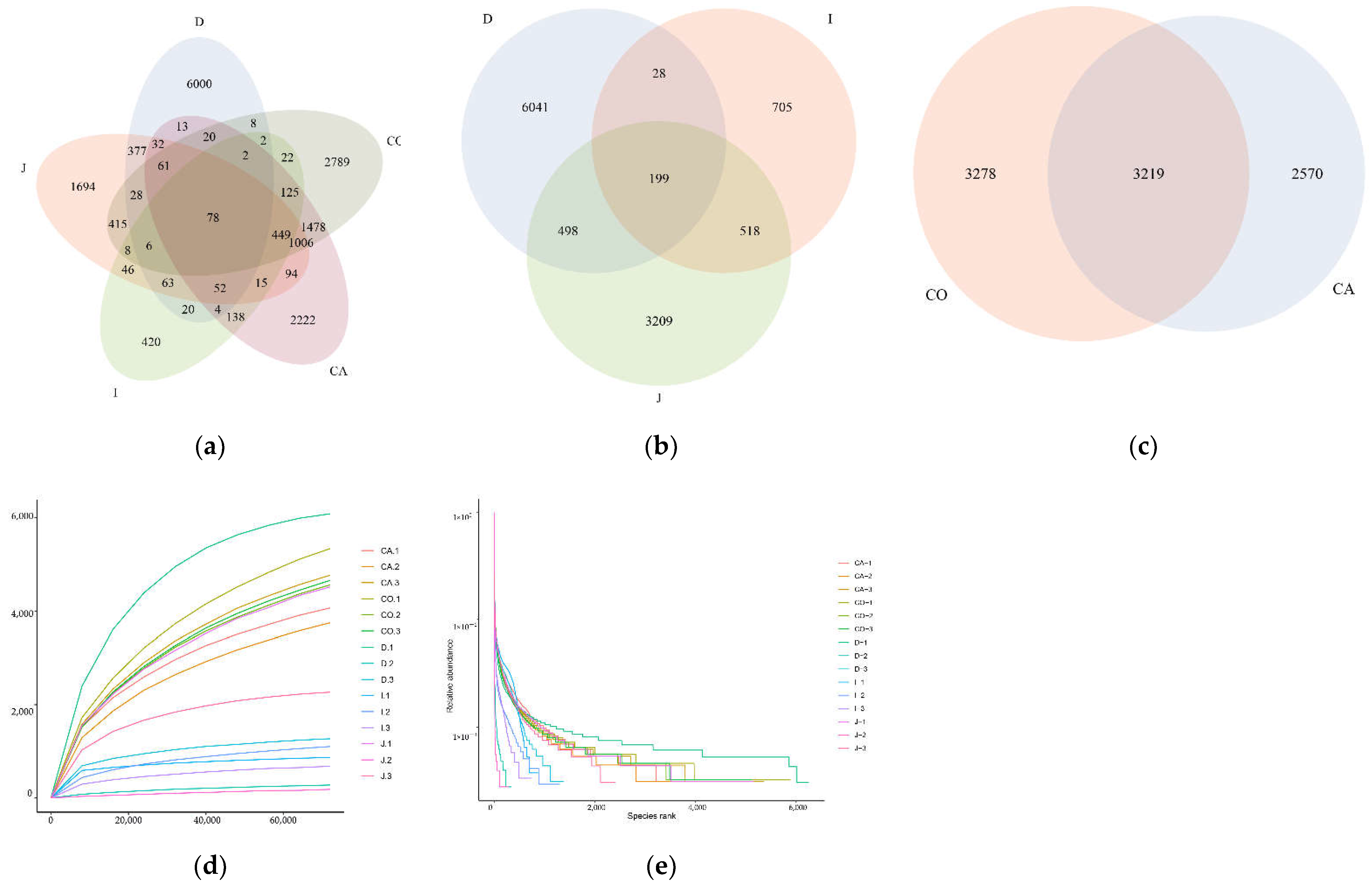
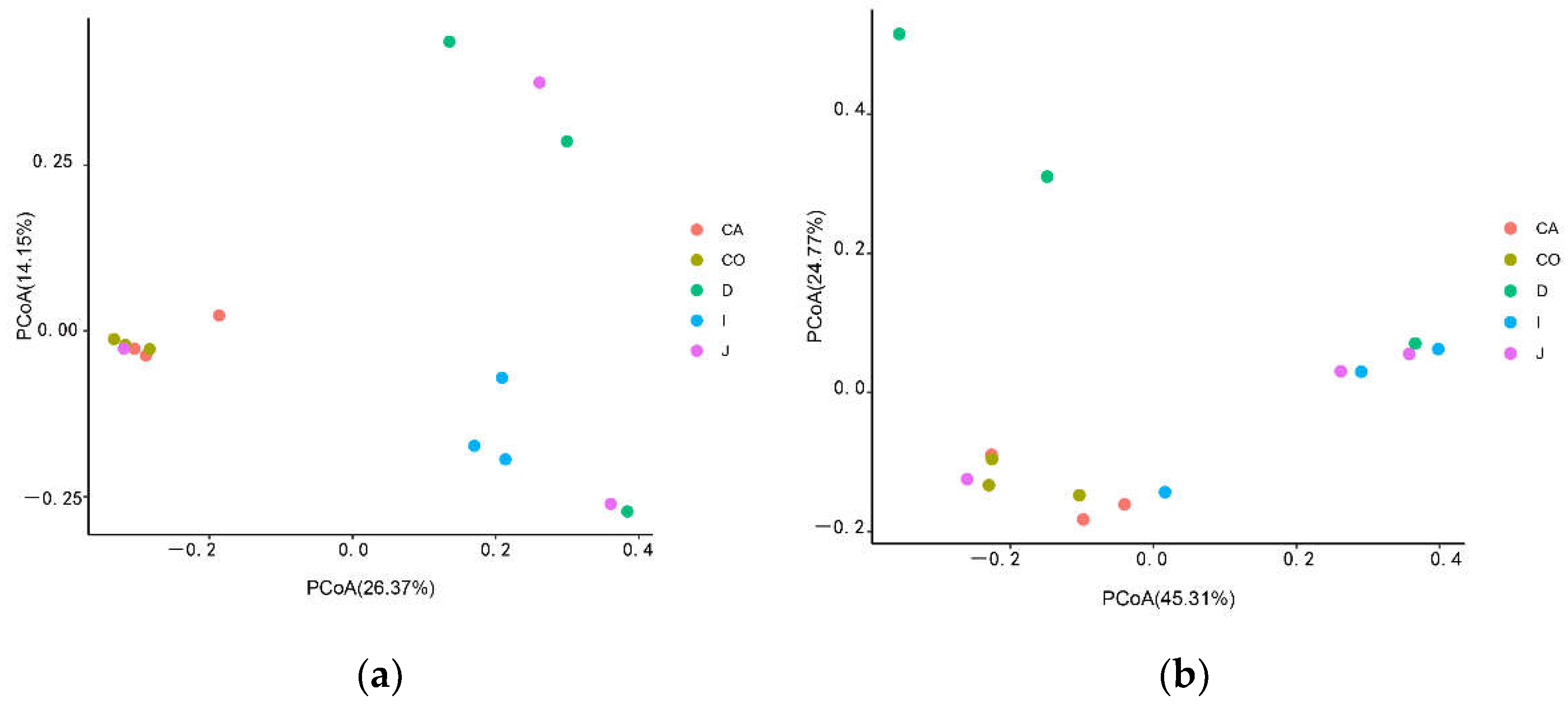
3.2. Analysis of Microbial Diversity
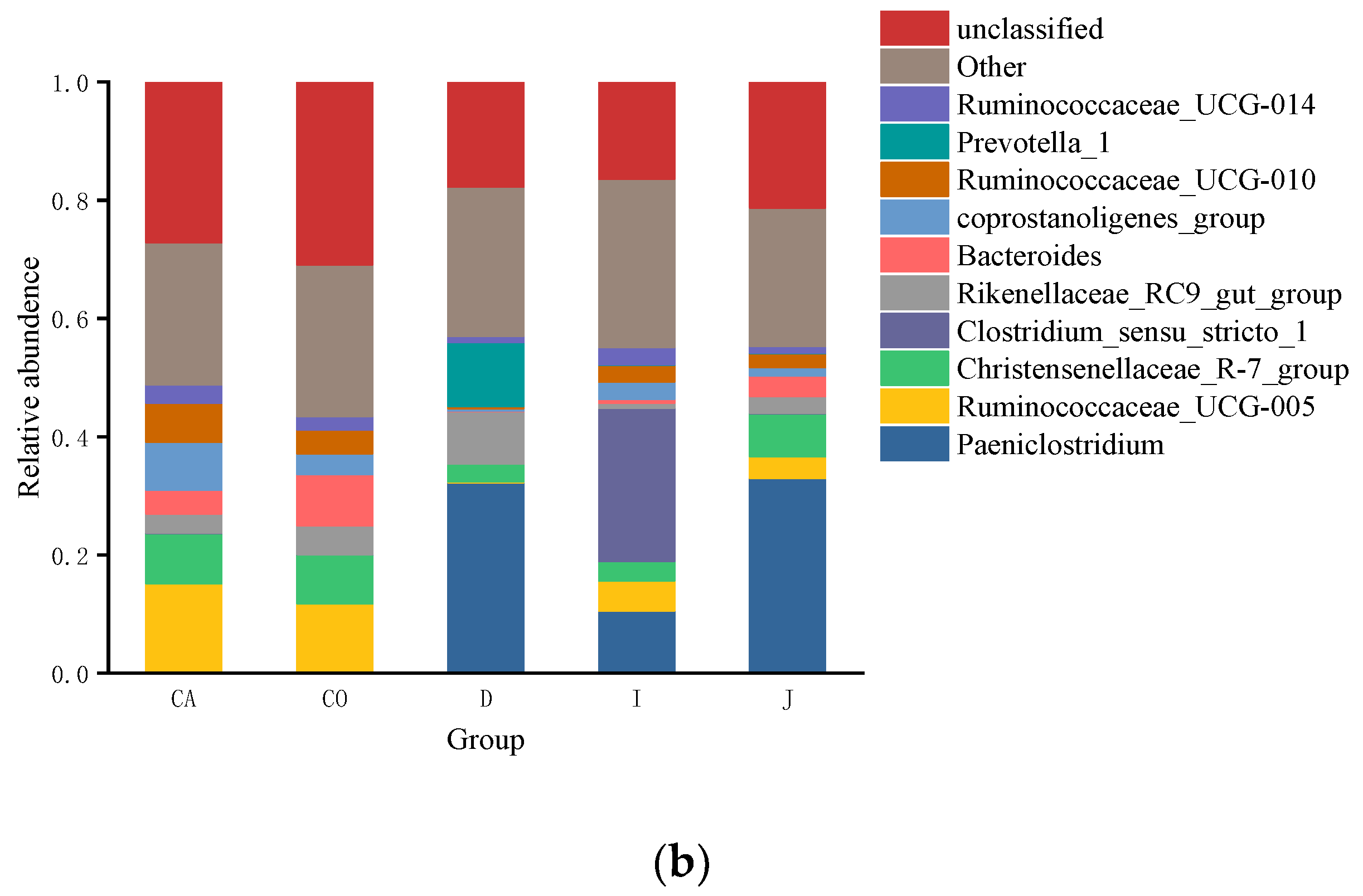
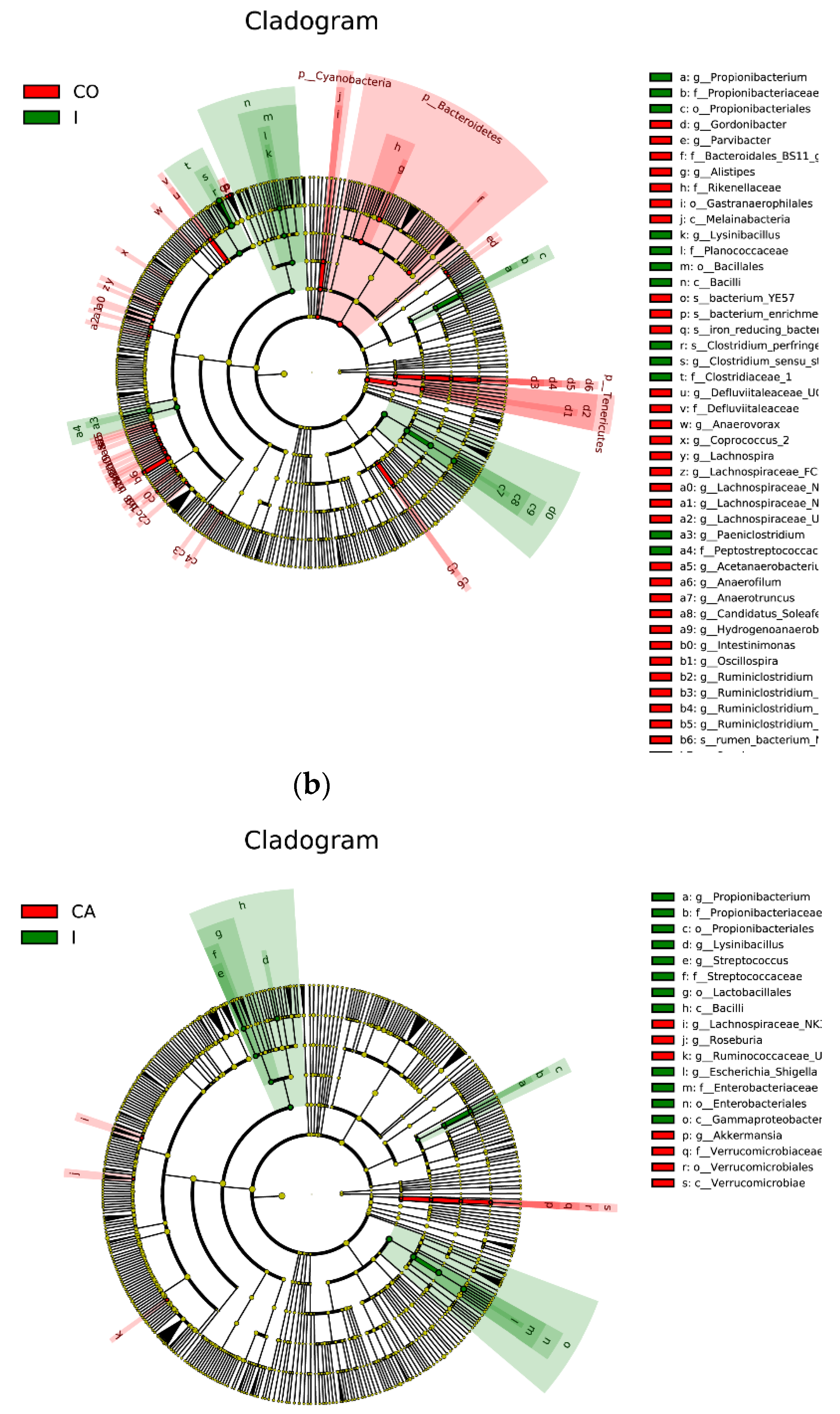
3.3. The Composition of Gut Microbiota
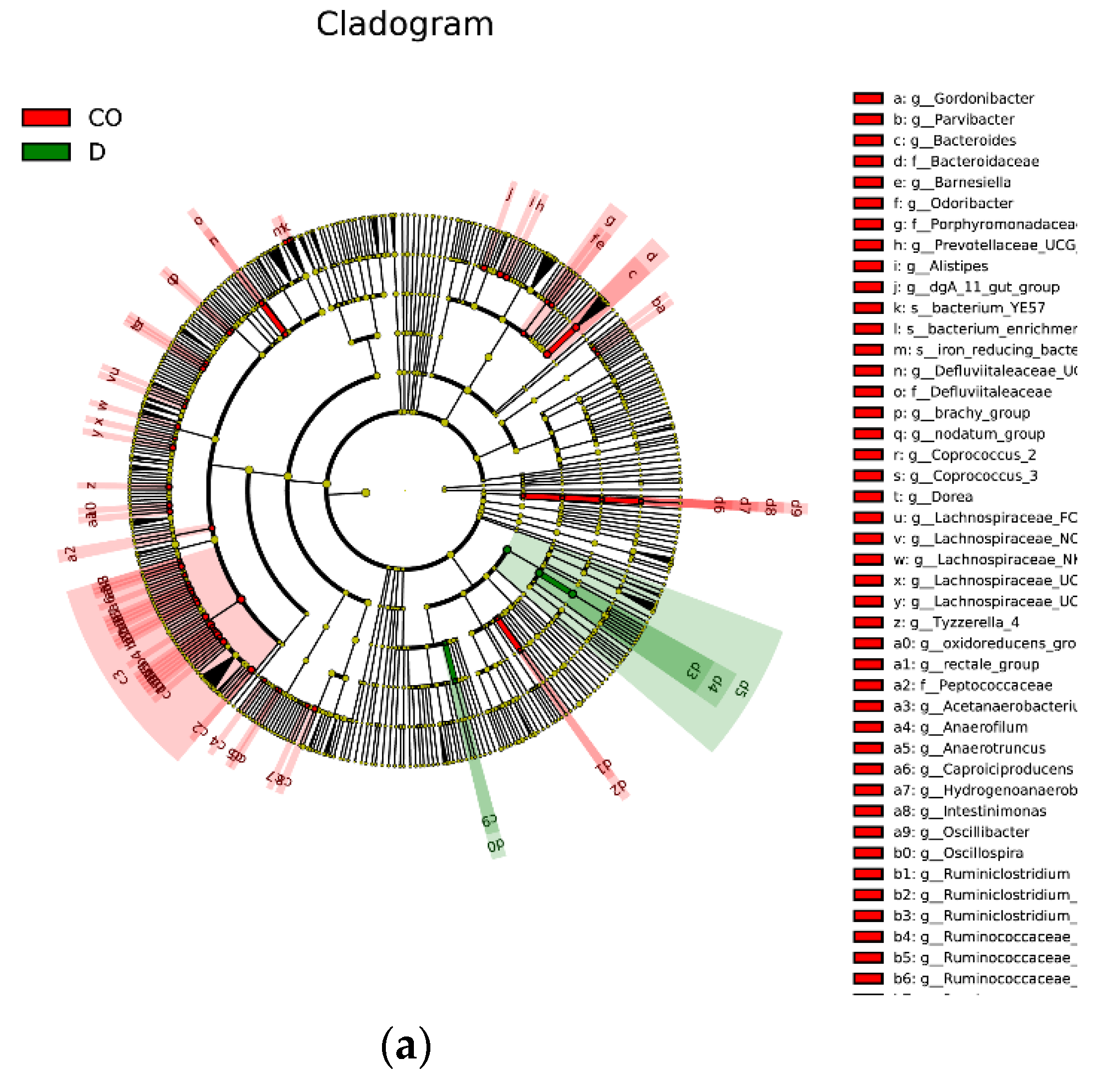
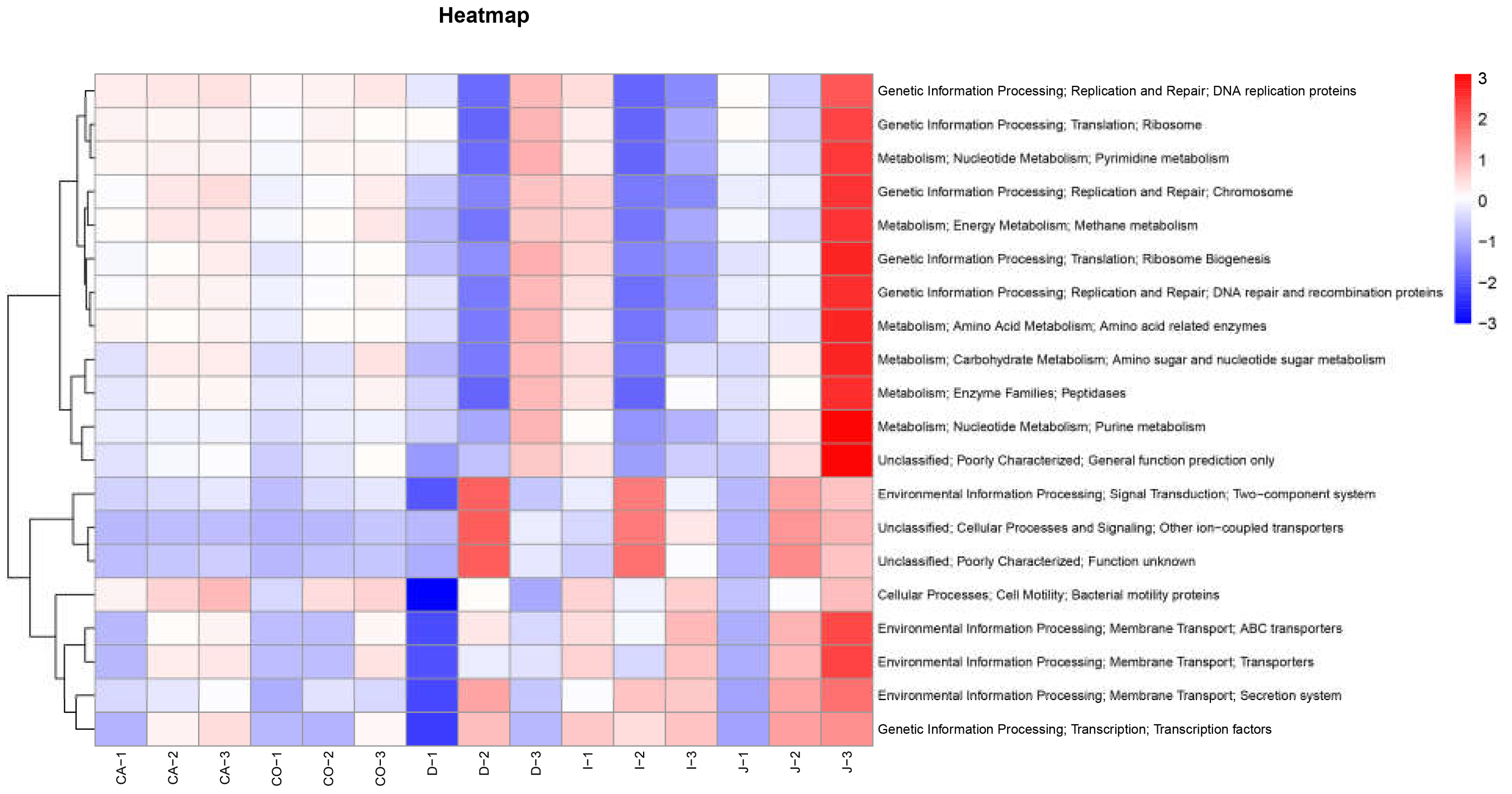
3.4. Functional Prediction of Intestinal Bacterial Flora
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ravichandran, V.; Yin, Y.; Yin, J.; Zhang, Y. Natural products from mammalian gut microbiota. Trends Biotechnol. 2019, 37, 492–504. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Kelly, W.J.; Janssen, P.H.; Attwood, G.T. Rumen microbial (meta)genomics and its application to ruminant production. Animal 2012, 7, 184–201. [Google Scholar] [CrossRef] [Green Version]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, D.; Ji, W.; Lin, B.; Chen, Y.; Huang, C.; Xiong, X.; Fu, M.; Mipam, T.D.; Ai, Y.; Zeng, B. Correlations between gut microbiota community structures of Tibetans and geography. Sci. Rep. 2017, 7, 16982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Yan, S.; Chen, Y.; Ross, R.P.; Stanton, C.; Zhao, J.; Zhang, H.; Chen, W. Diversity of gut microbiota and bifidobacterial community of Chinese subjects of different ages and from different regions. Microorganisms 2020, 8, 1108. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Desselberger, U. The mammalian intestinal microbiome: Composition, interaction with the immune system, significance for vaccine efficacy, and potential for disease therapy. Pathogens 2018, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Huo, D.; You, Z.; Peng, Q.; Ma, C.; Chang, H.; Lin, X.; Wang, L.; Zhang, J. The distal intestinal microbiome of hybrids of Hainan black goats and Saanen goats. PLoS ONE 2020, 15, e0228496. [Google Scholar] [CrossRef]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, K.P.; Bian, Z.Y.; Chen, Z.S.; Li, B.L.; Cui, K.; Wang, F.Y. Association between body weight and distal gut microbes in Hainan black goats at weaning age. Front. Microbiol. 2022, 13, 951473. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhou, L.; Zhou, H.; Hou, G.; Shi, L. Effects of dietary α-lipoic acid on carcass characteristics, antioxidant capability and meat quality in Hainan black goats. Ital. J. Anim. Sci. 2017, 16, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhou, L.; Zhou, H.; Hou, G.; Li, M.; Shi, L.; Huang, X.; Guan, S. Effects of nutrition level of concentrate-based diets on growth performance and carcass characteristics of Hainan black goats. Trop. Anim. Health Prod. 2014, 46, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhao, W.; Niu, L.; Wang, L.; Li, L.; Zhang, H.; Zhong, T. Gene organization and characterization of the complete mitochondrial genome of Hainan black goat (Capra hircus). Mitochondrial DNA Part A 2016, 27, 1656–1657. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Clarridge III, J.E. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; He, Z.; Yang, Y.; Deng, Y.; Tringe, S.G.; Alvarez-Cohen, L. High-throughput metagenomic technologies for complex microbial community analysis: Open and closed formats. MBio 2015, 6, e02288-14. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (Capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yang, Y.; Zhang, Y.; Lv, S.; Jin, T.; Li, K.; Han, Z.; Li, Y. Microbiome analysis reveals the alterations in gut microbiota in different intestinal segments of Yimeng black goats. Microb. Pathog. 2021, 155, 104900. [Google Scholar] [CrossRef]
- Martínez-Porchas, M.; Villalpando-Canchola, E.; Vargas-Albores, F. Significant loss of sensitivity and specificity in the taxonomic classification occurs when short 16S rRNA gene sequences are used. Heliyon 2016, 2, e00170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of Two 16S rRNA Primers (V3–V4 and V4–V5) for Studies of Arctic Microbial Communities. Front. Microbiol. 2021, 12, 637526. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, B.; Girard, I.; Jacotot, E.; Julliand, V. Effect of a preparation of Saccharomyces cerevisiae on microbial profiles and fermentation patterns in the large intestine of horses fed a high fiber or a high starch diet. J. Anim. Sci. 2002, 80, 2600–2609. [Google Scholar] [CrossRef]
- Zhang, D.; Ji, H.; Liu, H.; Wang, S.; Wang, J.; Wang, Y. Changes in the diversity and composition of gut microbiota of weaned piglets after oral administration of Lactobacillus or an antibiotic. Appl. Microbiol. Biotechnol. 2016, 100, 10081–10093. [Google Scholar] [CrossRef]
- Zeng, Y.; Zeng, D.; Zhang, Y.; Ni, X.; Tang, Y.; Zhu, H.; Wang, H.; Yin, Z.; Pan, K.; Jing, B. Characterization of the cellulolytic bacteria communities along the gastrointestinal tract of Chinese Mongolian sheep by using PCR-DGGE and real-time PCR analysis. World J. Microbiol. Biotechnol. 2015, 31, 1103–1113. [Google Scholar] [CrossRef]
- Gong, J.; Yang, C. Advances in the methods for studying gut microbiota and their relevance to the research of dietary fiber functions. Food Res. Int. 2012, 48, 916–929. [Google Scholar] [CrossRef]
- Zeng, Y.; Zeng, D.; Ni, X.; Zhu, H.; Jian, P.; Zhou, Y.; Xu, S.; Lin, Y.; Li, Y.; Yin, Z.; et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express 2017, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shao, M.; Huang, H.; Wang, S.; Ma, L.; Wang, H.; Hu, L.; Wei, K.; Zhu, R. The dynamic distribution of small-tail han sheep microbiota across different intestinal segments. Front. Microbiol. 2018, 9, 32. [Google Scholar] [CrossRef]
- Pham, T.P.; Tidjani Alou, M.; Bachar, D.; Levasseur, A.; Brah, S.; Alhousseini, D.; Sokhna, C.; Diallo, A.; Wieringa, F.; Million, M.; et al. Gut Microbiota Alteration is Characterized by a Pseudomonadota and Fusobacteria Bloom in Kwashiorkor and a Bacteroidota Paucity in Marasmus. Sci. Rep. 2019, 9, 9084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Pseudomonadota: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Li, A.Y.; Yang, Y.; Qin, S.K.; Lv, S.J.; Jin, T.H.; Li, K.; Han, Z.Q.; Li, Y.Z. Microbiome analysis reveals gut microbiota alteration of early-weaned Yimeng black goats with the effect of milk replacer and age. Microb. Cell Factories 2021, 20, 78. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cao, P.; Wang, L.; Zhao, Z.; Chen, Y.; Yang, Y. Bacterial community diversity associated with different levels of dietary nutrition in the rumen of sheep. Appl. Microbiol. Biotechnol. 2017, 101, 3717–3728. [Google Scholar] [CrossRef]
- Zhong, T.; Wang, C.; Wang, X.; Freitas-de-Melo, A.; Zeng, B.; Zhao, Q.; Zhan, S.; Wang, L.; Cao, J.; Dai, D.; et al. Early Weaning and Milk Substitutes Affect the Gut Microbiome, Metabolomics, and Antibody Profile in Goat Kids Suffering From Diarrhea. Front. Microbiol. 2022, 13, 904475. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Li, Z.; Feng, J.; Zhao, L.; Yu, J. Effects of digestate recirculation ratios on biogas production and methane yield of continuous dry anaerobic digestion. Bioresour. Technol. 2020, 316, 123963. [Google Scholar] [CrossRef]
- Waters, J.L.; Ley, R.E. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.Y.; You, H.J.; Yoon, H.S.; Kwon, B.; Lee, J.Y.; Lee, S.; Song, Y.-M.; Lee, K.; Sung, J.; Ko, G. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 2017, 66, 1031–1038. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Wu, S.; Zheng, H.-M.; Li, P.; Sheng, H.-F.; Chen, M.-X.; Chen, Z.-H.; Ji, G.-Y.; Zheng, Z.-D.-X. Linking gut microbiota, metabolic syndrome and economic status based on a population-level analysis. Microbiome 2018, 6, 172. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Dekker Nitert, M. Increased systolic and diastolic blood pressure is associated with altered gut microbiota composition and butyrate production in early pregnancy. Hypertension 2016, 68, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Hu, J.; Peng, H.; Li, B.; Xu, J.; Song, X.; Yu, C.; Zhang, Z.; Du, X.; Bu, G.; et al. Research Note: The gut microbiota varies with dietary fiber levels in broilers. Poult. Sci. 2022, 101, 101922. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.T.; Mongodin, E.F.; Davenport, K.P.; Fraser, C.M.; Sandler, A.D.; Zeichner, S.L. Culture-independent evaluation of the appendix and rectum microbiomes in children with and without appendicitis. PLoS ONE 2014, 9, e95414. [Google Scholar] [CrossRef] [PubMed]
- Cobo, F.; Foronda, C.; Perez-Carrasco, V.; Martin-Hita, L.; Garcia-Salcedo, J.A.; Navarro-Mari, J.M. First description of abdominal infection due to Alistipes onderdonkii. Anaerobe 2020, 66, 102283. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kwon, Y.M.; Kim, I.S.; Kim, J.A.; Yu, D.Y.; Adhikari, B.; Lee, S.S.; Choi, I.S.; Cho, K.K. Effects of the Brown Seaweed Laminaria japonica Supplementation on Serum Concentrations of IgG, Triglycerides, and Cholesterol, and Intestinal Microbiota Composition in Rats. Front. Nutr. 2018, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, Z.; Guo, H.; He, D.; Zhao, H.; Wang, Z.; Zhang, W.; Liao, L.; Zhang, C.; Ni, L. The modulatory effect of infusions of green tea, oolong tea, and black tea on gut microbiota in high-fat-induced obese mice. Food Funct. 2016, 7, 4869–4879. [Google Scholar] [CrossRef]
- Hasan, R.; Bose, S.; Roy, R.; Paul, D.; Rawat, S.; Nilwe, P.; Chauhan, N.K.; Choudhury, S. Tumor tissue-specific bacterial biomarker panel for colorectal cancer: Bacteroides massiliensis, Alistipes species, Alistipes onderdonkii, Bifidobacterium pseudocatenulatum, Corynebacterium appendicis. Arch. Microbiol. 2022, 204, 348. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.I.M.; Wassie, S.E.; Joergensen, R.G.; Korir, D.; Goopy, J.P.; Butterbach-Bahl, K.; Merbold, L.; Dickhoefer, U.; Schlecht, E. Feed Quality and Feeding Level Effects on Faecal Composition in East African Cattle Farming Systems. Animals 2021, 11, 564. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | D | J | I | CA | CO |
|---|---|---|---|---|---|
| Alistipes | 0.0002409 ± 0.0002362 c | 0.0114919 ± 0.0114387 b | 0.0029477 ± 0.0009794 b | 0.0290308 ± 0.0039248 a | 0.030547 4± 0.0031616 a |
| Anaerofilum | ND | 0.0000254 ± 0.0000254 b | ND | 0.0000744 ± 0.0000065 bc | 0.000129 ± 0.0000468 a |
| Bacteroides | 0.0015784 ± 0.0015367 c | 0.0348161 ± 0.0347024 bc | 0.0063273 ± 0.0022585 c | 0.0402566 ± 0.01842 b | 0.0870625 ± 0.0059029 a |
| Bifidobacterium | 0.0000543 ± 0.0000543 a | ND | 0.0000112 ± 0.0000015 a | ND | 0.0000348 ± 0.0000244 a |
| Ruminiclostridium | 0.0000062 ± 0.0000062 b | 0.000189 ± 0.0001767 b | 0.0000265 ± 0.0000212 b | 0.0004042 ± 0.000026 a | 0.0005055 ± 0.0001536 a |
| Ruminococcaceae_UCG-005 | 0.0016093 ± 0.0009648 c | 0.0368468 ± 0.0347901 bc | 0.0512074 ± 0.044556 bc | 0.1506268 ± 0.0100503 a | 0.1170258 ± 0.0102332 ab |
| Ruminococcaceae_UCG-009 | 0.0000093 ± 0.0000093 d | 0.0013664 ± 0.0013626 c | 0.0046782 ± 0.0045223 b | 0.0097851 ± 0.0026403 a | 0.0057623 ± 0.0005336 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhi, W.; Tang, K.; Yang, J.; Yang, T.; Chen, R.; Huang, J.; Tan, H.; Zhao, J.; Sheng, Z. Research on the Gut Microbiota of Hainan Black Goat. Animals 2022, 12, 3129. https://doi.org/10.3390/ani12223129
Zhi W, Tang K, Yang J, Yang T, Chen R, Huang J, Tan H, Zhao J, Sheng Z. Research on the Gut Microbiota of Hainan Black Goat. Animals. 2022; 12(22):3129. https://doi.org/10.3390/ani12223129
Chicago/Turabian StyleZhi, Wenbo, Kai Tang, Jinsong Yang, Tianshu Yang, Rong Chen, Jiaming Huang, Haisheng Tan, Jianguo Zhao, and Zhanwu Sheng. 2022. "Research on the Gut Microbiota of Hainan Black Goat" Animals 12, no. 22: 3129. https://doi.org/10.3390/ani12223129