
Genetic Structure and Genome-Wide Association Analysis of Growth and Reproductive Traits in Fengjing Pigs

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotype
2.2. Genotype Data
2.3. Population Genetic Diversity and Structure Analysis
2.4. Genome-Wide Association Study
2.5. Candidate Gene Annotation
3. Results
3.1. Genetic Diversity of Fengjing Pigs
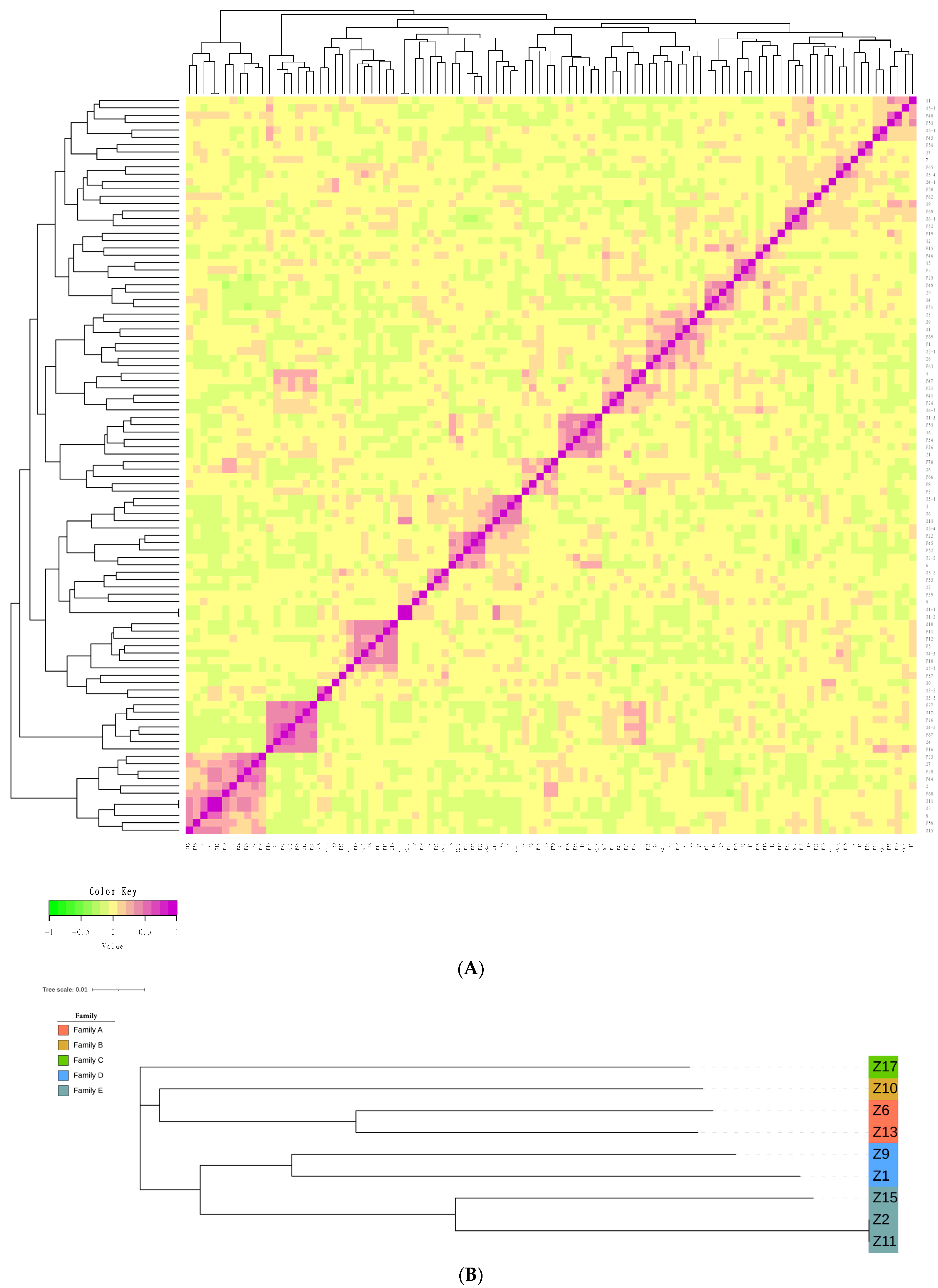
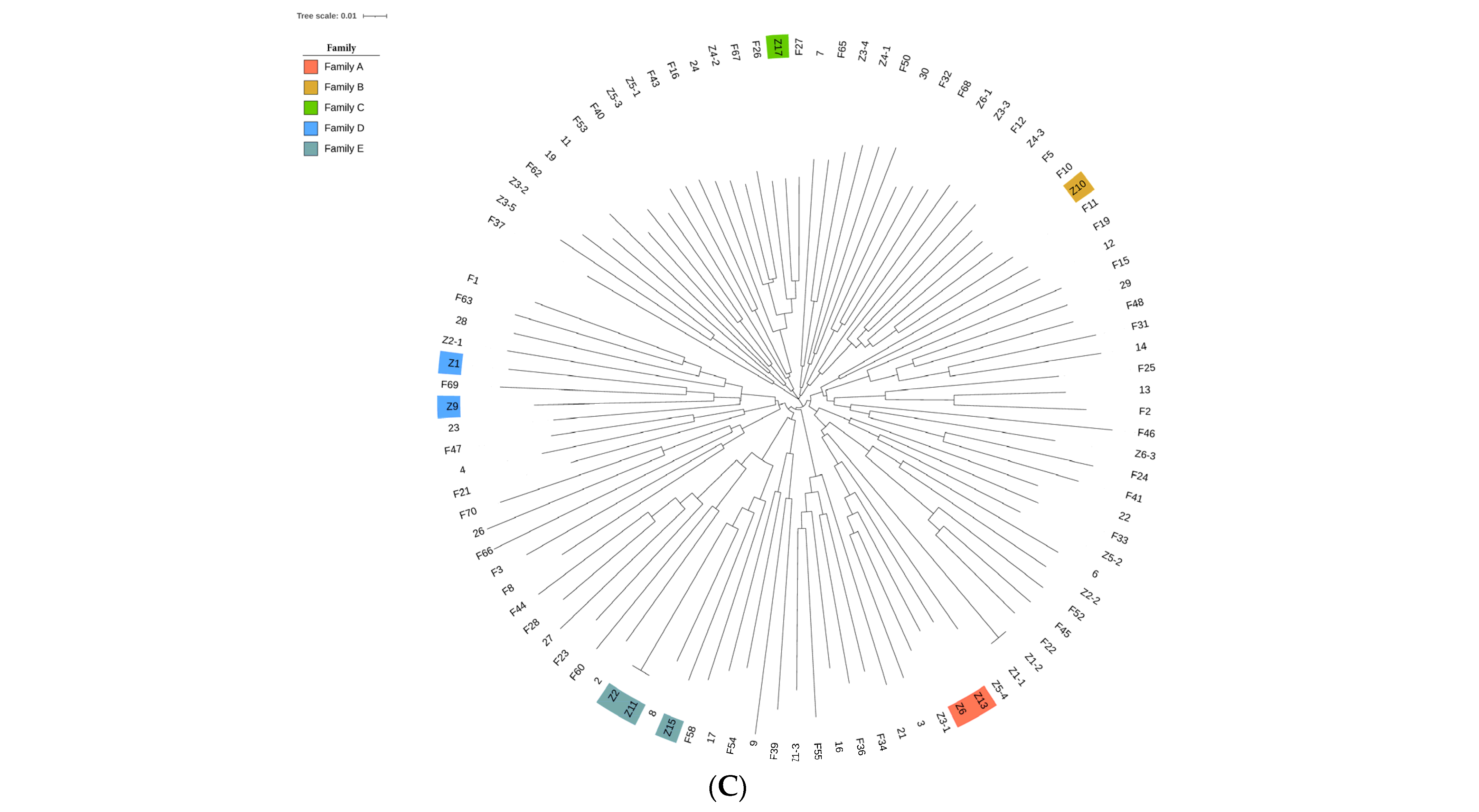
3.2. Genetic Relationships between and Population Structure of Fengjing Pigs
3.3. Inbreeding Coefficient Results
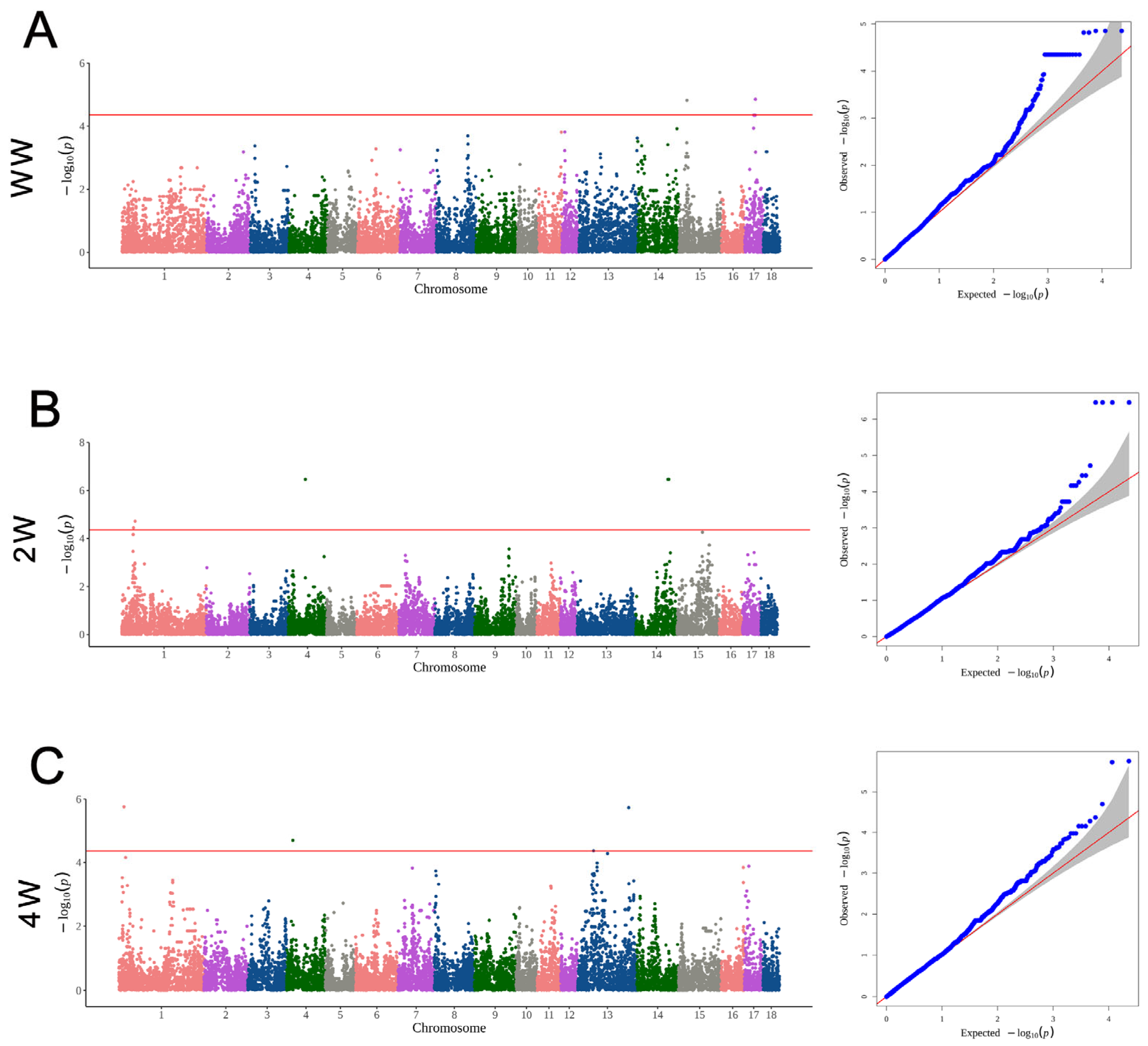
3.4. The Significant SNPs and Genes for Growth Traits
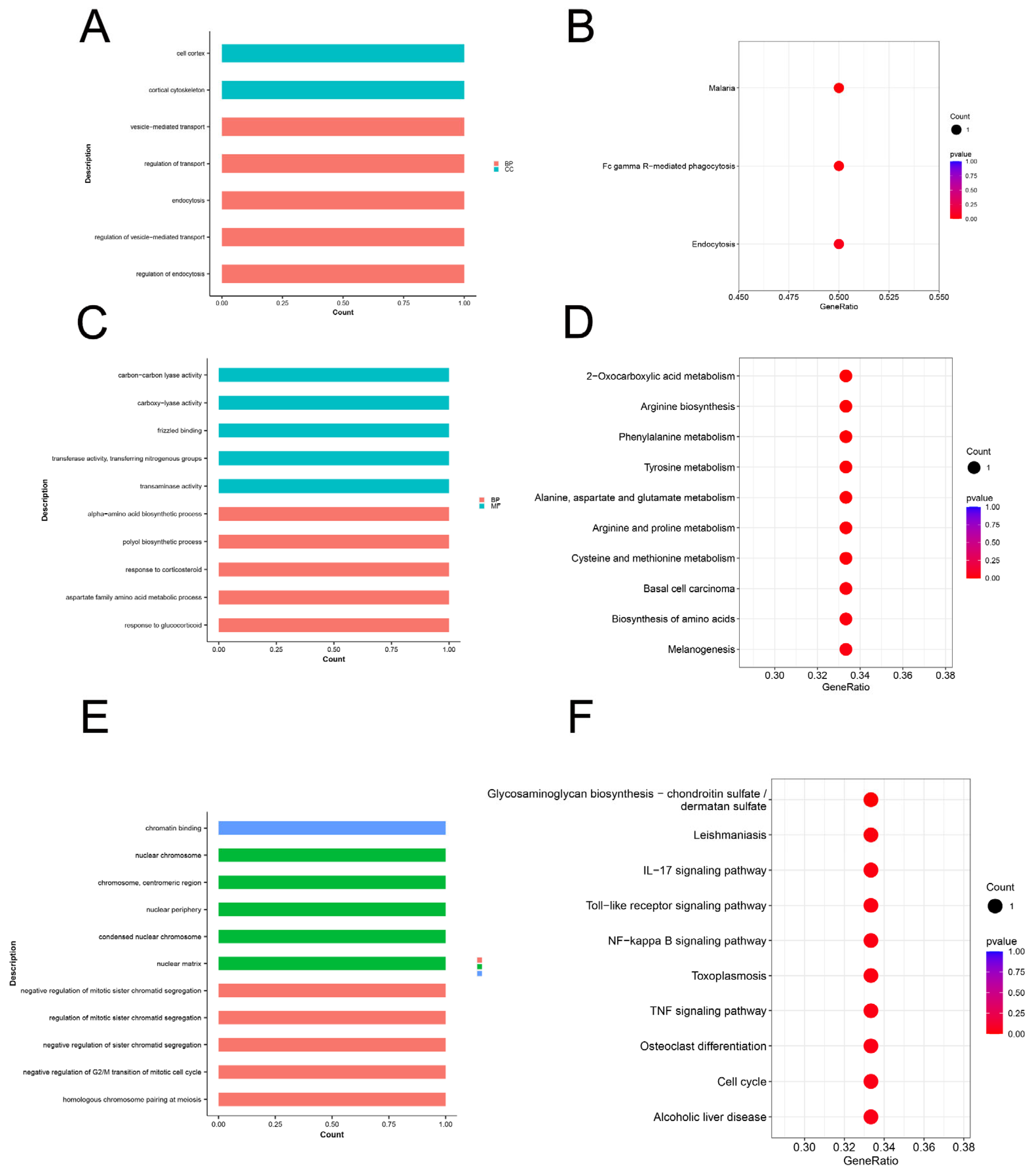
3.5. Enrichment Analysis for Growth Traits
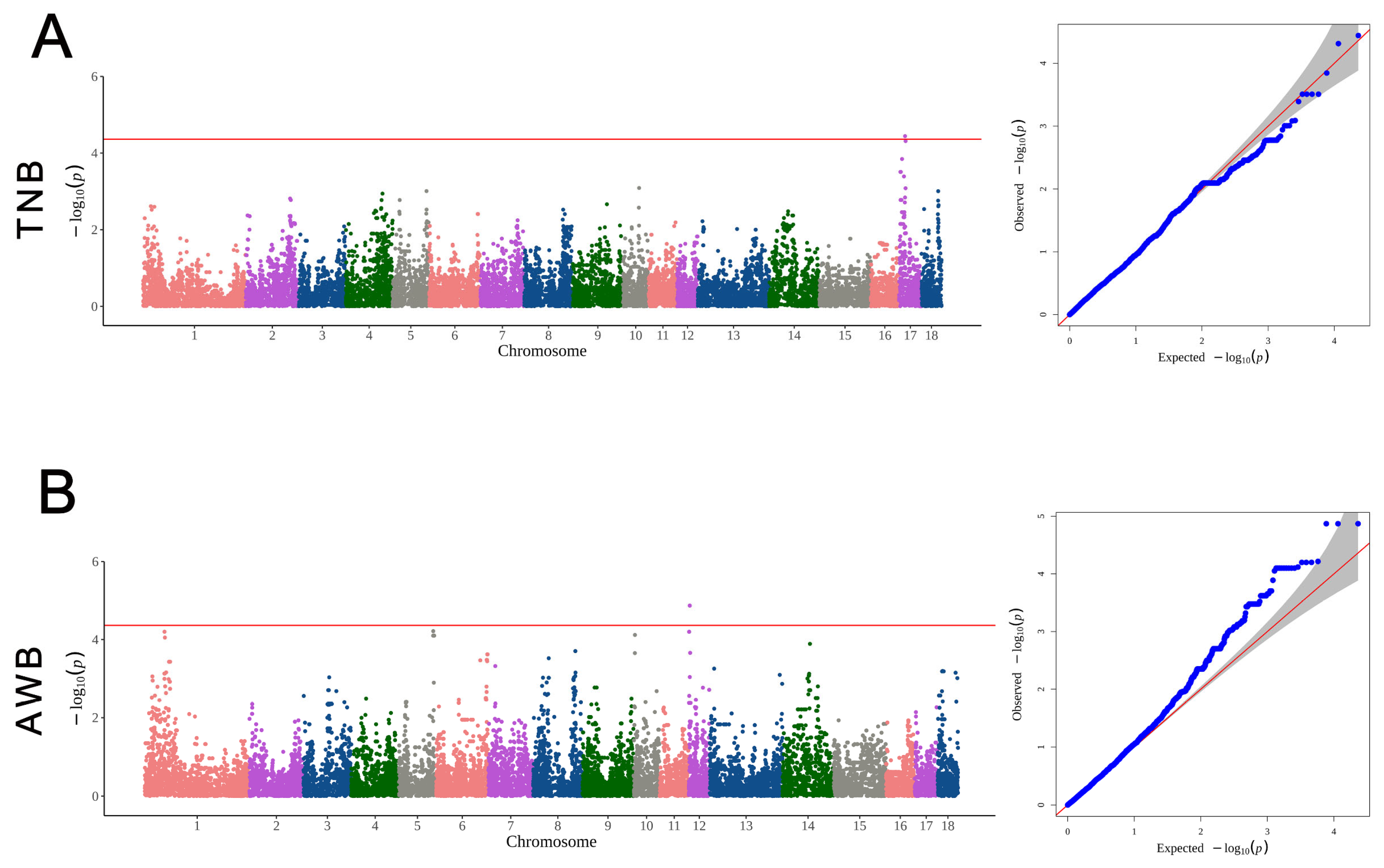
3.6. The Significant SNPs and Genes for Reproduction Traits
3.7. Enrichment Analysis for Reproduction Traits
4. Discussion
4.1. Analysis of Genetic Structure
4.2. Analysis of Candidate Genes for Growth
4.3. Analysis of Candidate Genes for Reproduction Traits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Wu, J.; Ding, W.; Chen, J.; Xing, J. The complete mitochondrial genome of the Fengjing pig. Mitochondrial DNA Part B 2019, 4, 443–445. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Li, S.-F.; Gao, X.-C.; Li, N.; Liu, Y.-H.; Lu, C.-G. Current status, conservation and exploitation of Fengjing pig resources. Shanghai Anim. Husb. Vet. Commun. 2021, 1, 46–47. [Google Scholar] [CrossRef]
- Ge, Q.; Gao, C.; Cai, Y.; Jiao, T.; Quan, J.; Guo, Y.; Zheng, W.; Zhao, S. Evaluating genetic diversity and identifying priority conservation for seven Tibetan pig populations in China based on the mtDNA D-loop. Asian-Australas. J. Anim. Sci. 2020, 33, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fu, Y.; Gu, P.; Zhang, Y.; Tu, W.; Chao, Z.; Wu, H.; Cao, J.; Zhou, X.; Liu, B.; et al. Genome-Wide Characterization and Comparative Analyses of Simple Sequence Repeats among Four Miniature Pig Breeds. Animals 2020, 10, 1792. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Hordofa, B.; Meressa, B.H.; Tamiru, M. Population structure and genetic diversity of Nile tilapia (Oreochromis niloticus) using microsatellite markers from selected water bodies in southwest Ethiopia. Vet. Med. Sci. 2023, 9, 2095–2106. [Google Scholar] [CrossRef]
- Qin, M.; Li, C.; Li, Z.; Chen, W.; Zeng, Y. Genetic Diversities and Differentially Selected Regions Between Shandong Indigenous Pig Breeds and Western Pig Breeds. Front. Genet. 2019, 10, 1351. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Sarup, P.; Ostersen, T.; Nielsen, B.; Fredholm, M.; Karlskov-Mortensen, P.; Sørensen, P.; Jensen, J.; Guldbrandtsen, B.; Lund, M.S.; et al. Genomic diversity revealed by whole-genome sequencing in three Danish commercial pig breeds. J. Anim. Sci. 2020, 98, skaa229. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.M.; Crooijmans, R.P.; Affara, N.A.; Amaral, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE 2009, 4, e6524. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, R.; Li, X.; Cui, C.; Yu, G. Analysis of the Genetic Diversity and Family Structure of the Licha Black Pig Population on Jiaodong Peninsula, Shandong Province, China. Animals 2022, 12, 1045. [Google Scholar] [CrossRef]
- Liu, B.; Shen, L.; Guo, Z.; Gan, M.; Chen, Y.; Yang, R.; Niu, L.; Jiang, D.; Zhong, Z.; Li, X.; et al. Single nucleotide polymorphism-based analysis of the genetic structure of Liangshan pig population. Anim. Biosci. 2021, 34, 1105–1115. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Huang, M.; Tang, J.; Yang, L.; Yu, Z.; Li, D.; Li, G.; Jiang, Y.; Sun, Y.; et al. Whole-genome SNP markers reveal conservation status, signatures of selection, and introgression in Chinese Laiwu pigs. Evol. Appl. 2021, 14, 383–398. [Google Scholar] [CrossRef]
- He, J.; Wu, X.L.; Zeng, Q.; Li, H.; Ma, H.; Jiang, J.; Rosa, G.J.M.; Gianola, D.; Tait, R.G., Jr.; Bauck, S. Genomic mating as sustainable breeding for Chinese indigenous Ningxiang pigs. PLoS ONE 2020, 15, e0236629. [Google Scholar] [CrossRef]
- Xu, P.; Wang, X.; Ni, L.; Zhang, W.; Lu, C.; Zhao, X.; Zhao, X.; Ren, J. Genome-wide genotyping uncovers genetic diversity, phylogeny, signatures of selection, and population structure of Chinese Jiangquhai pigs in a global perspective1. J. Anim. Sci. 2019, 97, 1491–1500. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Wang, Y.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Wu, Z.; Ren, J.; Huang, Z.; Liu, D.; He, X.; Prakapenka, D.; Zhang, R.; Li, N.; Da, Y.; et al. Genome-wide association study and accuracy of genomic prediction for teat number in Duroc pigs using genotyping-by-sequencing. Genet. Sel. Evol. 2017, 49, 35. [Google Scholar] [CrossRef] [PubMed]
- Duijvesteijn, N.; Veltmaat, J.M.; Knol, E.F.; Harlizius, B. High-resolution association mapping of number of teats in pigs reveals regions controlling vertebral development. BMC Genom. 2014, 15, 542. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wang, K.; Zhou, J.; Yang, Q.; Yang, X.; Jiang, A.; Jiang, Y.; Li, M.; Zhu, L.; Bai, L.; et al. A genome wide association study for the number of animals born dead in domestic pigs. BMC Genet. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Grindflek, E.; Lien, S.; Hamland, H.; Hansen, M.H.; Kent, M.; van Son, M.; Meuwissen, T.H. Large scale genome-wide association and LDLA mapping study identifies QTLs for boar taint and related sex steroids. BMC Genom. 2011, 12, 362. [Google Scholar] [CrossRef]
- Luo, W.; Cheng, D.; Chen, S.; Wang, L.; Li, Y.; Ma, X.; Song, X.; Liu, X.; Li, W.; Liang, J.; et al. Genome-wide association analysis of meat quality traits in a porcine Large White × Minzhu intercross population. Int. J. Biol. Sci. 2012, 8, 580–595. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, W.; Zhang, Z.; Fan, Y.; Xie, X.; Ai, H.; Ma, J.; Xiao, S.; Huang, L.; Ren, J. Genome-wide association analyses for fatty acid composition in porcine muscle and abdominal fat tissues. PLoS ONE 2013, 8, e65554. [Google Scholar] [CrossRef]
- Lee, T.; Shin, D.H.; Cho, S.; Kang, H.S.; Kim, S.H.; Lee, H.K.; Kim, H.; Seo, K.S. Genome-wide Association Study of Integrated Meat Quality-related Traits of the Duroc Pig Breed. Asian-Australas. J. Anim. Sci. 2014, 27, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Z.; Bruce, H.; Kemp, R.A.; Charagu, P.; Miar, Y.; Yang, T.; Plastow, G. Genome-wide association studies (GWAS) identify a QTL close to PRKAG3 affecting meat pH and colour in crossbred commercial pigs. BMC Genet. 2015, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Kim, Y.S.; Kang, J.M.; Lee, Y.M.; Rai, R.; Jung, J.H.; Oh, D.Y.; Nam, K.C.; Lee, H.K.; Kim, J.J. Genome-wide Association Study to Identify Quantitative Trait Loci for Meat and Carcass Quality Traits in Berkshire. Asian-Australas. J. Anim. Sci. 2015, 28, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Gao, N.; Li, S.; Kuang, W.; Zhu, L.; Jiang, W.; Yu, W.; Guo, J.; Li, Z.; Yang, C.; et al. Genome-Wide Association Study of Meat Quality Traits in a Three-Way Crossbred Commercial Pig Population. Front. Genet. 2021, 12, 614087. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Scotti, E.; Martelli, P.L.; Buttazzoni, L.; Casadio, R.; Russo, V. A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genom. 2012, 13, 583. [Google Scholar] [CrossRef]
- Fabbri, M.C.; Zappaterra, M.; Davoli, R.; Zambonelli, P. Genome-wide association study identifies markers associated with carcass and meat quality traits in Italian Large White pigs. Anim. Genet. 2020, 51, 950–952. [Google Scholar] [CrossRef]
- Ren, J.; Mao, H.; Zhang, Z.; Xiao, S.; Ding, N.; Huang, L. A 6-bp deletion in the TYRP1 gene causes the brown colouration phenotype in Chinese indigenous pigs. Heredity 2011, 106, 862–868. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Tang, J.; Chen, H.; Zhang, J.; Li, Y.; Lei, S.; Ji, H.; Yang, B.; Ren, J.; et al. Genome-wide association studies for two exterior traits in Chinese Dongxiang spotted pigs. Anim. Sci. J. 2018, 89, 868–875. [Google Scholar] [CrossRef]
- Liu, Q.; Yue, J.; Niu, N.; Liu, X.; Yan, H.; Zhao, F.; Hou, X.; Gao, H.; Shi, L.; Wang, L.; et al. Genome-Wide Association Analysis Identified BMPR1A as a Novel Candidate Gene Affecting the Number of Thoracic Vertebrae in a Large White × Minzhu Intercross Pig Population. Animals 2020, 10, 2186. [Google Scholar] [CrossRef]
- Ren, D.R.; Ren, J.; Ruan, G.F.; Guo, Y.M.; Wu, L.H.; Yang, G.C.; Zhou, L.H.; Li, L.; Zhang, Z.Y.; Huang, L.S. Mapping and fine mapping of quantitative trait loci for the number of vertebrae in a White Duroc × Chinese Erhualian intercross resource population. Anim. Genet. 2012, 43, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, J.; Yu, J.; Zheng, X.; Kang, H.; Wang, Z.; Zhang, S.; Zhou, L.; Liu, J. A genome-wide association study reveals additive and dominance effects on growth and fatness traits in large white pigs. Anim. Genet. 2021, 52, 749–753. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Ning, C.; Wang, D.; Zhou, L.; Wei, J.; Liu, Y.; Kang, H.; Zhang, S.; Zhou, X.; Xu, S.; Liu, J.F. Efficient multivariate analysis algorithms for longitudinal genome-wide association studies. Bioinformatics 2019, 35, 4879–4885. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yuan, J.; Zhou, X.; Xu, G.; Xu, S.; Liu, B. Genetic diversity and population structure of Tongcheng pigs in China using whole-genome SNP chip. Front. Genet. 2022, 13, 910521. [Google Scholar] [CrossRef] [PubMed]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Zinovieva, N.; Yang, B.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 16. [Google Scholar] [CrossRef]
- Silió, L.; Rodríguez, M.C.; Fernández, A.; Barragán, C.; Benítez, R.; Óvilo, C.; Fernández, A.I. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J. Anim. Breed. Genet. 2013, 130, 349–360. [Google Scholar] [CrossRef]
- Légaré, C.; Akintayo, A.; Blondin, P.; Calvo, E.; Sullivan, R. Impact of male fertility status on the transcriptome of the bovine epididymis. Mol. Hum. Reprod. 2017, 23, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Binns, K.; Lemberg, M.K.; Ashman, K.; Martoglio, B. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 2002, 296, 2215–2218. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Conserved POU/OCT- and GATA-binding sites in 5′-flanking promoter region of mammalian WNT8B orthologs. Int. J. Oncol. 2007, 30, 1273–1277. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Williams, B.O. Wnt signaling in bone and muscle. Bone 2015, 80, 60–66. [Google Scholar] [CrossRef]
- Habib, S.J.; Chen, B.C.; Tsai, F.C.; Anastassiadis, K.; Meyer, T.; Betzig, E.; Nusse, R. A localized Wnt signal orients asymmetric stem cell division in vitro. Science 2013, 339, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Gros, J.; Serralbo, O.; Marcelle, C. WNT11 acts as a directional cue to organize the elongation of early muscle fibres. Nature 2009, 457, 589–593. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, W.; Lin, Q.; Gao, Y.; Teng, J.; Xu, Z.; Cai, X.; Zhong, Z.; Wu, J.; Liu, Y.; et al. PigBiobank: A valuable resource for understanding genetic and biological mechanisms of diverse complex traits in pigs. Nucleic Acids Res. 2023, 52, D980–D989. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Qi, X.; Liu, G.; Li, X.; Gao, X.; Bou, G.; Bai, D.; Zhao, Y.; Du, M.; Dugarjaviin, M.; et al. A Transcriptomic Regulatory Network among miRNAs, lncRNAs, circRNAs, and mRNAs Associated with L-leucine-induced Proliferation of Equine Satellite Cells. Animals 2023, 13, 208. [Google Scholar] [CrossRef]
- Starčević, K.; Roškarić, P.; Šperanda, M.; Đidara, M.; Kurilj, A.G.; Maurić, M.; Mašek, T. High dietary n6/n3 ratio decreases eicosapentaenoic to arachidonic acid ratios and upregulates NFκB/p50 expression in short-term low-dose streptozotocin and high-fructose rat model of diabetes. Prostaglandins Leukot. Essent. Fatty Acids 2019, 149, 1–7. [Google Scholar] [CrossRef]
- Urlić, M.; Urlić, I.; Urlić, H.; Mašek, T.; Benzon, B.; Vitlov Uljević, M.; Vukojević, K.; Filipović, N. Effects of Different n6/n3 PUFAs Dietary Ratio on Cardiac Diabetic Neuropathy. Nutrients 2020, 12, 2761. [Google Scholar] [CrossRef]
- Murru, E.; Carta, G.; Manca, C.; Saebo, A.; Santoni, M.; Mostallino, R.; Pistis, M.; Banni, S. Dietary Phospholipid-Bound Conjugated Linoleic Acid and Docosahexaenoic Acid Incorporation Into Fetal Liver and Brain Modulates Fatty Acid and N-Acylethanolamine Profiles. Front. Nutr. 2022, 9, 834066. [Google Scholar] [CrossRef]
- Cussotto, S.; Delgado, I.; Oriolo, G.; Kemper, J.; Begarie, D.; Dexpert, S.; Sauvant, J.; Leboyer, M.; Aouizerate, B.; Martin-Santos, R.; et al. Low omega-3 polyunsaturated fatty acids predict reduced response to standard antidepressants in patients with major depressive disorder. Depress. Anxiety 2022, 39, 407–418. [Google Scholar] [CrossRef]
- Roškarić, P.; Šperanda, M.; Mašek, T.; Verbanac, D.; Starčević, K. Low Dietary n6/n3 Ratio Attenuates Changes in the NRF 2 Gene Expression, Lipid Peroxidation, and Inflammatory Markers Induced by Fructose Overconsumption in the Rat Abdominal Adipose Tissue. Antioxidants 2021, 10, 2005. [Google Scholar] [CrossRef] [PubMed]
- Starčević, K.; Maurić, M.; Galan, A.; Gudan Kurilj, A.; Mašek, T. Effects of different n6/n3 ratios and supplementation with DHA and EPA on the testicular histology and lipogenesis in streptozotocin-treated rats. Andrologia 2018, 50, e13067. [Google Scholar] [CrossRef]
- Fowler, L.A.; Dennis-Cornelius, L.N.; Dawson, J.A.; Barry, R.J.; Davis, J.L.; Powell, M.L.; Yuan, Y.; Williams, M.B.; Makowsky, R.; D’Abramo, L.R.; et al. Both Dietary Ratio of n−6 to n−3 Fatty Acids and Total Dietary Lipid Are Positively Associated with Adiposity and Reproductive Health in Zebrafish. Curr. Dev. Nutr. 2020, 4, nzaa034. [Google Scholar] [CrossRef]
- Health effects of dietary risks in 195 countries, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [CrossRef]
- Yadav, D.; Singh, A.K.; Kumar, B.; Mahla, A.S.; Singh, S.K.; Patra, M.K.; Kumar, H.; Kumar, S.; Tyagi, B.; Verma, M.R.; et al. Effect of n-3 PUFA-rich fish oil supplementation during late gestation on kidding, uterine involution and resumption of follicular activity in goat. Reprod. Domest. Anim. 2019, 54, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Borges, J.B.S.; Gonsioroski, A.V.; Silva, E.P.D. Effects of polyunsaturated fatty acids (PUFA) supplementation on reproductive performance of beef heifers submitted to fixed-time artificial insemination (FTAI) protocol. Acta Sci. Vet. 2019, 47, 1675. [Google Scholar] [CrossRef]
- Srivastava, A.; Srivastava, N.; Mittal, B. Genetics of Obesity. Indian J. Clin. Biochem. IJCB 2016, 31, 361–371. [Google Scholar] [CrossRef]
- Minchenko, D.; Ratushna, O.; Bashta, Y.; Herasymenko, R.; Minchenko, O. The Expression of TIMP1, TIMP2, VCAN, SPARC, CLEC3B and E2F1 in Subcutaneous Adipose Tissue of Obese Males and Glucose Intolerance. Cellbio 2013, 2, 45–53. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | N | Mean | SD | Minimum | Maximum | CV (%) |
|---|---|---|---|---|---|---|
| WW | 62 | 5.99 | 0.26 | 5.5 | 6.5 | 4.35% |
| 2W | 102 | 12.72 | 0.85 | 11 | 17 | 6.67% |
| 4W | 102 | 20.64 | 1.12 | 19 | 26 | 5.45% |
| TNB | 90 | 11.79 | 1.90 | 5 | 15.2 | 16.16% |
| AWB | 90 | 1.10 | 0.08 | 0.96 | 1.33 | 7.00% |
| Trait | SSC | SNPs | Position | Allele | MAF | p-Value | Candidate Genes | Distance (bp) |
|---|---|---|---|---|---|---|---|---|
| WW | 17 | CNC10170722 | 39945627 | C/A | 0.15 | 1.4 × 10−5 | DEFB119 DEFB116 | 13,972 51,035 |
| 17 | CNC10170723 | 39980954 | T/C | 0.15 | 1.4 × 10−5 | DEFB119 DEFB116 | 49,299 15,708 | |
| 17 | CNC10170724 | 40081486 | C/T | 0.15 | 1.4 × 10−5 | HM13 | within | |
| 15 | CNC10150572 | 29322127 | C/T | 0.18 | 1.52 × 10−5 | BIN1 ENSSSCG00000015724 | 264,892 79,072 | |
| 15 | CNC10150582 | 29648112 | G/A | 0.18 | 1.52 × 10−5 | GYPC ENSSSCG00000023800 | 22,639 861,666 | |
| 2W | 4 | CNC10041249 | 63954894 | G/A | 0.02 | 3.44 × 10−7 | WNT8B | 652 |
| 14 | CNC10142321 | 120080503 | C/T | 0.02 | 3.44 × 10−7 | GOT1 | within | |
| 14 | CNC10142322 | 120080803 | G/A | 0.02 | 3.44 × 10−7 | GOT1 | within | |
| 14 | CNC10142394 | 123290079 | C/T | 0.02 | 3.44 × 10−7 | PSD | within | |
| 1 | CNC10011036 | 49820676 | T/C | 0.22 | 1.91 × 10−5 | ROS1 | within | |
| 1 | CNC10010898 | 43672115 | G/A | 0.20 | 3.57 × 10−5 | ENSSSCG00000027613 ENSSSCG00000004231 | 140,659 115,540 | |
| 1 | CNC10010900 | 43741515 | C/T | 0.20 | 3.57 × 10−5 | ENSSSCG00000027613 ENSSSCG00000004231 | 210,059 46,140 | |
| 4W | 1 | CNC10010417 | 18998864 | T/C | 0.02 | 1.75 × 10−6 | TAB2 UST | 95,072 296,182 |
| 13 | CNC10133689 | 188376810 | C/T | 0.26 | 1.86 × 10−6 | ENSSSCG00000012002 LIPI | 525,216 956,415 | |
| 4 | CNC10040479 | 23082238 | A/G | 0.01 | 2 × 10–5 | RAD21 | within | |
| 13 | CNC10131137 | 58006732 | A/G | 0.46 | 4.27 × 10–5 | ENSSSCG00000027675 | within |
| Trait | SSC | SNP | Position | Allele | MAF | p-Value | Candidate Genes | Distance (bp) |
|---|---|---|---|---|---|---|---|---|
| TNB | 17 | CNC10170350 | 18770198 | G/T | 0.12 | 3.61 × 10−5 | ENSSSCG00000025527 HAO1 | 1,086,671 44,229 |
| AWB | 12 | CNC10120065 | 3097741 | T/C | 0.14 | 1.35 × 10−5 | ENPP7 TIMP2 | 410,670 196,212 |
| AWB | 12 | CNC10120066 | 3106253 | G/A | 0.14 | 1.35 × 10−5 | ENPP7 TIMP2 | 419,182 187,700 |
| AWB | 12 | CNC10120068 | 3119720 | C/T | 0.14 | 1.35 × 10−5 | ENPP7 TIMP2 | 432,649 174,233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, L.; Lu, X.; Zhang, W.; Wang, Q.; Zhang, W. Genetic Structure and Genome-Wide Association Analysis of Growth and Reproductive Traits in Fengjing Pigs. Animals 2024, 14, 2449. https://doi.org/10.3390/ani14172449
Xing L, Lu X, Zhang W, Wang Q, Zhang W. Genetic Structure and Genome-Wide Association Analysis of Growth and Reproductive Traits in Fengjing Pigs. Animals. 2024; 14(17):2449. https://doi.org/10.3390/ani14172449
Chicago/Turabian StyleXing, Lei, Xuelin Lu, Wengang Zhang, Qishan Wang, and Weijian Zhang. 2024. "Genetic Structure and Genome-Wide Association Analysis of Growth and Reproductive Traits in Fengjing Pigs" Animals 14, no. 17: 2449. https://doi.org/10.3390/ani14172449