
Effect of Fibroblast Growth Factor 10 and an Interacting Non-Coding RNA on Secondary Hair Follicle Dermal Papilla Cells in Cashmere Goats’ Follicle Development Assessed by Whole-Transcriptome Sequencing Technology
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Library Preparation and Illumina Hiseq xten/NovaSeq6000 Sequencing
2.3. Sequencing Data Quality Control
2.4. Sequence Alignment
2.5. Transcript Assembly and Functional Annotation
2.6. Identification and Prediction of ncRNAs
2.7. Differential Expression Analysis
2.8. ncRNA Target Gene Prediction
2.9. Functional Enrichment Analysis
2.10. Construction of a ceRNA Network
2.11. Construction of Vectors
2.12. Morphological Observation
2.13. Target Verification
2.14. Assessment of Cell Proliferation
2.15. Detection of Apoptosis
2.16. Cell Cycle Analysis
2.17. Reverse-Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) Validation
2.18. Western Blotting
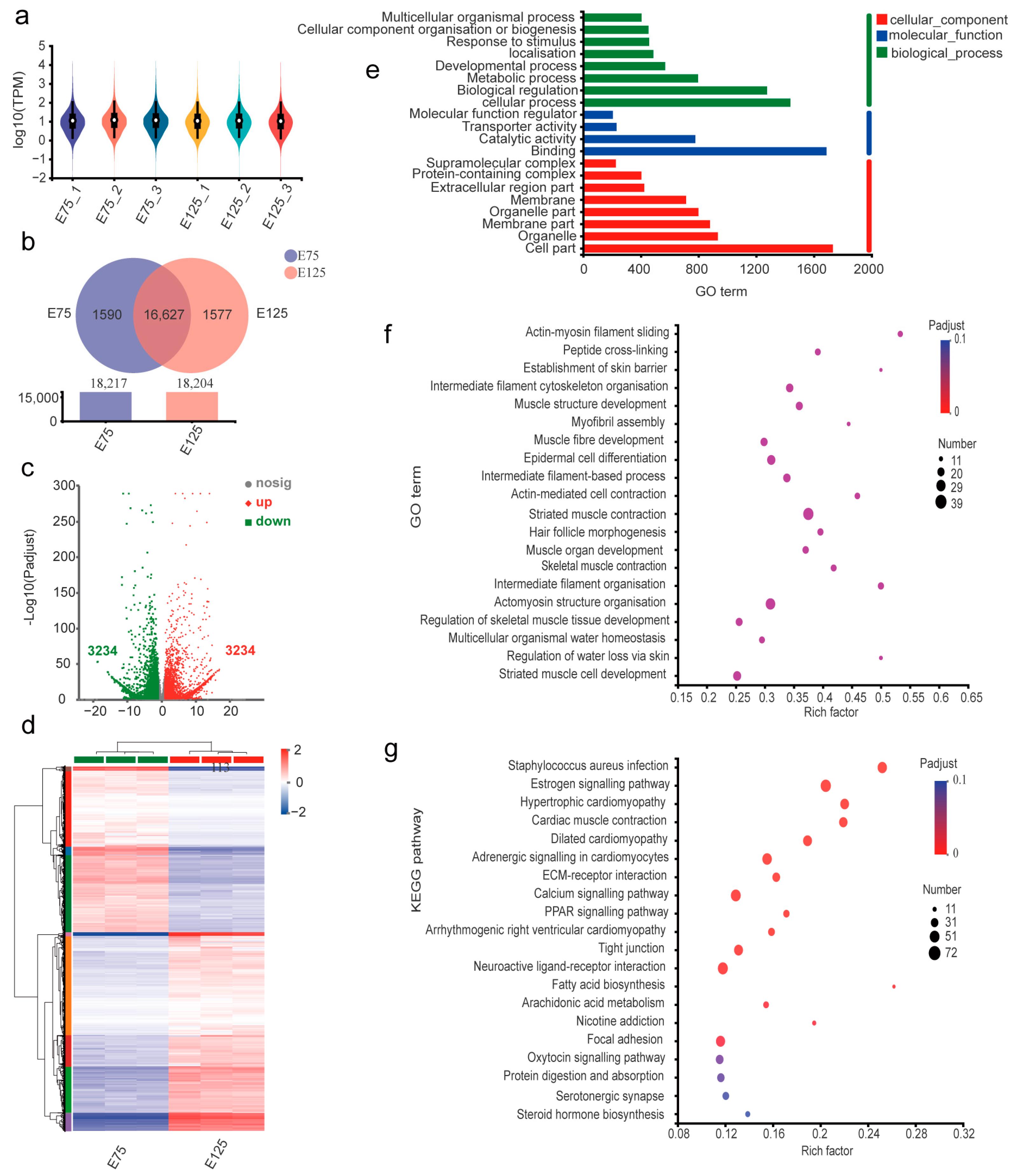
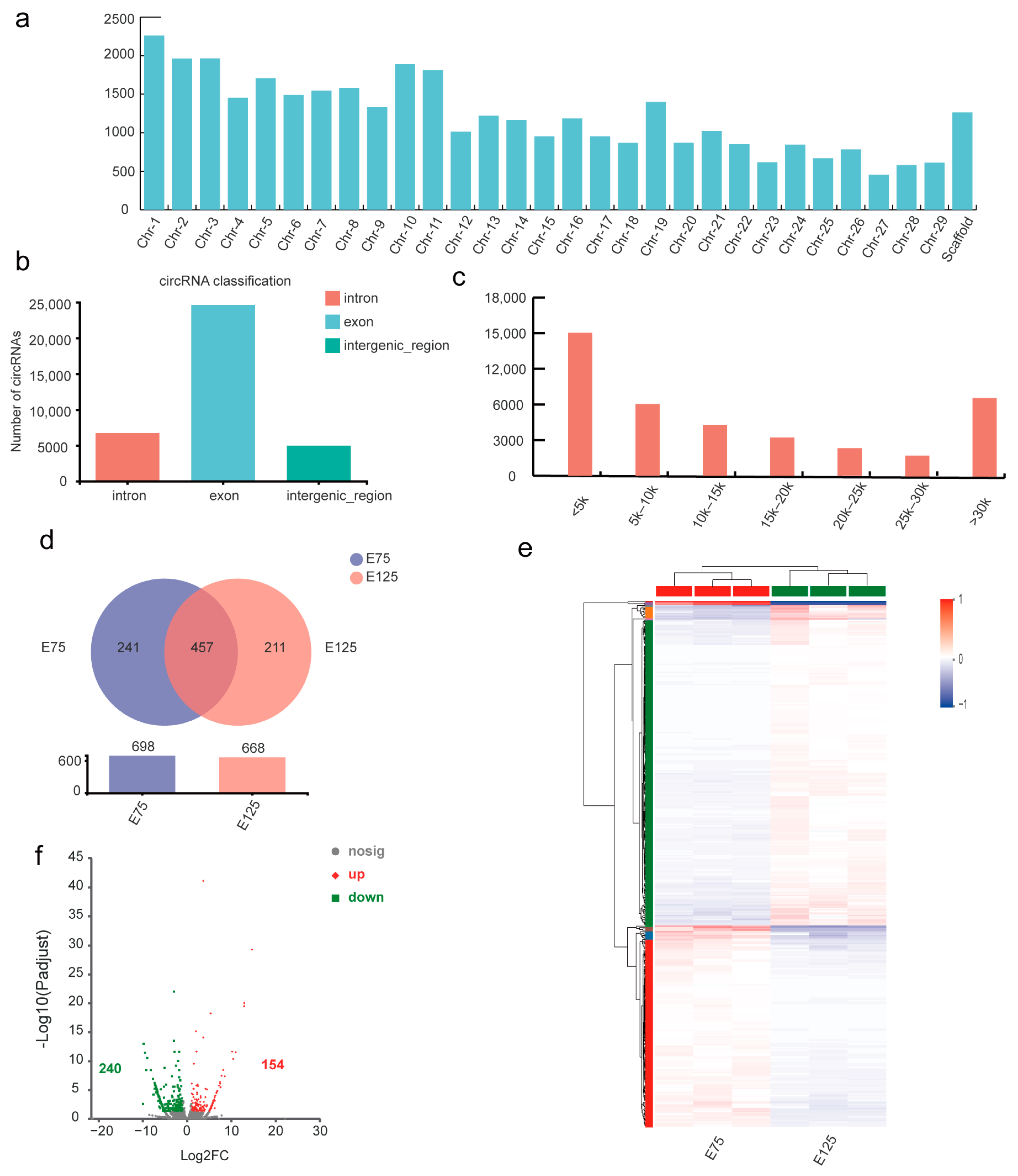
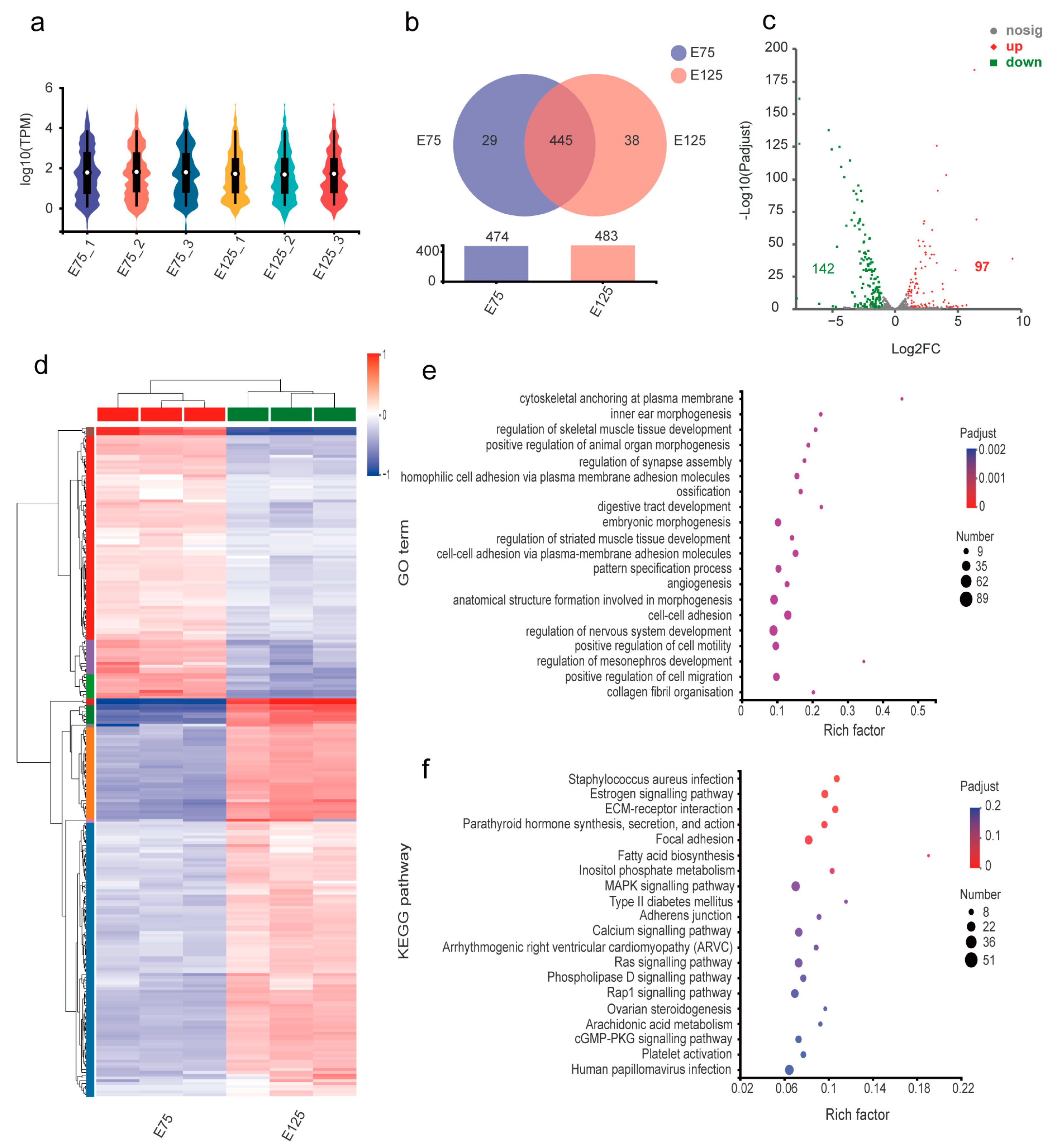
3. Results
3.1. HF Morphogenesis
3.2. RNA Sequencing and cDNA Library Construction
3.3. Expression Analysis and Functional Annotation of mRNAs
3.4. Expression Analysis and Functional Annotation of circRNAs
3.5. Expression Analysis and Functional Annotation of miRNAs
3.6. IPath Metabolic Pathway Analysis
3.7. Construction of ceRNA Network
3.8. RT-qPCR Validation
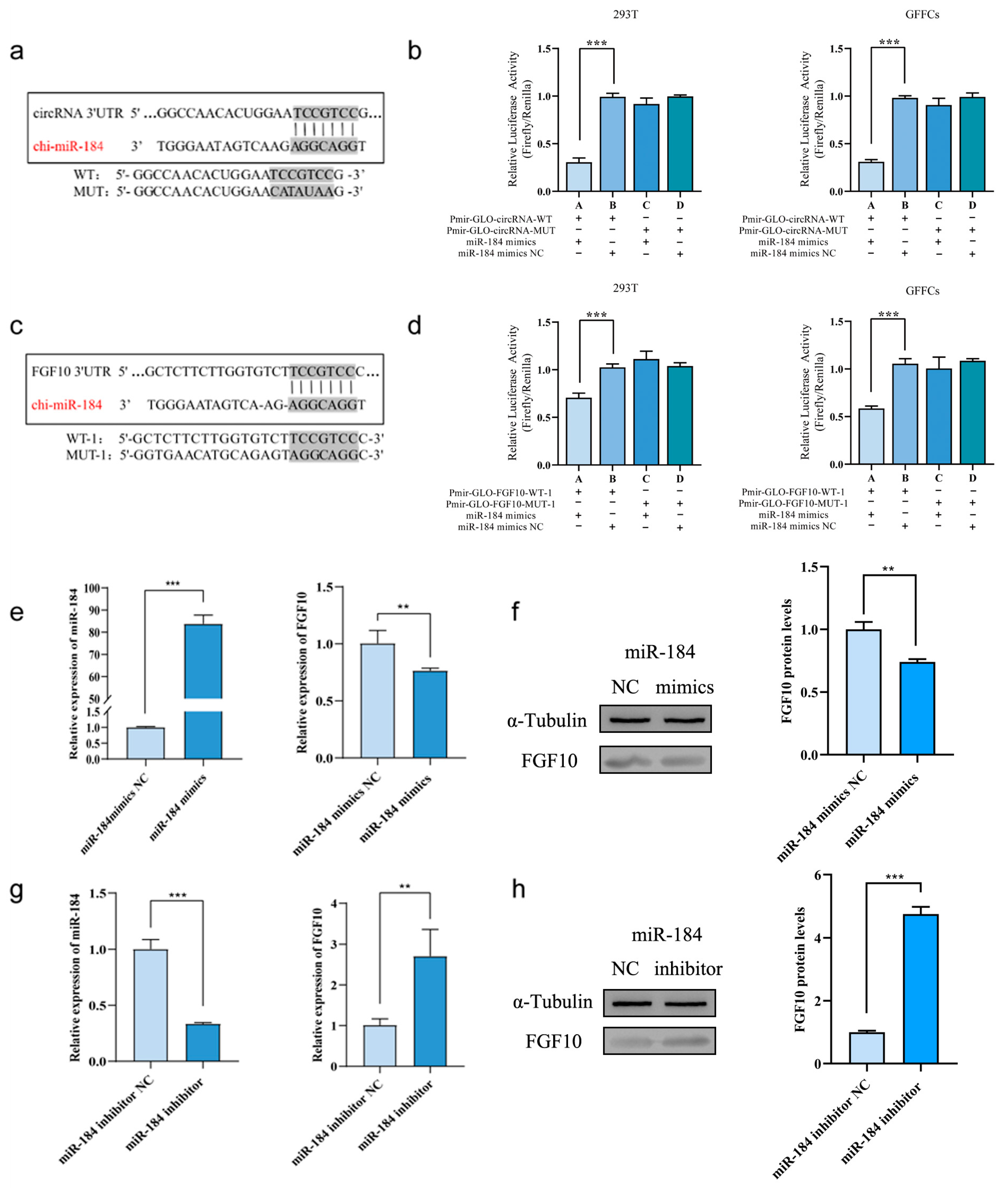
3.9. Verification of the Targeting Relationship between FGF10 and ncRNA
3.10. miR-184 Inhibits the Proliferation of SHF-DPCs by Targeting FGF10
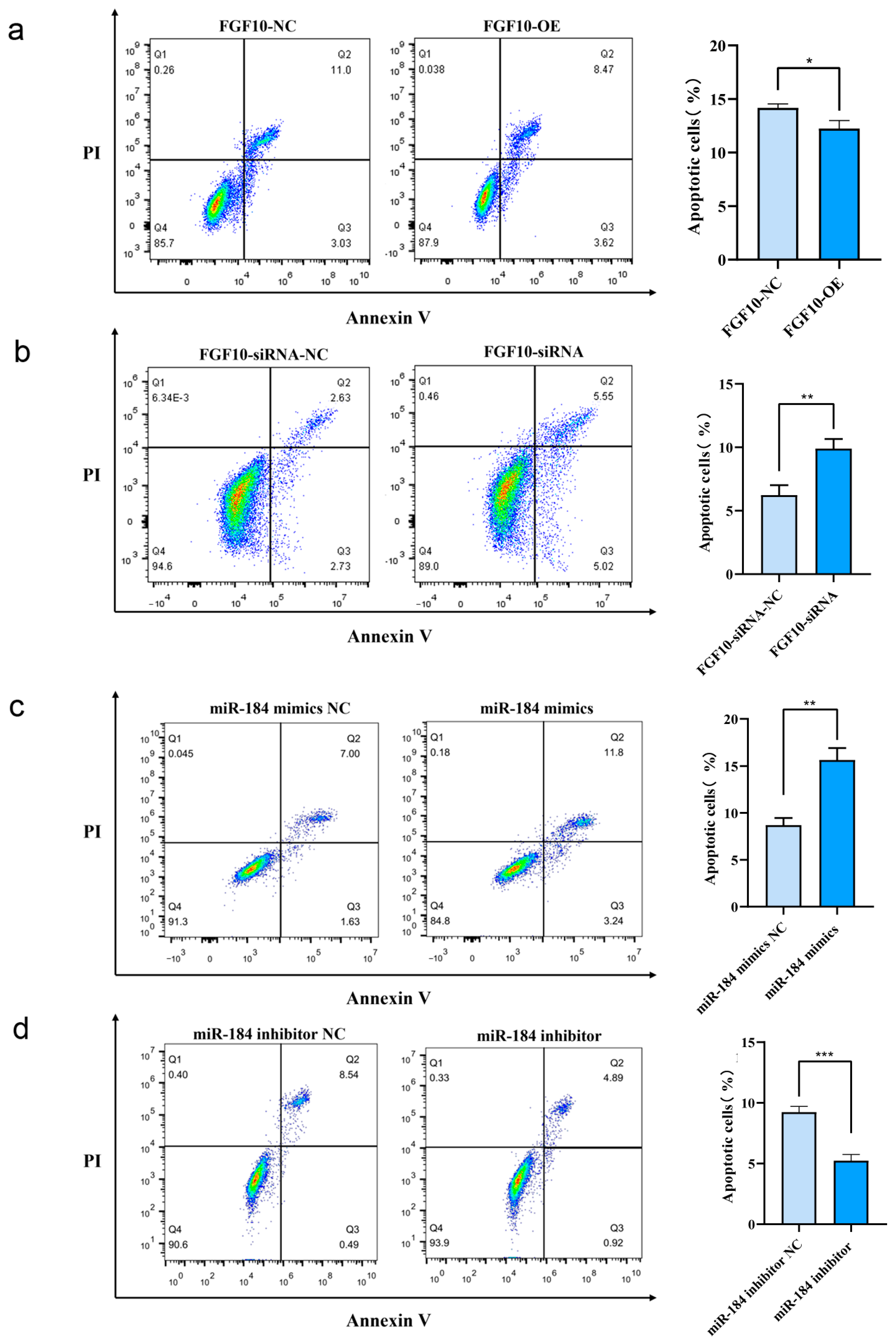
3.11. miR-184 Inhibits Apoptosis in SHF-DPCs by Targeting FGF10
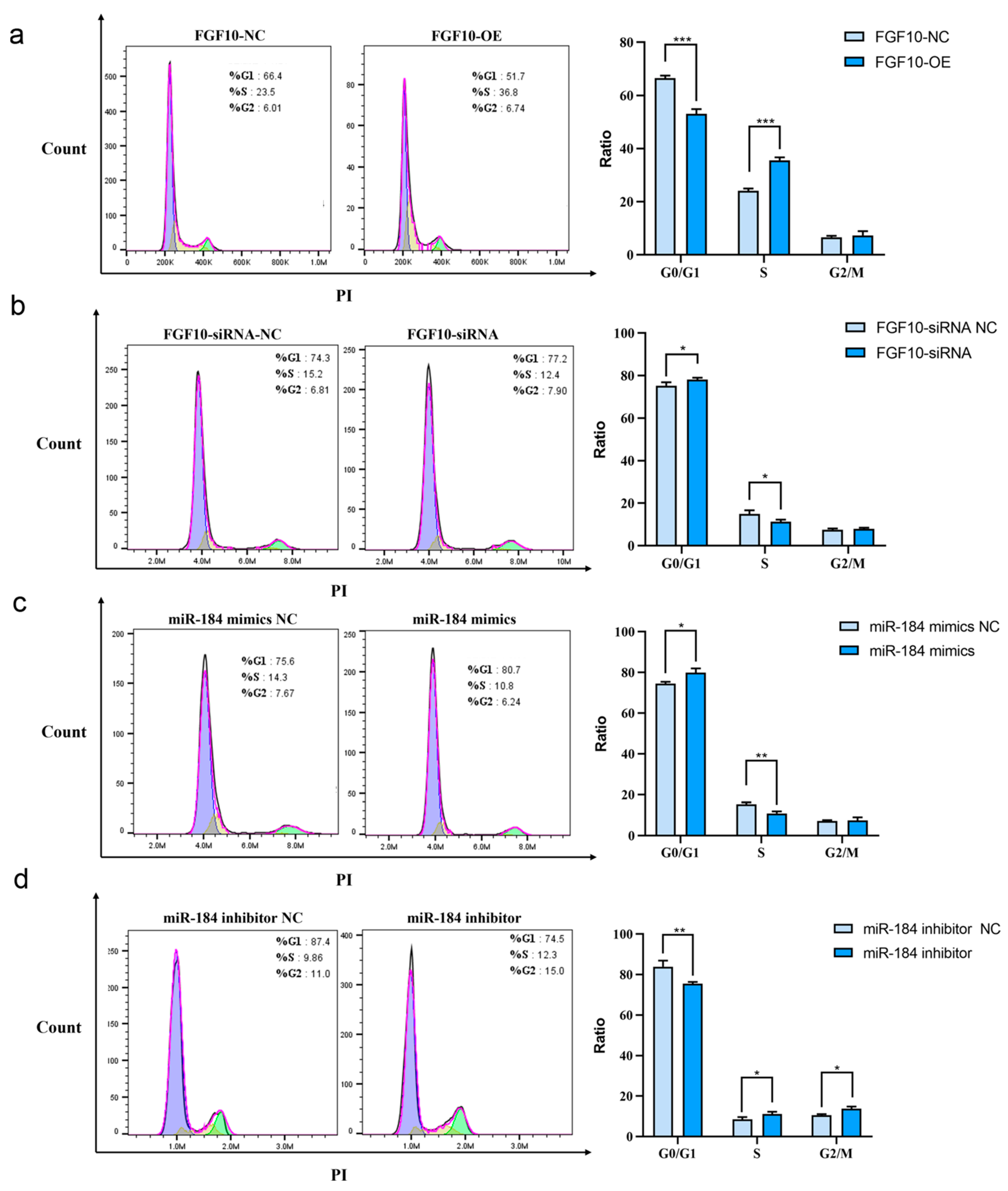
3.12. miR-184 Affects Cell Cycle Progression in SHF-DPCs by Targeting FGF10
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhao, B.; Li, J.; Zhang, X.; Dai, Y.; Yang, N.; Bao, Z.; Chen, Y.; Wu, X. Exosomal miRNA-181a-5p from the cells of the hair follicle dermal papilla promotes the hair follicle growth and development via the Wnt/β-catenin signaling pathway. Int. J. Biol. Macromol. 2022, 207, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Liu, C.; Li, L.; Lan, M.; Yu, Y.; Gu, L.; Su, Y.; Zhang, K.; Zhang, Y.; Wang, T. miR-29a/b1 inhibits hair follicle stem cell lineage progression by spatiotemporally suppressing WNT and BMP signaling. Cell Rep. 2019, 29, 2489–2504.4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Gao, Y.; Fan, Z.; Xiao, X.; Chen, Y.; Si, Y.; Kong, D.; Wang, S.; Liao, M.; Chen, X. Amphiregulin promotes hair regeneration of skin-derived precursors via the PI3K and MAPK pathways. Cell Prolif. 2021, 54, e13106. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; He, J.; Wang, J.; Chen, X.; Yang, R. Regulation of signaling pathways in hair follicle stem cells. Burns Trauma 2022, 10, tkac022. [Google Scholar] [CrossRef]
- Millar, S.E. Molecular mechanisms regulating hair follicle development. J. Investig. Dermatol. 2002, 118, 216–225. [Google Scholar] [CrossRef]
- Lin, B.-J.; Lin, G.-Y.; Zhu, J.-Y.; Yin, G.-Q.; Huang, D.; Yan, Y.-Y. LncRNA-PCAT1 maintains characteristics of dermal papilla cells and promotes hair follicle regeneration by regulating miR-329/Wnt10b axis. Exp. Cell Res. 2020, 394, 112031. [Google Scholar] [CrossRef]
- Ma, T.; Li, J.; Li, J.; Wu, S.; Xiangba; Jiang, H.; Zhang, Q. Expression of miRNA-203 and its target gene in hair follicle cycle development of Cashmere goat. Cell Cycle 2021, 20, 204–210. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Shi, Y.; Zhang, T.; Wang, X. Comprehensive Transcriptome Analysis of Hair Follicle Morphogenesis Reveals That lncRNA-H19 Promotes Dermal Papilla Cell Proliferation through the Chi-miR-214-3p/β-Catenin Axis in Cashmere Goats. Int. J. Mol. Sci. 2022, 23, 10006. [Google Scholar] [CrossRef]
- Wu, Z.; Hai, E.; Di, Z.; Ma, R.; Shang, F.; Wang, M.; Liang, L.; Rong, Y.; Pan, J.; Su, R. Chi-miR-130b-3p regulates Inner Mongolia cashmere goat skin hair follicles in fetuses by targeting Wnt family member 10A. G3 2021, 11, jkaa023. [Google Scholar] [CrossRef]
- Ma, R.; Shang, F.; Rong, Y.; Pan, J.; Wang, M.; Niu, S.; Qi, Y.; Li, Y.; Lv, Q.; Wang, Z. Expression profile of long non-coding RNA in inner Mongolian cashmere goat with putative roles in hair follicles development. Front. Vet. Sci. 2022, 9, 995604. [Google Scholar] [CrossRef]
- Shang, F.; Wang, Y.; Ma, R.; Rong, Y.; Wang, M.; Wu, Z.; Hai, E.; Pan, J.; Liang, L.; Wang, Z. Screening of microRNA and mRNA related to secondary hair follicle morphogenesis and development and functional analysis in cashmere goats. Funct. Integr. Genom. 2022, 22, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Fan, Y.; Yan, X.; Li, W.; Yan, X.; Liu, H.; Zhang, L.; Su, Y.; Zhang, J.; Jiang, W. Identification of genes related to hair follicle cycle development in inner Mongolia cashmere goat by WGCNA. Front. Vet. Sci. 2022, 9, 894380. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Fan, Y.; Zhang, Y.; Yan, X.; Li, W.; Yan, X.; He, L.; Wang, N.; Chen, O.; He, D. The regulation mechanism of different hair types in inner Mongolia cashmere goat based on PI3K-AKT pathway and FGF21. J. Anim. Sci. 2022, 100, skac292. [Google Scholar] [CrossRef] [PubMed]
- Soon, W.W.; Hariharan, M.; Snyder, M.P. High-throughput sequencing for biology and medicine. Mol. Syst. Biol. 2013, 9, 640. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, R.; Wu, T.; Ma, Y.; Gao, Y.; Wu, Y.; Hao, B.; Yin, J.; Li, Y. Transcriptomes reveal microRNAs and mRNAs in different photoperiods influencing cashmere growth in goat. PLoS ONE 2023, 18, e0282772. [Google Scholar] [CrossRef]
- Wang, W.; Li, Z.; Xie, G.; Li, X.; Wu, Z.; Li, M.; Liu, A.; Xiong, Y.; Wang, Y. Convergent Genomic Signatures of Cashmere Traits: Evidence for Natural and Artificial Selection. Int. J. Mol. Sci. 2023, 24, 1165. [Google Scholar] [CrossRef]
- Zhang, C.; Qin, Q.; Liu, Z.; Xu, X.; Lan, M.; Xie, Y.; Wang, Z.; Li, J.; Liu, Z. Identification of the key proteins associated with different hair types in sheep and goats. Front. Genet. 2022, 13, 993192. [Google Scholar] [CrossRef]
- Gong, G.; Fan, Y.; Li, W.; Yan, X.; Yan, X.; Zhang, L.; Wang, N.; Chen, O.; Zhang, Y.; Wang, R. Identification of the key genes associated with different hair types in the inner Mongolia cashmere goat. Animals 2022, 12, 1456. [Google Scholar] [CrossRef]
- Jin, M.; Fan, W.; Piao, J.; Zhao, F.; Piao, J.A. Effects of lncRNA MTC on protein expression in skin fibroblasts of Liaoning Cashmere goat based on iTRAQ technique. Anim. Biotechnol. 2022; online ahead of print. [Google Scholar]
- Zhang, Y.; Qin, Y.; Gu, M.; Xu, Y.; Dou, X.; Han, D.; Lin, G.; Wang, L.; Wang, Z.; Wang, J. Association between the cashmere production performance, milk production performance, and body size traits and polymorphism of COL6A5 and LOC102181374 genes in Liaoning cashmere goats. Anim. Biotechnol. 2022; online ahead of print. [Google Scholar]
- Cao, J.; Wang, J.; Zhou, H.; Hu, J.; Liu, X.; Li, S.; Luo, Y.; Hickford, J.G. Sequence Variation in Caprine KRTAP6-2 Affects Cashmere Fiber Diameter. Animals 2022, 12, 2040. [Google Scholar] [CrossRef]
- Jiang, L.; Schlesinger, F.; Davis, C.A.; Zhang, Y.; Li, R.; Salit, M.; Gingeras, T.R.; Oliver, B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011, 21, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Xu, J.H.; Ren, Q.C.; Duan, T.; Mo, F.; Zhang, W. Melatonin promotes secondary hair follicle development of early postnatal cashmere goat and improves cashmere quantity and quality by enhancing antioxidant capacity and suppressing apoptosis. J. Pineal Res. 2019, 67, e12569. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, X.; Hui, T.; Sun, J.; Cai, W.; Lin, G.; Wang, L.; Dou, X.; Wang, Z.; Han, D. Association analysis for SNPs of KRT26 and TCHH genes with cashmere production performance, body measurement traits and milk production traits in Liaoning cashmere goats. Anim. Biotechnol. 2021, 34, 698–708. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, L.; Gong, G.; Yan, X.; Zhang, L.; Zhang, F.; Liu, H.; Lv, Q.; Wang, Z.; Wang, R. Genome-wide association study of fleece traits in Inner Mongolia Cashmere goats. Anim. Genet. 2021, 52, 375–379. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Y.; Li, S.; Guo, S.; Wang, J.; Luo, Y.; Hu, J.; Liu, X.; Li, S.; Hao, Z. RNA-Seq Reveals the Roles of Long Non-Coding RNAs (lncRNAs) in Cashmere Fiber Production Performance of Cashmere Goats in China. Genes 2023, 14, 384. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, F.; Zhao, M.; Ma, L.; Li, H.; Xie, Y.; Nai, R.; Che, T.; Su, R.; Zhang, Y. The intragenic mRNA-microRNA regulatory network during telogen-anagen hair follicle transition in the cashmere goat. Sci. Rep. 2018, 8, 14227. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Liu, Z.; Zhao, M.; Mu, Q.; Che, T.; Xie, Y.; Ma, L.; Mi, L.; Li, J.; Zhao, Y. Skin transcriptome reveals the periodic changes in genes underlying cashmere (ground hair) follicle transition in cashmere goats. BMC Genom. 2020, 21, 392. [Google Scholar] [CrossRef]
- Diao, X.; Yao, L.; Duan, T.; Qin, J.; He, L.; Zhang, W. Melatonin Regulates the Mitogen-Activated Protein Kinase (MAPK) Pathway via the circMPP5-miR-211-MAPK3 Axis to Induce Secondary Hair Follicle Differentiation and Development in Newborn Cashmere Goat Kids. 2022. Available online: https://assets.researchsquare.com/files/rs-2121117/v1/387d0068-537d-4752-8295-2d46cbaad72e.pdf?c=1665581693 (accessed on 15 January 2023).
- Wang, J.; Sui, J.; Mao, C.; Li, X.; Chen, X.; Liang, C.; Wang, X.; Wang, S.-H.; Jia, C. Identification of key pathways and genes related to the development of hair follicle cycle in cashmere goats. Genes 2021, 12, 180. [Google Scholar] [CrossRef]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Kawano, M.; Komi-Kuramochi, A.; Asada, M.; Suzuki, M.; Oki, J.; Jiang, J.; Imamura, T. Comprehensive analysis of FGF and FGFR expression in skin: FGF18 is highly expressed in hair follicles and capable of inducing anagen from telogen stage hair follicles. J. Investig. Dermatol. 2005, 124, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozeki, M.; Tabata, Y. In vivo promoted growth of mice hair follicles by the controlled release of growth factors. Biomaterials 2003, 24, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, H.; Wang, L.; Guo, K.; Jing, R.; Li, X.; Chen, Y.; Hu, Z.; Gao, S.; Xu, N. Suppression of FGF5 and FGF18 expression by cholesterol-modified siRNAs promotes hair growth in mice. Front. Pharmacol. 2021, 12, 666860. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, P.; Zhang, X.; Li, X.; Bai, Y.; Ao, Y.; Hexig, B.; Guo, X.; Liu, D. Fgf21 knockout mice generated using CRISPR/Cas9 reveal genetic alterations that may affect hair growth. Gene 2020, 733, 144242. [Google Scholar] [CrossRef] [PubMed]
- Hoefert, J.E.; Bjerke, G.A.; Wang, D.; Yi, R. The microRNA-200 family coordinately regulates cell adhesion and proliferation in hair morphogenesis. J. Cell Biol. 2018, 217, 2185–2204. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Lena, A.M.; Bonanno, E.; Melino, G.; Candi, E. miR-24 affects hair follicle morphogenesis targeting Tcf-3. Cell Death Dis. 2013, 4, e922. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fu, S.; Li, C.; Bai, X.; Liu, B.; Ji, X.; Liu, Y.; Zhang, W. Effects of melatonin on let-7 family and its target genes in skin of Inner Mongolian Cashmere goats. Acta Vet. Zootech. Sin. 2019, 50, 1367–1379. [Google Scholar]
- Shang, F.; Wang, Y.; Ma, R.; Di, Z.; Wu, Z.; Hai, E.; Rong, Y.; Pan, J.; Liang, L.; Wang, Z. Expression profiling and functional analysis of circular RNAs in inner Mongolian cashmere goat hair follicles. Front. Genet. 2021, 12, 678825. [Google Scholar] [CrossRef]
- Abreu, C.M.; Marques, A.P. Recreation of a hair follicle regenerative microenvironment: Successes and pitfalls. Bioeng. Trans. Med. 2022, 7, e10235. [Google Scholar] [CrossRef]
- Malzkorn, B.; Wolter, M.; Liesenberg, F.; Grzendowski, M.; Stühler, K.; Meyer, H.E.; Reifenberger, G. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol. 2010, 20, 539–550. [Google Scholar] [CrossRef]
- Foley, N.H.; Bray, I.M.; Tivnan, A.; Bryan, K.; Murphy, D.M.; Buckley, P.G.; Ryan, J.; O’meara, A.; O’sullivan, M.; Stallings, R.L. MicroRNA-184 inhibits neuroblastoma cell survival through targeting the serine/threonine kinase AKT2. Mol. Cancer 2010, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Zhang, D.; Geng, Y.; Li, R.; Zhang, Y. Circular RNA-ZNF609 regulates corneal neovascularization by acting as a sponge of miR-184. Exp. Eye Res. 2020, 192, 107937. [Google Scholar] [CrossRef]
- Park, J.K.; Peng, H.; Yang, W.; Katsnelson, J.; Volpert, O.; Lavker, R.M. miR-184 exhibits angiostatic properties via regulation of Akt and VEGF signaling pathways. FASEB J. 2017, 31, 256. [Google Scholar] [CrossRef] [Green Version]
- Nagosa, S.; Leesch, F.; Putin, D.; Bhattacharya, S.; Altshuler, A.; Serror, L.; Amitai-Lange, A.; Nasser, W.; Aberdam, E.; Rouleau, M. microRNA-184 induces a commitment switch to epidermal differentiation. Stem Cell Rep. 2017, 9, 1991–2004. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Song, W.; Hao, F.; Duo, L.; Zhe, X.; Gao, C.; Guo, X.; Liu, D. Effect of Fibroblast Growth Factor 10 and an Interacting Non-Coding RNA on Secondary Hair Follicle Dermal Papilla Cells in Cashmere Goats’ Follicle Development Assessed by Whole-Transcriptome Sequencing Technology. Animals 2023, 13, 2234. https://doi.org/10.3390/ani13132234
Gao Y, Song W, Hao F, Duo L, Zhe X, Gao C, Guo X, Liu D. Effect of Fibroblast Growth Factor 10 and an Interacting Non-Coding RNA on Secondary Hair Follicle Dermal Papilla Cells in Cashmere Goats’ Follicle Development Assessed by Whole-Transcriptome Sequencing Technology. Animals. 2023; 13(13):2234. https://doi.org/10.3390/ani13132234
Chicago/Turabian StyleGao, Yuan, Weiguo Song, Fei Hao, Lei Duo, Xiaoshu Zhe, Chunyan Gao, Xudong Guo, and Dongjun Liu. 2023. "Effect of Fibroblast Growth Factor 10 and an Interacting Non-Coding RNA on Secondary Hair Follicle Dermal Papilla Cells in Cashmere Goats’ Follicle Development Assessed by Whole-Transcriptome Sequencing Technology" Animals 13, no. 13: 2234. https://doi.org/10.3390/ani13132234