Ningxiang Pig-Derived Microbiota Affects the Growth Performance, Gut Microbiota, and Serum Metabolome of Nursery Pigs

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Sample Collection
2.3. Measurement of Apparent Total Tract Digestibility
2.4. Measurement of Antioxidant Indicators
2.5. DNA Extraction, 16S rRNA Sequencing of Fecal Microbiota and Data Analysis
2.6. Untargeted Metabolomics Study of Serum
2.7. Statistical Analysis
3. Results
3.1. Growth Performance, Apparent Digestibility, and Serum Antioxidant Activity
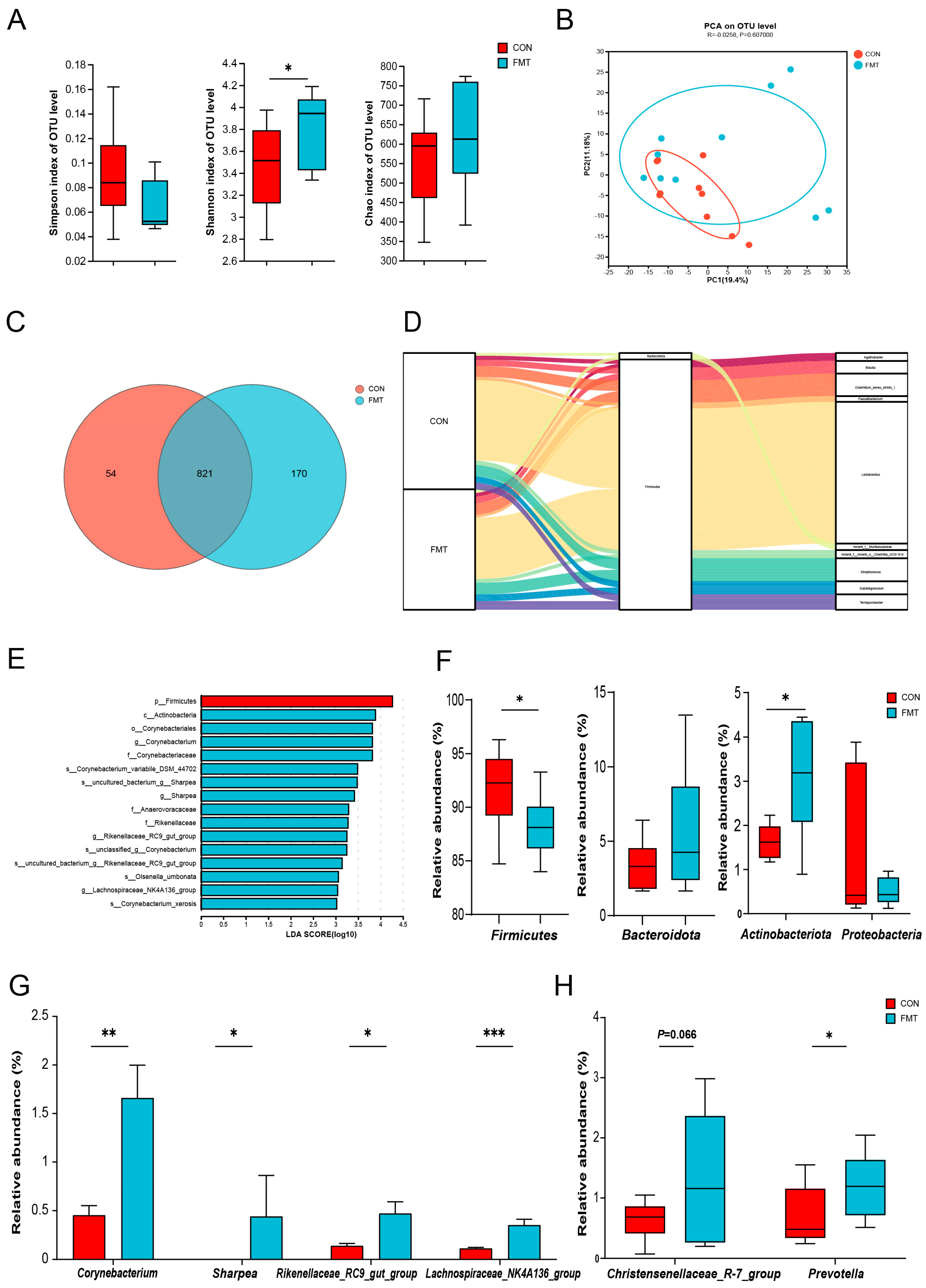
3.2. Ningxiang Pig-Derived Microbiota Shaped the Gut Microbiome in DLY Pigs
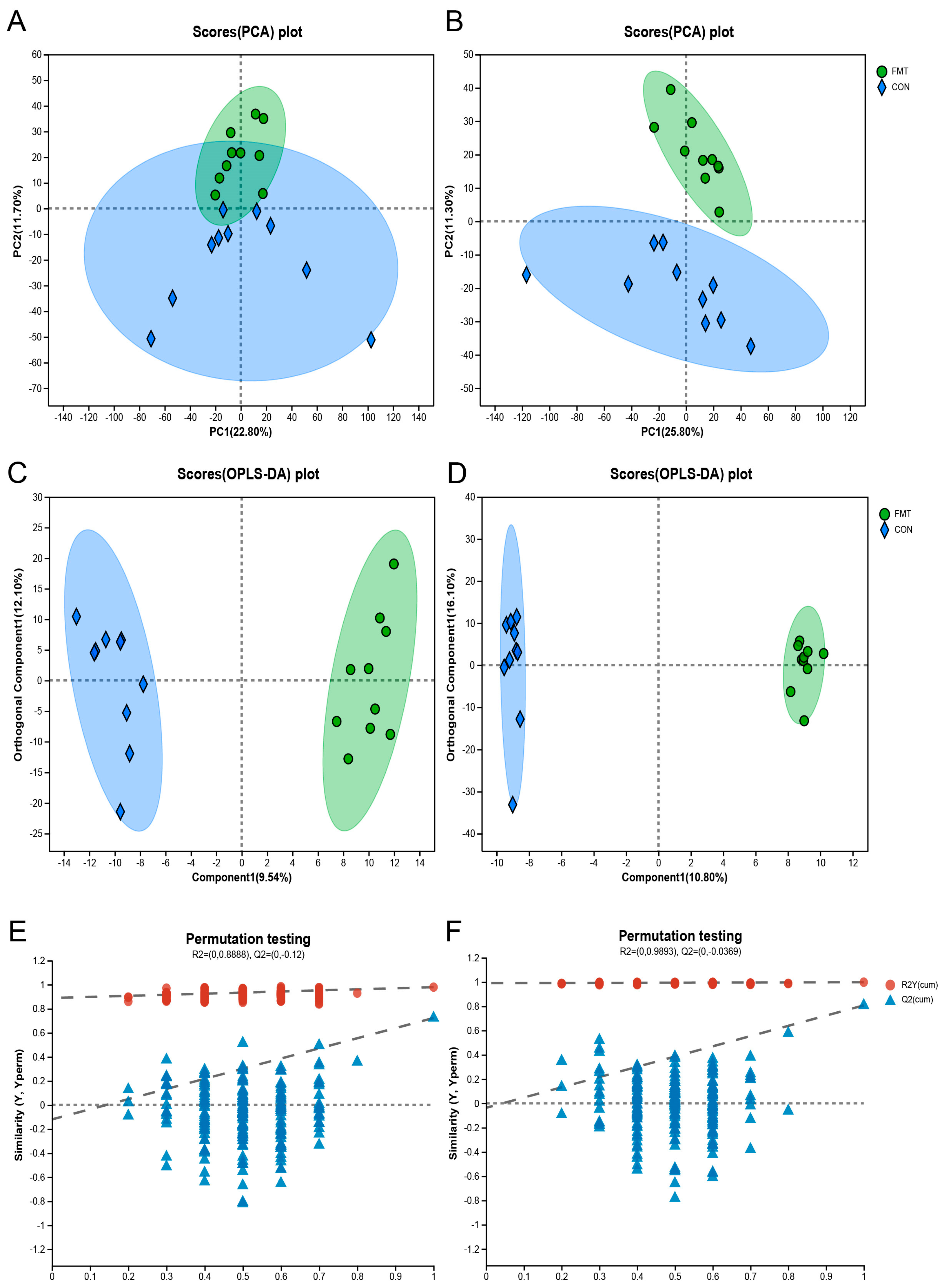
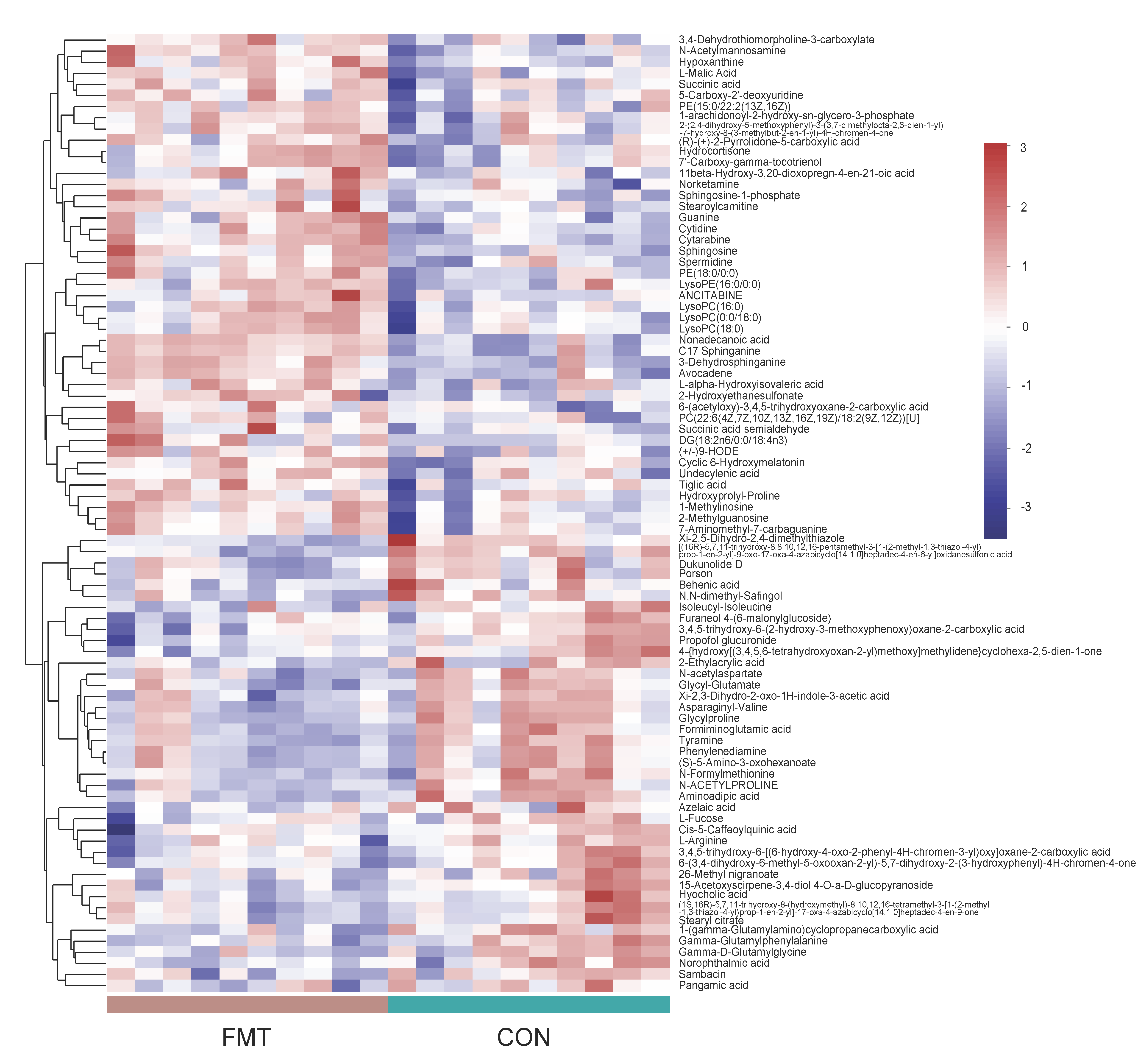
3.3. Ningxiang Pig-Derived Microbiota Altered the Serum Metabolism of DLY Pigs
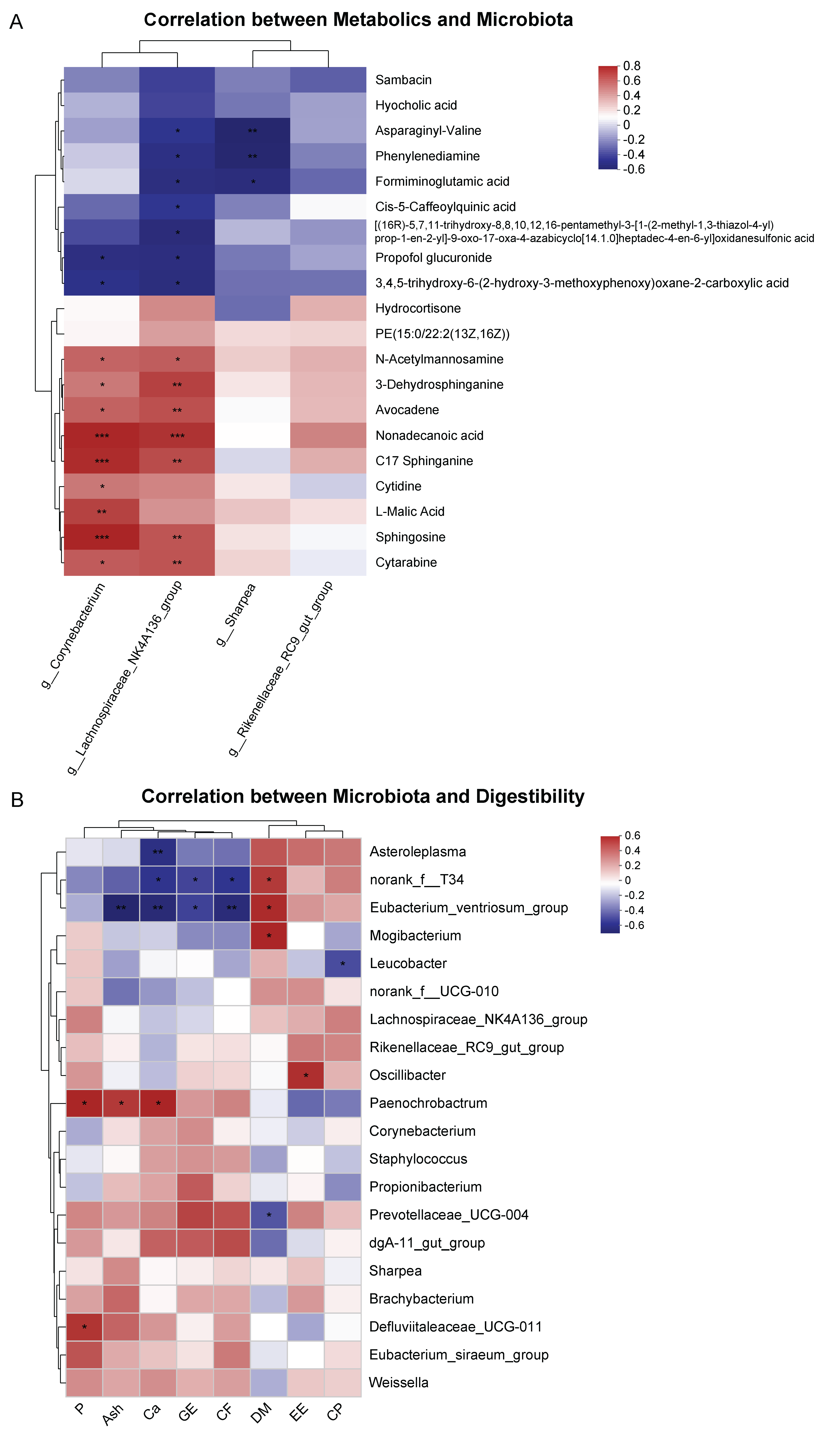
3.4. Correlation between Serum Metabolites and Fecal Microbiota, Fecal Microbiota, and Digestibility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, X.; Li, L. Human gut microbiome: The second genome of human body. Protein Cell 2010, 1, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W.; Munro, K.; Harmsen, H.J.; Welling, G.W.; Smart, J.; Gopal, P.K. Analysis of the fecal microflora of human subjects consuming a probiotic product containing lactobacillus rhamnosus dr20. Appl. Environ. Microb. 2000, 66, 2578–2588. [Google Scholar] [CrossRef]
- Drekonja, D.; Reich, J.; Gezahegn, S.; Greer, N.; Shaukat, A.; MacDonald, R.; Rutks, I.; Wilt, T.J. Fecal microbiota transplantation for clostridium difficile infection a systematic review. Ann. Intern. Med. 2015, 162, 630–638. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.B.; Jones, M.R.; Taylor, K.D.; Marciante, K.; Yan, X.; Dubinsky, M.; Ippoliti, A.; Vasiliauskas, E.; Berel, D.; Derkowski, C.; et al. Fucosyltransferase 2 (fut2) non-secretor status is associated with crohn’s disease. Hum. Mol. Genet. 2010, 19, 3468–3476. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Geng, S.; Li, Y.; Cheng, S.; Fu, X.; Yue, X.; Han, X. Exogenous fecal microbiota transplantation from local adult pigs to crossbred newborn piglets. Front. Microbiol. 2018, 8, 2663. [Google Scholar] [CrossRef]
- McCormack, U.M.; Curião, T.; Wilkinson, T.; Metzler-Zebeli, B.U.; Reyer, H.; Ryan, T.; Calderon-Diaz, J.A.; Crispie, F.; Cotter, P.D.; Creevey, C.J.; et al. Fecal microbiota transplantation in gestating sows and neonatal offspring alters lifetime intestinal microbiota and growth in offspring. Msystems 2018, 3, e117–e134. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef]
- Zhang, W.; Zou, G.; Li, B.; Du, X.; Sun, Z.; Sun, Y.; Jiang, X. Fecal microbiota transplantation (fmt) alleviates experimental colitis in mice by gut microbiota regulation. J. Microbiol. Biotechnol. 2020, 30, 1132–1141. [Google Scholar] [CrossRef]
- Feng, Z.; Guo, J.; Kong, X.; Wang, W.; Li, F.; Nyachoti, M.; Yin, Y. Molecular cloning and expression profiling of g protein coupled receptor 120 in landrace pig and different chinese indigenous pig breeds. J. Food Agric. Environ. 2012, 10, 809–814. [Google Scholar]
- Yan, H.; Diao, H.; Xiao, Y.; Li, W.; Yu, B.; He, J.; Yu, J.; Zheng, P.; Mao, X.; Luo, Y.; et al. Gut microbiota can transfer fiber characteristics and lipid metabolic profiles of skeletal muscle from pigs to germ-free mice. Sci. Rep. 2016, 6, 31786. [Google Scholar] [CrossRef]
- Lei, L.; Wang, Z.; Li, J.; Yang, H.; Yin, Y.; Tan, B.; Chen, J. Comparative microbial profiles of colonic digesta between ningxiang pig and large white pig. Animals 2021, 11, 1862. [Google Scholar] [CrossRef]
- Hu, J.; Ma, L.; Nie, Y.; Chen, J.; Zheng, W.; Wang, X.; Xie, C.; Zheng, Z.; Wang, Z.; Yang, T.; et al. A microbiota-derived bacteriocin targets the host to confer diarrhea resistance in early-weaned piglets. Cell Host Microbe 2018, 24, 817–832. [Google Scholar] [CrossRef] [PubMed]
- de Groot, P.F.; Frissen, M.N.; de Clercq, N.C.; Nieuwdorp, M. Fecal microbiota transplantation in metabolic syndrome: History, present and future. Gut Microbes 2017, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mocanu, V.; Cai, C.; Dang, J.; Slater, L.; Deehan, E.C.; Walter, J.; Madsen, K.L. Impact of fecal microbiota transplantation on obesity and metabolic syndrome—A systematic review. Nutrients 2019, 11, 2291. [Google Scholar] [CrossRef]
- Yin, S.; Li, Z.; Yang, F.; Guo, H.; Zhao, Q.; Zhang, Y.; Yin, Y.; Wu, X.; He, J. A comprehensive genomic analysis of chinese indigenous ningxiang pigs: Genomic breed compositions, runs of homozygosity, and beyond. Int. J. Mol. Sci. 2023, 24, 14550. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ma, X.; Geng, S.; Jiang, X.; Li, Y.; Hu, L.; Li, J.; Wang, Y.; Han, X. Fecal microbiota transplantation beneficially regulates intestinal mucosal autophagy and alleviates gut barrier injury. Msystems 2018, 3, e118–e137. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Wu, X.; Pan, Y.; Wang, L.; Cui, C.; Guo, Y.; Zhu, L.; Peng, J.; Wei, H. Early-life intervention using fecal microbiota combined with probiotics promotes gut microbiota maturation, regulates immune system development, and alleviates weaning stress in piglets. Int. J. Mol. Sci. 2020, 21, 503. [Google Scholar] [CrossRef]
- Niu, Q.; Li, P.; Hao, S.; Zhang, Y.; Kim, S.W.; Li, H.; Ma, X.; Gao, S.; He, L.; Wu, W.; et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 2015, 5, 9938. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, A.; et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 2019, 26, 252–264. [Google Scholar] [CrossRef]
- Liu, B.; Ye, D.; Yang, H.; Song, J.; Sun, X.; Mao, Y.; He, Z. Two-Sample Mendelian Randomization Analysis Investigates Causal Associations Between Gut Microbial Genera and Inflammatory Bowel Disease, and Specificity Causal Associations in Ulcerative Colitis or Crohn’s Disease. Front. Immunol. 2022, 13, 921546. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.T.; Bhat, B.; Gupta, M.; Bajaj, B.K. Phytase-producing potential and other functional attributes of lactic acid bacteria isolates for prospective probiotic applications. Probiotics Antimicro 2016, 8, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vega, J.C.; Stein, H.H. -Invited review—Calcium digestibility and metabolism in pigs. Asian Austral. J. Anim. Sci. 2014, 27, 1–9. [Google Scholar] [CrossRef]
- Maurya, R.P.; Prajapat, M.K.; Singh, V.P.; Roy, M.; Todi, R.; Bosak, S.; Singh, S.K.; Chaudhary, S.; Kumar, A.; Morekar, S.R. Serum malondialdehyde as a biomarker of oxidative stress in patients with primary ocular carcinoma: Impact on response to chemotherapy. Clin. Ophthalmol. 2021, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, L.; Zhang, W.; Yang, Z.; Ding, B.; Zhu, H.; Liu, Y.; Qiu, Y.; Yin, Y.; Wu, G. Protective effects of n-acetylcysteine on intestinal functions of piglets challenged with lipopolysaccharide. Amino Acids 2012, 43, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Novais, A.K.; Deschêne, K.; Martel-Kennes, Y.; Roy, C.; Laforest, J.; Lessard, M.; Matte, J.J.; Lapointe, J.; Óvilo, C. Weaning differentially affects mitochondrial function, oxidative stress, inflammation and apoptosis in normal and low birth weight piglets. PLoS ONE 2021, 16, e247188. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Sign. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Shang, Q.; Gao, Z.; Hao, F.; Guo, H.; Guo, C. Fecal microbiota transplantation (fmt) could reverse the severity of experimental necrotizing enterocolitis (nec) via oxidative stress modulation. Free Radic. Bio Med. 2017, 108, 32–43. [Google Scholar] [CrossRef]
- Liu, M.; Ma, J.; Xu, J.; Huangfu, W.; Zhang, Y.; Ali, Q.; Liu, B.; Li, D.; Cui, Y.; Wang, Z.; et al. Fecal microbiota transplantation alleviates intestinal inflammatory diarrhea caused by oxidative stress and pyroptosis via reducing gut microbiota-derived lipopolysaccharides. Int. J. Biol. Macromol. 2024, 261, 129696. [Google Scholar] [CrossRef]
- Hong, J.; Ndou, S.P.; Adams, S.; Scaria, J.; Woyengo, T.A. Canola meal in nursery pig diets: Growth performance and gut health. J. Anim. Sci. 2020, 98, 338. [Google Scholar] [CrossRef]
- Jo, H.E.; Kwon, M.; Whon, T.W.; Kim, D.W.; Yun, M.; Lee, J.; Shin, M.; Kim, S.; Choi, H. Alteration of gut microbiota after antibiotic exposure in finishing swine. Front. Microbiol. 2021, 12, 596002. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, C.; Grimaud, G.M.; Coakley, M.; OConnor, P.M.; Mathur, H.; Peterson, V.L.; ODonovan, C.M.; Lawlor, P.G.; Cotter, P.D.; Stanton, C.; et al. Modulation of the gut microbiome with nisin. Sci. Rep. 2023, 13, 7899. [Google Scholar] [CrossRef]
- Molist, F.; Manzanilla, E.G.; Pérez, J.F.; Nyachoti, C.M. Coarse, but not finely ground, dietary fibre increases intestinal firmicutes bacteroidetes ratio and reduces diarrhoea induced by experimental infection in piglets. Brit. J. Nutr. 2012, 108, 9–15. [Google Scholar] [CrossRef]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Guan, L.L.; Liu, J.X. Assessment of rumen bacteria in dairy cows with varied milk protein yield. J. Dairy Sci. 2019, 102, 5031–5041. [Google Scholar] [CrossRef]
- Kamke, J.; Kittelmann, S.; Soni, P.; Li, Y.; Tavendale, M.; Ganesh, S.; Janssen, P.H.; Shi, W.; Froula, J.; Rubin, E.M.; et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a sharpea-enriched microbiome characterised by lactic acid formation and utilisation. Microbiome 2016, 4, 56. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Zhang, J.; Liang, Z.; Du, M.; Yang, Y.; Zheng, J.; Yan, P.; Long, R.; Tong, B.; Han, J.; et al. Age-dependent variations in rumen bacterial community of mongolian cattle from weaning to adulthood. BMC Microbiol. 2022, 22, 213. [Google Scholar] [CrossRef]
- Yan, C.; Huang, S.H.; Ding, H.F.; Kwek, E.; Liu, J.H.; Chen, Z.X.; Ma, K.Y.; Chen, Z.Y. Adverse effect of oxidized cholesterol exposure on colitis is mediated by modulation of gut microbiota. J. Hazard. Mater. 2023, 459, 132057. [Google Scholar] [CrossRef]
- Ma, L.; Ni, Y.; Wang, Z.; Tu, W.; Ni, L.; Zhuge, F.; Zheng, A.; Hu, L.; Zhao, Y.; Zheng, L.; et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes 2020, 12, 1832857. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.; Gooneratne, R.; Ju, X.H. Interactions between host and gut microbiota in domestic pigs: A review. Gut Microbes 2020, 11, 310–334. [Google Scholar] [CrossRef]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-utilizing capacity varies in prevotella-versus bacteroides-dominated gut microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Zhang, Z.; Wang, J.; Qiu, X.; Wang, Q.; Yang, F.; Huang, J.; Liu, Z. Introduction of colonic and fecal microbiota from an adult pig differently affects the growth, gut health, intestinal microbiota and blood metabolome of newborn piglets. Front. Microbiol. 2021, 12, 623673. [Google Scholar] [CrossRef]
- Chen, C.; Fang, S.; Wei, H.; He, M.; Fu, H.; Xiong, X.; Zhou, Y.; Wu, J.; Gao, J.; Yang, H.; et al. Prevotella copri increases fat accumulation in pigs fed with formula diets. Microbiome 2021, 9, 175. [Google Scholar] [CrossRef]
- Jia, B.; Lin, H.; Yu, S.; Liu, N.; Yu, D.; Wu, A. Mycotoxin deoxynivalenol-induced intestinal flora disorders, dysfunction and organ damage in broilers and pigs. J. Hazard. Mater. 2023, 451, 131172. [Google Scholar] [CrossRef]
- Kiros, T.G.; Luise, D.; Derakhshani, H.; Petri, R.; Trevisi, P.; D’Inca, R.; Auclair, E.; van Kessel, A.G. Effect of live yeast saccharomyces cerevisiae supplementation on the performance and cecum microbial profile of suckling piglets. PLoS ONE 2019, 14, e219557. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Gu, X.; Lei, C.; Chen, L.; Chu, S.; Xu, G.; Doll, M.A.; Tan, Y.; Feng, W.; Siskind, L.; et al. Neutral ceramidase-dependent regulation of macrophage metabolism directs intestinal immune homeostasis and controls enteric infection. Cell Rep. 2022, 38, 110560. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Pugin, B.; Barcik, W.; Westermann, P.; Heider, A.; Wawrzyniak, M.; Hellings, P.; Akdis, C.A.; O’Mahony, L. A wide diversity of bacteria from the human gut produces and degrades biogenic amines. Microb. Ecol. Health Dis. 2017, 28, 1353881. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, M.; Yan, E.; Wang, Y.; Ma, C.; Zhang, P.; Yin, J. Dietary malic acid supplementation induces skeletal muscle fiber-type transition of weaned piglets and further improves meat quality of finishing pigs. Front. Nutr. 2022, 8, 825495. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, Y.; Li, S.; Chang, Z.; Liu, H.; Zhang, D.; Wang, S.; Zhang, X.; Wang, J. Maternal malic acid may ameliorate oxidative stress and inflammation in sows through modulating gut microbiota and host metabolic profiles during late pregnancy. Antioxidants 2024, 13, 253. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | CON 1 | FMT 2 | p-Value |
|---|---|---|---|
| Initial weight (kg) | 19.79 ± 0.31 | 19.58 ± 0.30 | 0.620 |
| Final weight (kg) | 35.41 ± 0.89 | 38.60 ± 1.31 | 0.055 |
| ADG (g/d) | 558.10 ± 33.87 | 679.52 ± 47.15 | 0.046 |
| Items | CON 1 | FMT 2 | p-Value |
|---|---|---|---|
| DM | 86.8 ± 0.09 | 86.08 ± 0.27 | 0.039 |
| GE | 87.96 ± 0.06 | 88.65 ± 0.10 | <0.001 |
| EE | 86.31 ± 0.58 | 84.6 ± 0.84 | 0.122 |
| CP | 91.33 ± 0.22 | 90.93 ± 0.15 | 0.178 |
| CF | 42.08 ± 0.83 | 47.76 ± 1.62 | 0.011 |
| ASH | 64.06 ± 0.53 | 67.42 ± 0.40 | <0.001 |
| Ca | 50.59 ± 0.87 | 55.12 ± 1.56 | 0.026 |
| P | 48.95 ± 0.75 | 51.77 ± 1.44 | 0.102 |
| Items | CON 1 | FMT 2 | p-Value |
|---|---|---|---|
| CAT (U/mL) | 20.00 ± 2.86 | 30.79 ± 3.66 | 0.039 |
| TAOC (U/mL) | 2.38 ± 0.48 | 3.58 ± 1.14 | 0.351 |
| TSOD (U/mL) | 56.43 ± 5.05 | 70.97 ± 3.09 | 0.034 |
| MDA (nmol/mL) | 15.05 ± 3.31 | 5.21 ± 0.63 | 0.027 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Han, L.; Zhou, F.; Wu, Z.; Zhang, L.; Xie, R.; Jiang, F.; Tian, Q.; Huang, X. Ningxiang Pig-Derived Microbiota Affects the Growth Performance, Gut Microbiota, and Serum Metabolome of Nursery Pigs. Animals 2024, 14, 2450. https://doi.org/10.3390/ani14172450
Li H, Han L, Zhou F, Wu Z, Zhang L, Xie R, Jiang F, Tian Q, Huang X. Ningxiang Pig-Derived Microbiota Affects the Growth Performance, Gut Microbiota, and Serum Metabolome of Nursery Pigs. Animals. 2024; 14(17):2450. https://doi.org/10.3390/ani14172450
Chicago/Turabian StyleLi, Hongkun, Li Han, Feng Zhou, Zichen Wu, Longlin Zhang, Renjie Xie, Feng Jiang, Qiyu Tian, and Xingguo Huang. 2024. "Ningxiang Pig-Derived Microbiota Affects the Growth Performance, Gut Microbiota, and Serum Metabolome of Nursery Pigs" Animals 14, no. 17: 2450. https://doi.org/10.3390/ani14172450
APA StyleLi, H., Han, L., Zhou, F., Wu, Z., Zhang, L., Xie, R., Jiang, F., Tian, Q., & Huang, X. (2024). Ningxiang Pig-Derived Microbiota Affects the Growth Performance, Gut Microbiota, and Serum Metabolome of Nursery Pigs. Animals, 14(17), 2450. https://doi.org/10.3390/ani14172450