
Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal and Phenotypic Data
2.2. Genotyping and Quality Control
2.3. Detection and Classification of ROH
2.4. Estimation of Pedigree Inbreeding Coefficients (FPED)
2.5. Estimation of Genomic Inbreeding Coefficient
3. Results and Discussion
3.1. Equivalent Complete Generations (ECG)
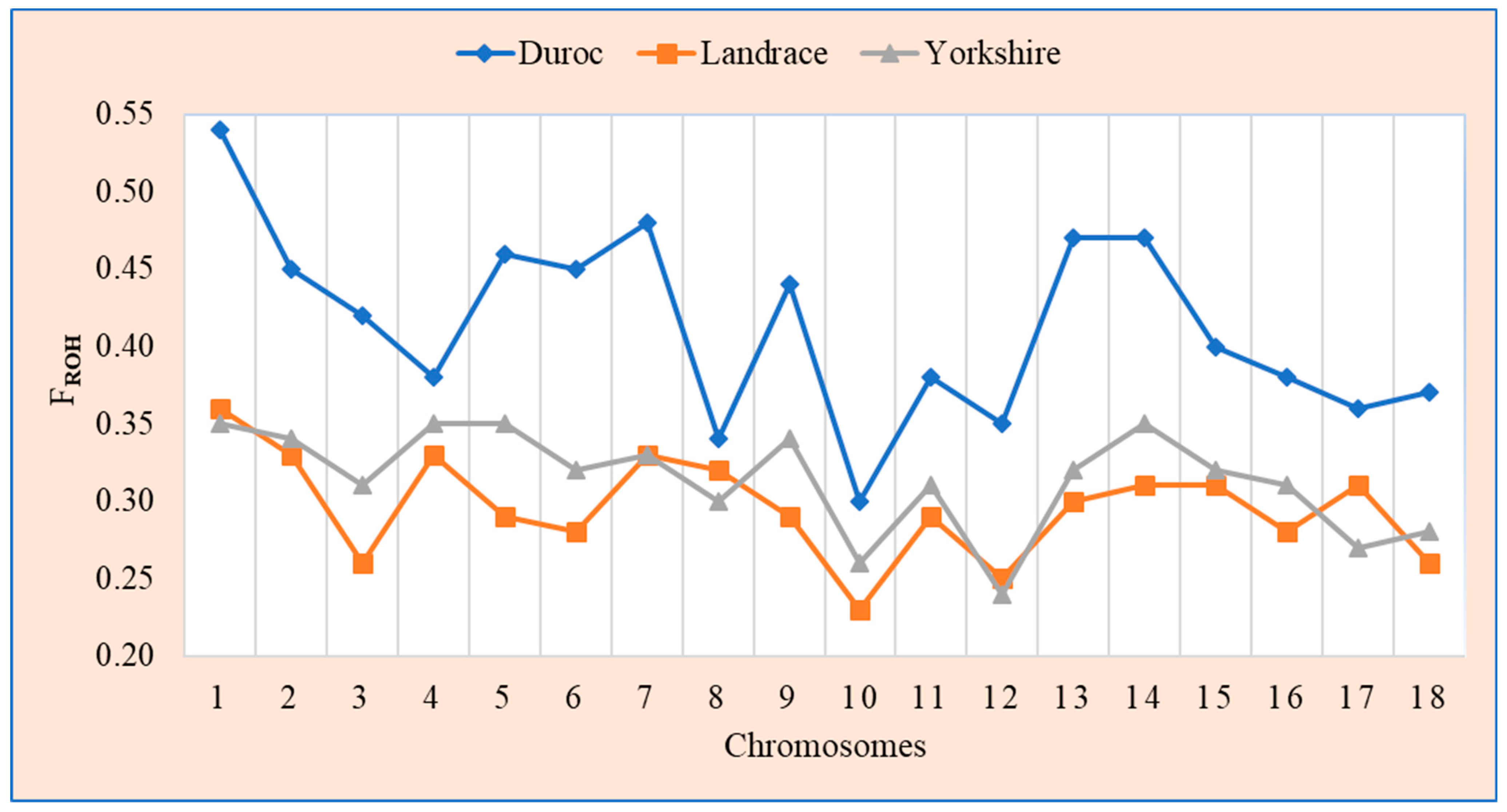
3.2. Genomic Distribution of Runs of Homozygosity
3.3. Genomic and Pedigree Inbreeding Coefficient
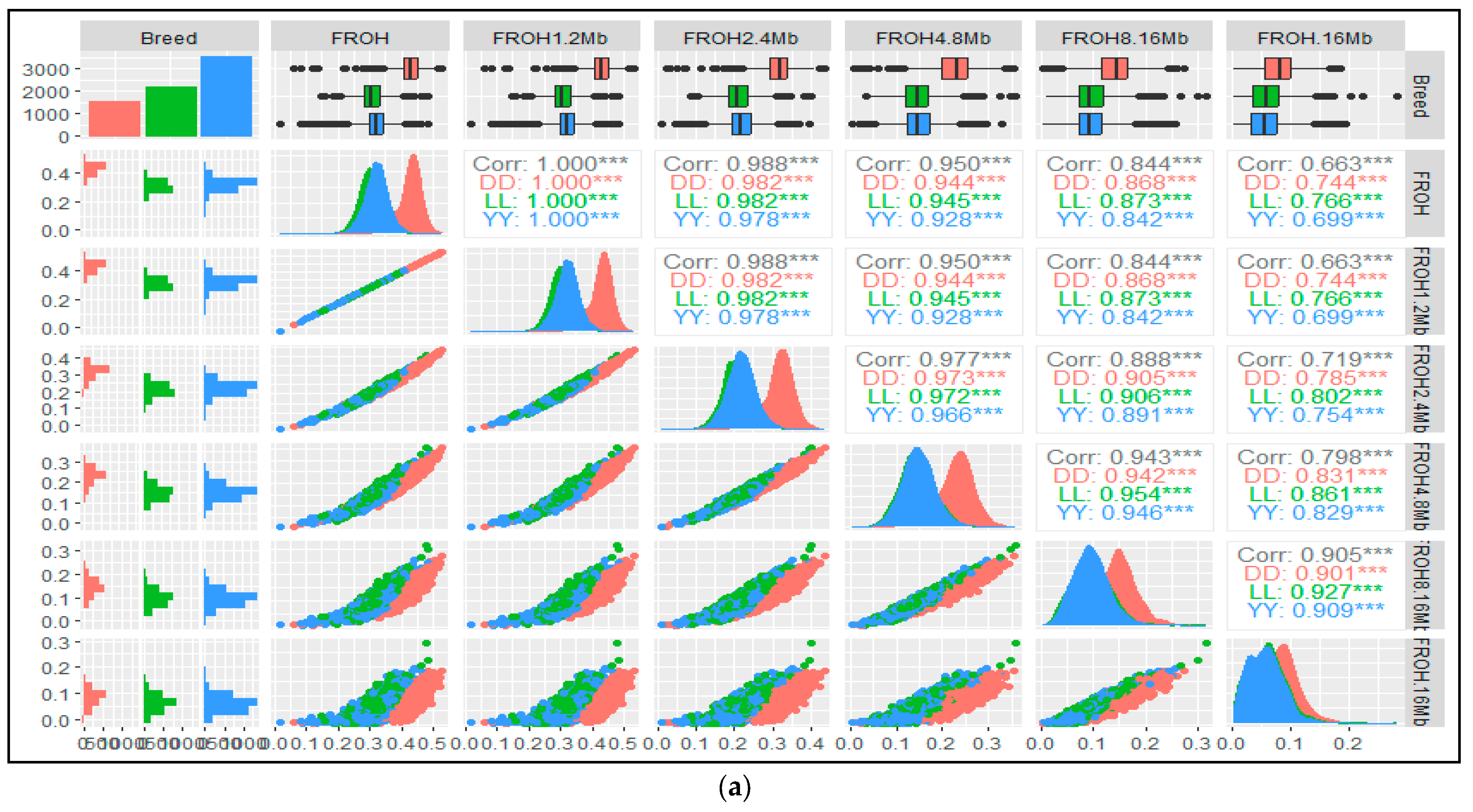
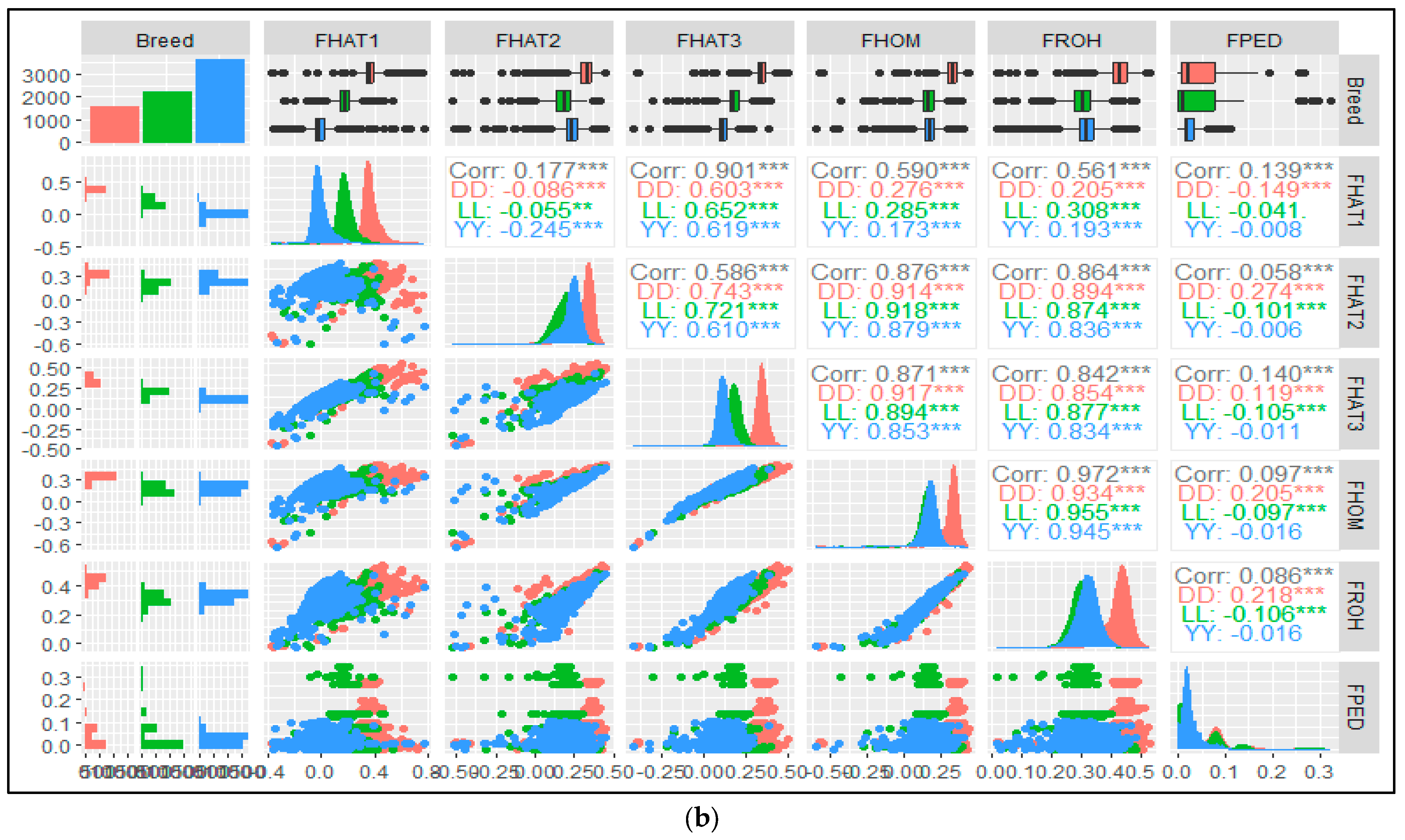
3.4. Genomic and Pedigree Inbreeding Coefficients Correlations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Polašek, O.; Hayward, C.; Bellenguez, C.; Vitart, V.; Kolčić, I.; McQuillan, R.; Saftić, V.; Gyllensten, U.; Wilson, J.F.; Rudan, I. Comparative assessment of methods for estimating individual genome-wide homozygosity-by-descent from human genomic data. BMC Genom. 2010, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Ciani, E.; Sardina, M.; Sottile, G.; Pilla, F.; Portolano, B.; Consortium, B.O.I. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-wide scan for runs of homozygosity identifies candidate genes in three pig breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, T.; Liu, Y.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; Gao, H.; Liu, G.E. Genome-wide assessment of runs of homozygosity in Chinese wagyu beef cattle. Animals 2020, 10, 1425. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.; Megens, H.-J.; Madsen, O.; Paudel, Y.; Frantz, L.A.; Schook, L.B.; Crooijmans, R.P.; Groenen, M.A. Regions of homozygosity in the porcine genome: Consequence of demography and the recombination landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Ząbek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef]
- Kim, E.-S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar]
- Pryce, J.E.; Haile-Mariam, M.; Goddard, M.E.; Hayes, B.J. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet. Sel. Evol. 2014, 46, 71. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Balmain, A. New-age tumour suppressors. Nature 2002, 417, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Ismail, J.; Jafar, T.; Jafary, F.; White, F.; Faruqui, A.; Chaturvedi, N. Risk factors for non-fatal myocardial infarction in young South Asian adults. Heart 2004, 90, 259–263. [Google Scholar] [CrossRef]
- Garrod, A. The incidence of alkaptonuria: A study in chemical individuality. Lancet 1902, 160, 1616–1620. [Google Scholar] [CrossRef]
- Woods, C.G.; Cox, J.; Springell, K.; Hampshire, D.J.; Mohamed, M.D.; McKibbin, M.; Stern, R.; Raymond, F.L.; Sandford, R.; Sharif, S.M. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 2006, 78, 889–896. [Google Scholar] [CrossRef]
- Fang, Y.; Hao, X.; Xu, Z.; Sun, H.; Zhao, Q.; Cao, R.; Zhang, Z.; Ma, P.; Sun, Y.; Qi, Z. Genome-wide detection of runs of homozygosity in Laiwu pigs revealed by sequencing data. Front. Genet. 2021, 12, 629966. [Google Scholar] [CrossRef]
- Dixit, S.; Singh, S.; Ganguly, I.; Bhatia, A.K.; Sharma, A.; Kumar, N.A.; Dang, A.K.; Jayakumar, S. Genome-wide runs of homozygosity revealed selection signatures in Bos indicus. Front. Genet. 2020, 11, 92. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.d.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishifar, S.M.; Moradi-Shahrbabak, H.; Fallahi, M.H.; Jalil Sarghale, A.; Moradi-Shahrbabak, M.; Abdollahi-Arpanahi, R.; Khansefid, M. Genomic measures of inbreeding coefficients and genome-wide scan for runs of homozygosity islands in Iranian river buffalo, Bubalus bubalis. BMC Genet. 2020, 21, 16. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Australia, 2013. [Google Scholar]
- Coster, A. Pedigree: Pedigree Functions. R Package Version 1.4.2. 2013. Available online: https://cran.r-project.org/web/packages/pedigree/index.html (accessed on 25 April 2024).
- Maignel, L.; Boichard, D.; Verrier, E. Genetic variability of French dairy breeds estimated from pedigree information. Interbull Bull. 1996, 49. [Google Scholar]
- Nishio, M.; Inoue, K.; Ogawa, S.; Ichinoseki, K.; Arakawa, A.; Fukuzawa, Y.; Okamura, T.; Kobayashi, E.; Taniguchi, M.; Oe, M. Comparing pedigree and genomic inbreeding coefficients, and inbreeding depression of reproductive traits in Japanese Black cattle. BMC Genom. 2023, 24, 376. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Leutenegger, A.-L.; Prum, B.; Génin, E.; Verny, C.; Lemainque, A.; Clerget-Darpoux, F.; Thompson, E.A. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003, 73, 516–523. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Yengo, L.; Zhu, Z.; Wray, N.R.; Weir, B.S.; Yang, J.; Robinson, M.R.; Visscher, P.M. Detection and quantification of inbreeding depression for complex traits from SNP data. Proc. Natl. Acad. Sci. USA 2017, 114, 8602–8607. [Google Scholar] [CrossRef]
- Alemu, S.W.; Kadri, N.K.; Harland, C.; Faux, P.; Charlier, C.; Caballero, A.; Druet, T. An evaluation of inbreeding measures using a whole-genome sequenced cattle pedigree. Heredity 2021, 126, 410–423. [Google Scholar] [CrossRef]
- Nietlisbach, P.; Muff, S.; Reid, J.M.; Whitlock, M.C.; Keller, L.F. Nonequivalent lethal equivalents: Models and inbreeding metrics for unbiased estimation of inbreeding load. Evol. Appl. 2019, 12, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Gazal, S.; Sahbatou, M.; Perdry, H.; Letort, S.; Génin, E.; Leutenegger, A.-L. Inbreeding coefficient estimation with dense SNP data: Comparison of strategies and application to HapMap III. Hum. Hered. 2014, 77, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Systems of mating. I. The biometric relations between parent and offspring. Genetics 1921, 6, 111. [Google Scholar] [CrossRef]
- Xu, Z.; Mei, S.; Zhou, J.; Zhang, Y.; Qiao, M.; Sun, H.; Li, Z.; Li, L.; Dong, B.; Oyelami, F.O. Genome-wide assessment of runs of homozygosity and estimates of genomic inbreeding in a chinese composite pig breed. Front. Genet. 2021, 12, 720081. [Google Scholar] [CrossRef]
- Shi, L.; Wang, L.; Liu, J.; Deng, T.; Yan, H.; Zhang, L.; Liu, X.; Gao, H.; Hou, X.; Wang, L. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population. J. Anim. Sci. Biotechnol. 2020, 11, 46. [Google Scholar] [CrossRef]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q. Assessment of autozygosity derived from runs of homozygosity in Jinhua pigs disclosed by sequencing data. Front. Genet. 2019, 10, 274. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Zhang, S.; Zhang, K.; Peng, X.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Genome-wide patterns of homozygosity and relevant characterizations on the population structure in Piétrain pigs. Genes 2020, 11, 577. [Google Scholar] [CrossRef]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Curik, I.; Sölkner, J.; Stipic, N. Effects of models with finite loci, selection, dominance, epistasis and linkage on inbreeding coefficients based on pedigree and genotypic information. J. Anim. Breed. Genet. 2002, 119, 101–115. [Google Scholar] [CrossRef]
- Carothers, A.D.; Rudan, I.; Kolcic, I.; Polasek, O.; Hayward, C.; Wright, A.F.; Campbell, H.; Teague, P.; Hastie, N.; Weber, J.L. Estimating human inbreeding coefficients: Comparison of genealogical and marker heterozygosity approaches. Ann. Hum. Genet. 2006, 70, 666–676. [Google Scholar] [CrossRef]
- Zavarez, L.B.; Utsunomiya, Y.T.; Carmo, A.S.; Neves, H.H.; Carvalheiro, R.; Ferenčaković, M.; Pérez O’Brien, A.M.; Curik, I.; Cole, J.B.; Van Tassell, C.P. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 2015, 6, 125037. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Medland, S.E.; Ferreira, M.A.R.; Morley, K.I.; Zhu, G.; Cornes, B.K.; Montgomery, G.W.; Martin, N.G. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006, 2, e41. [Google Scholar] [CrossRef]
- Villanueva, B.; Fernández, A.; Saura, M.; Caballero, A.; Fernández, J.; Morales-González, E.; Toro, M.A.; Pong-Wong, R. The value of genomic relationship matrices to estimate levels of inbreeding. Genet. Sel. Evol. 2021, 53, 42. [Google Scholar] [CrossRef] [PubMed]
- Dadousis, C.; Ablondi, M.; Cipolat-Gotet, C.; Van Kaam, J.-T.; Marusi, M.; Cassandro, M.; Sabbioni, A.; Summer, A. Genomic inbreeding coefficients using imputed genotypes: Assessing different estimators in Holstein-Friesian dairy cows. J. Dairy Sci. 2022, 105, 5926–5945. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Mastrangelo, S.; Catillo, G.; Senczuk, G.; Ciampolini, R. Insights into genetic diversity, runs of homozygosity and heterozygosity-rich regions in Maremmana semi-feral cattle using pedigree and genomic data. Animals 2020, 10, 2285. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Pedigree | FROH | FHOM | FHAT1 | FHAT2 | FHAT3 |
|---|---|---|---|---|---|---|
| Duroc | 76,987 | 1586 | 1586 | 1586 | 1586 | 1586 |
| Landrace | 155,628 | 2256 | 2556 | 2256 | 2256 | 2256 |
| Yorkshire | 409,018 | 3646 | 3646 | 3646 | 3646 | 3646 |
| Breed | N | Mean | SD | CV | Min | Max | Range | Median |
|---|---|---|---|---|---|---|---|---|
| Duroc | 76,980 | 14.89 | 0.68 | 4.55 | 3 | 15 | 12 | 15 |
| Landrace | 155,569 | 14.94 | 0.44 | 2.95 | 4 | 15 | 11 | 15 |
| Yorkshire | 408,890 | 14.92 | 0.54 | 3.61 | 1 | 15 | 14 | 15 |
| All genotype | 641,439 | 14.92 | 0.54 | 3.60 | 1 | 15 | 14 | 15 |
| ROH_C | Duroc | Landrace | Yorkshire | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ROH_N | ML(Mb) | P (%) | ROH_N | ML(Mb) | P (%) | ROH_N | ML(Mb) | P (%) | |
| 1–2 Mb | 462,355 | 1.09 | 65.18 | 576,546 | 1.072 | 70.58 | 983,942 | 1.085 | 70.19 |
| 2–4 Mb | 143,300 | 2.78 | 20.20 | 149,871 | 2.739 | 18.35 | 264,790 | 2.736 | 18.89 |
| 4–8 Mb | 68,029 | 5.52 | 9.59 | 59,050 | 5.444 | 7.23 | 99,388 | 5.463 | 7.09 |
| 8–16 Mb | 24,820 | 10.87 | 3.50 | 20,723 | 10.789 | 2.54 | 35,851 | 10.855 | 2.56 |
| >16 Mb | 10,880 | 33.33 | 1.53 | 10,708 | 36.161 | 1.31 | 17,810 | 33.321 | 1.27 |
| Total (>1) | 709,384 | 53.59 | 100 | 816,898 | 56.205 | 100 | 1,401,781 | 53.461 | 100 |
| SSC | Duroc | Landrace | Yorkshire | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ROH_N | ML (Mb) | P (%) | ROH_N | ML (Mb) | P (%) | ROH_N | ML (Mb) | P (%) | |
| 1 | 97,815 | 2.77 | 14.31 | 1,100,18 | 2.31 | 14.05 | 175,188 | 2.29 | 13.12 |
| 2 | 41,290 | 2.81 | 6.04 | 47,899 | 2.48 | 6.12 | 80,453 | 2.50 | 6.02 |
| 3 | 37,143 | 2.61 | 5.43 | 39,549 | 2.10 | 5.05 | 71,973 | 2.26 | 5.39 |
| 4 | 43,602 | 1.96 | 6.38 | 52,394 | 2.01 | 6.69 | 94,402 | 1.94 | 7.07 |
| 5 | 37,686 | 2.13 | 5.51 | 42,342 | 1.74 | 5.41 | 75,504 | 1.87 | 5.65 |
| 6 | 42,007 | 2.91 | 6.15 | 45,206 | 2.36 | 5.77 | 80,958 | 2.47 | 6.06 |
| 7 | 37,957 | 2.69 | 5.55 | 46,366 | 2.14 | 5.92 | 77,746 | 2.11 | 5.82 |
| 8 | 33,403 | 2.40 | 4.89 | 41,007 | 2.58 | 5.24 | 65,949 | 2.48 | 4.94 |
| 9 | 42,225 | 2.53 | 6.18 | 42,991 | 2.37 | 5.49 | 77,653 | 2.46 | 5.81 |
| 10 | 25,340 | 1.47 | 3.71 | 26,857 | 1.52 | 3.43 | 51,211 | 1.47 | 3.83 |
| 11 | 25,444 | 2.06 | 3.72 | 28,588 | 2.00 | 3.65 | 52,749 | 1.85 | 3.95 |
| 12 | 21,676 | 1.76 | 3.17 | 23,394 | 1.61 | 2.99 | 36,595 | 1.65 | 2.74 |
| 13 | 50,978 | 3.21 | 7.46 | 56,999 | 2.61 | 7.28 | 90,905 | 2.76 | 6.81 |
| 14 | 39,929 | 2.89 | 5.84 | 46,477 | 2.32 | 5.94 | 92,222 | 2.10 | 6.90 |
| 15 | 39,661 | 2.71 | 5.80 | 51,741 | 2.29 | 6.61 | 79,743 | 2.46 | 5.97 |
| 16 | 23,152 | 2.27 | 3.39 | 25,944 | 2.13 | 3.31 | 47,058 | 2.09 | 3.52 |
| 17 | 24,985 | 1.60 | 3.66 | 33,491 | 1.44 | 4.28 | 49,064 | 1.42 | 3.67 |
| 18 | 19,252 | 1.87 | 2.82 | 21,610 | 1.69 | 2.76 | 36,265 | 1.74 | 2.72 |
| FROH Class | Duroc | Landrace | Yorkshire | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean ± SD | CV | n | Mean ± SD | CV | n | Mean ± SD | CV | |
| FROH1–2Mb | 1584 | 0.42 ± 0.05 | 12.63 | 2251 | 0.30 ± 0.04 | 13.62 | 3643 | 0.32 ± 0.04 | 13.94 |
| FROH2–4Mb | 1582 | 0.31 ± 0.05 | 16.12 | 2251 | 0.21 ± 0.04 | 19.8 | 3643 | 0.22 ± 0.04 | 19.44 |
| FROH4–8Mb | 1579 | 0.22 ± 0.05 | 20.85 | 2249 | 0.14 ± 0.04 | 27.34 | 3641 | 0.15 ± 0.04 | 26.39 |
| FROH8–16Mb | 1577 | 0.14 ± 0.04 | 29.02 | 2245 | 0.09 ± 0.04 | 38.48 | 3636 | 0.09 ± 0.03 | 36.86 |
| FROH > 16Mb | 1567 | 0.08 ± 0.03 | 40.79 | 2212 | 0.06 ± 0.03 | 52.14 | 3587 | 0.06 ± 0.03 | 51.32 |
| FPED | 76,980 | 0.02 ± 0.03 | 186.69 | 155,569 | 0.01 ± 0.02 | 178.01 | 408,891 | 0.02 ± 0.02 | 146.43 |
| Estimator | Duroc | Landrace | Yorkshire | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean ± SD | CV | n | Mean ± SD | CV | n | Mean ± SD | CV | |
| FROH | 1584 | 0.42 ± 0.05 | 12.63 | 2251 | 0.30 ± 0.04 | 13.62 | 3643 | 0.32 ± 0.04 | 13.94 |
| FHOM | 1586 | 0.31 ± 0.09 | 29.28 | 2256 | 0.16 ± 0.09 | 55.49 | 3646 | 0.17 ± 0.17 | 42.17 |
| FHAT1 | 1586 | 0.50 ± 2.42 | 485.78 | 2256 | 0.28 ± 2.04 | 718.68 | 3646 | 0.04 ± 1.26 | 3565.36 |
| FHAT2 | 1586 | 0.23 ± 1.46 | 631.28 | 2256 | 0.09 ± 1.74 | 1855.5 | 3646 | 0.20 ± 0.97 | 490.98 |
| FHAT3 | 1586 | 0.36 ± 0.59 | 162.03 | 2256 | 0.19 ± 0.21 | 113.31 | 3646 | 0.12 ± 0.16 | 141.08 |
| FPED | 76,980 | 0.02 ± 0.03 | 186.69 | 155,569 | 0.01 ± 0.02 | 178.01 | 408,891 | 0.02 ± 0.02 | 146.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekonnen, K.T.; Lee, D.-H.; Cho, Y.-G.; Son, A.-Y.; Seo, K.-S. Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip. Animals 2024, 14, 2621. https://doi.org/10.3390/ani14172621
Mekonnen KT, Lee D-H, Cho Y-G, Son A-Y, Seo K-S. Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip. Animals. 2024; 14(17):2621. https://doi.org/10.3390/ani14172621
Chicago/Turabian StyleMekonnen, Kefala Taye, Dong-Hui Lee, Young-Gyu Cho, Ah-Yeong Son, and Kang-Seok Seo. 2024. "Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip" Animals 14, no. 17: 2621. https://doi.org/10.3390/ani14172621
APA StyleMekonnen, K. T., Lee, D.-H., Cho, Y.-G., Son, A.-Y., & Seo, K.-S. (2024). Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip. Animals, 14(17), 2621. https://doi.org/10.3390/ani14172621