
Genome-Wide Association Study of Body Conformation Traits in Tashi Goats (Capra hircus)

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
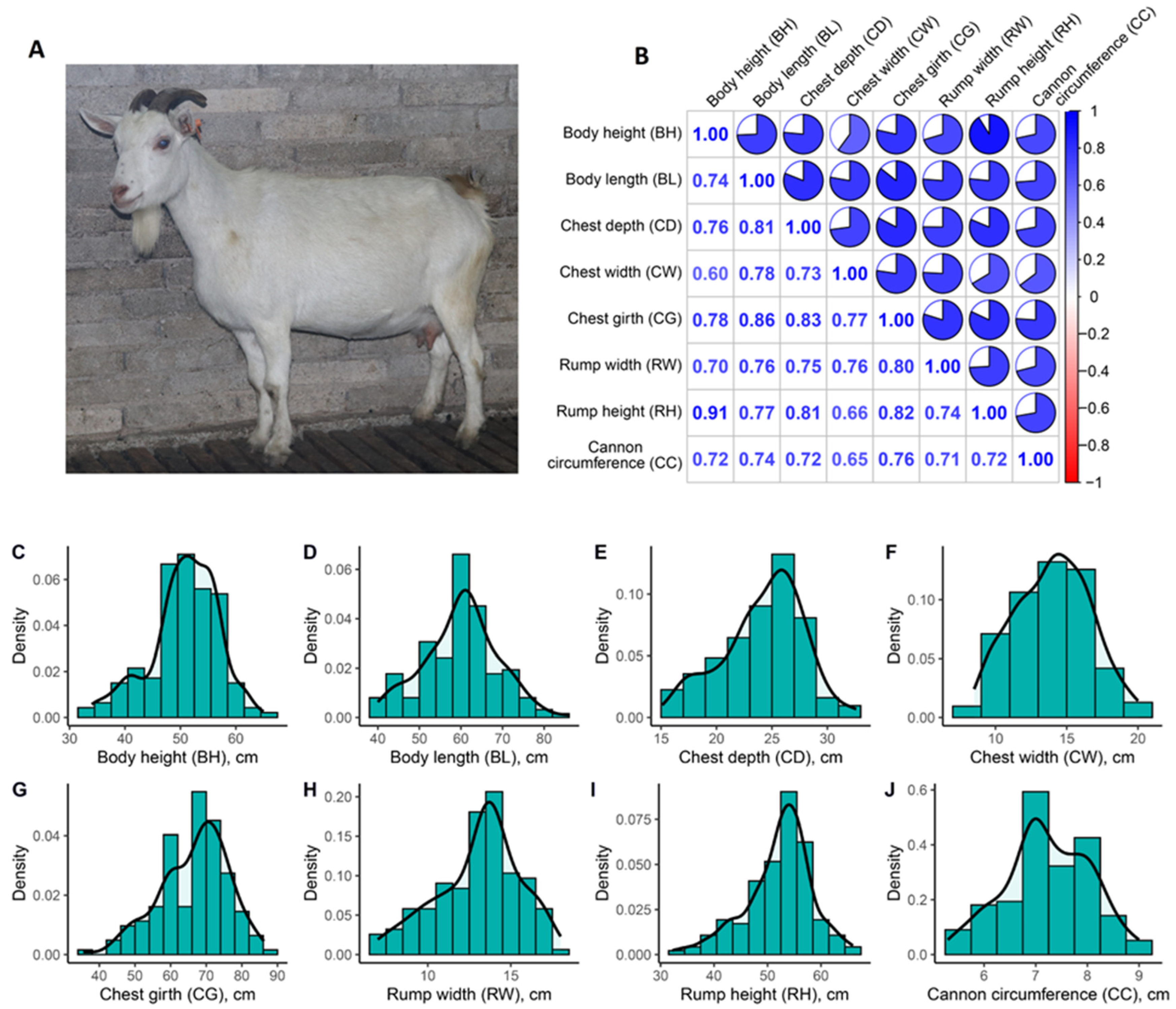
2.1. Animals and Phenotyping
- (1)
- Body height (BH) was measured from the ground to the top of the withers.
- (2)
- Body length (BL) was measured from the point of the shoulder to the pin bone.
- (3)
- Chest depth (CD) was measured as the distance between the top of the spine and the bottom of the body at the beginning of the last rib.
- (4)
- Chest width (CW) was measured as the inside surface of the chest between the tops of the front legs.
- (5)
- Chest girth (CG) was measured as the chest circumference behind the shoulder blades in a vertical plane.
- (6)
- Rump width (RW) was measured as the distance between the most posterior points of the pin bones (ischial tuberosities).
- (7)
- Rump height (RH) was measured from the ground to the top of the pin bones.
- (8)
- Cannon bone circumference (CC) was measured as the horizontal circumference at the thinnest point of the left forelimb.
2.2. Genome Sequencing and Genotyping
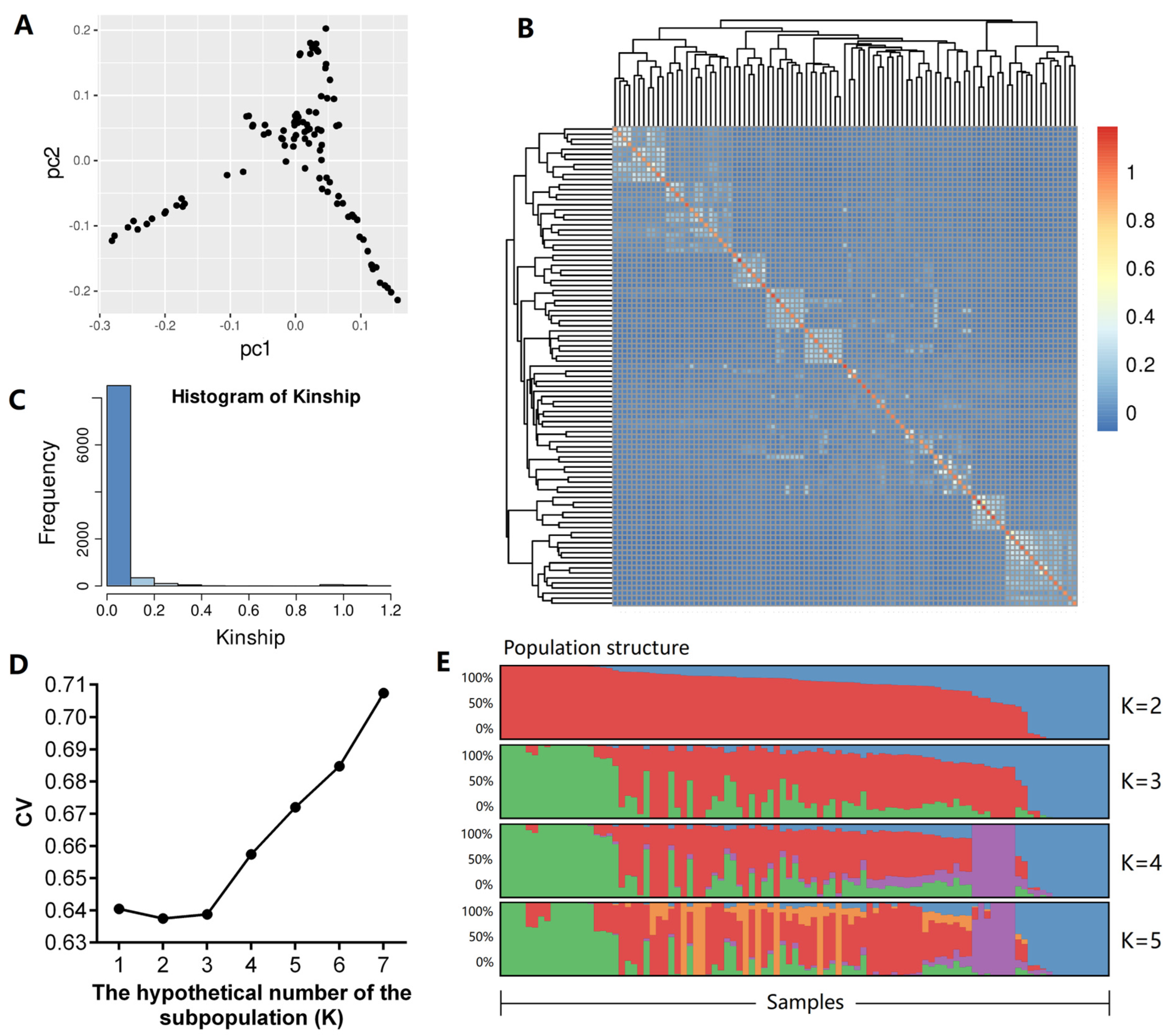
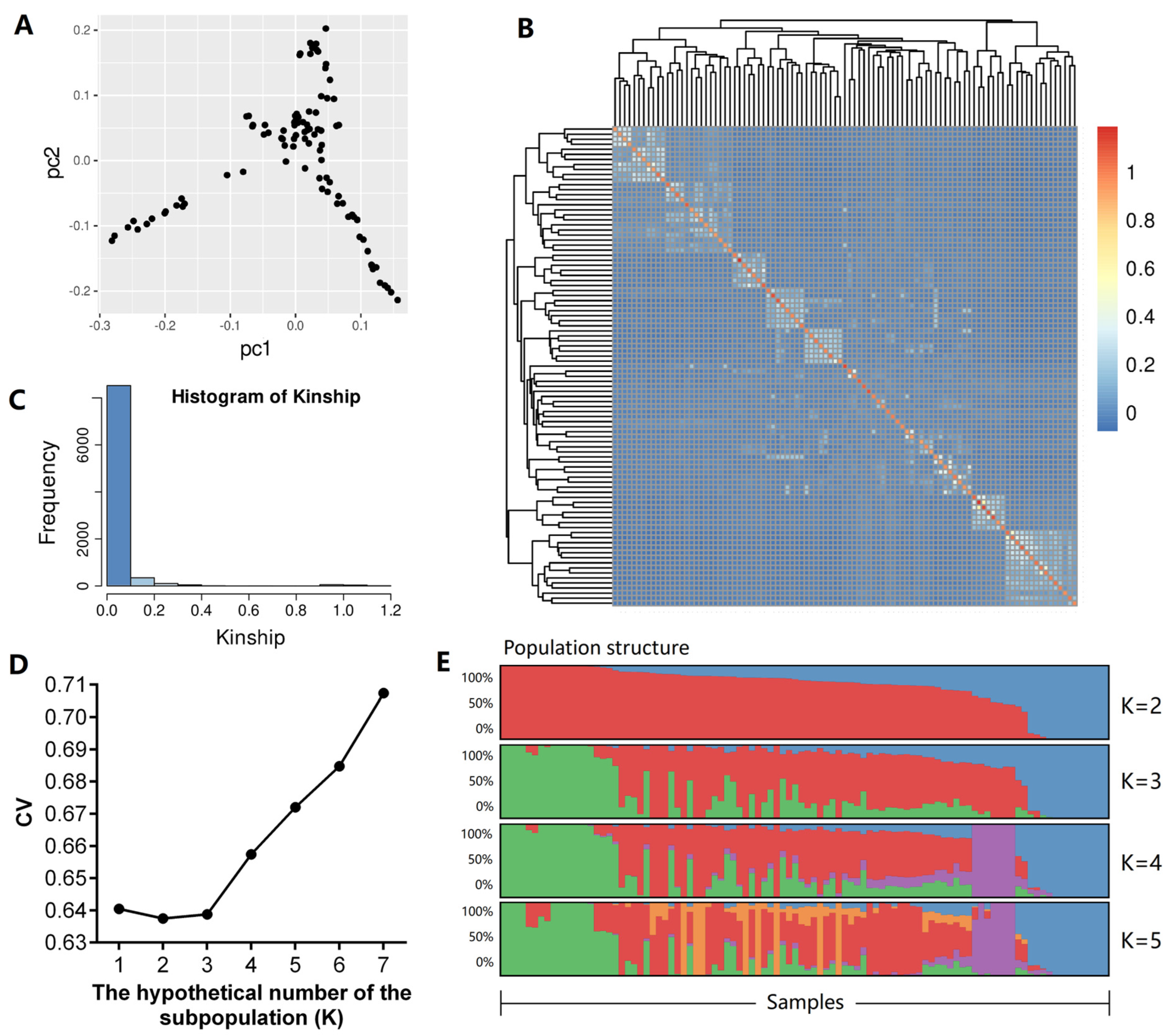
2.3. Population Structure Analysis
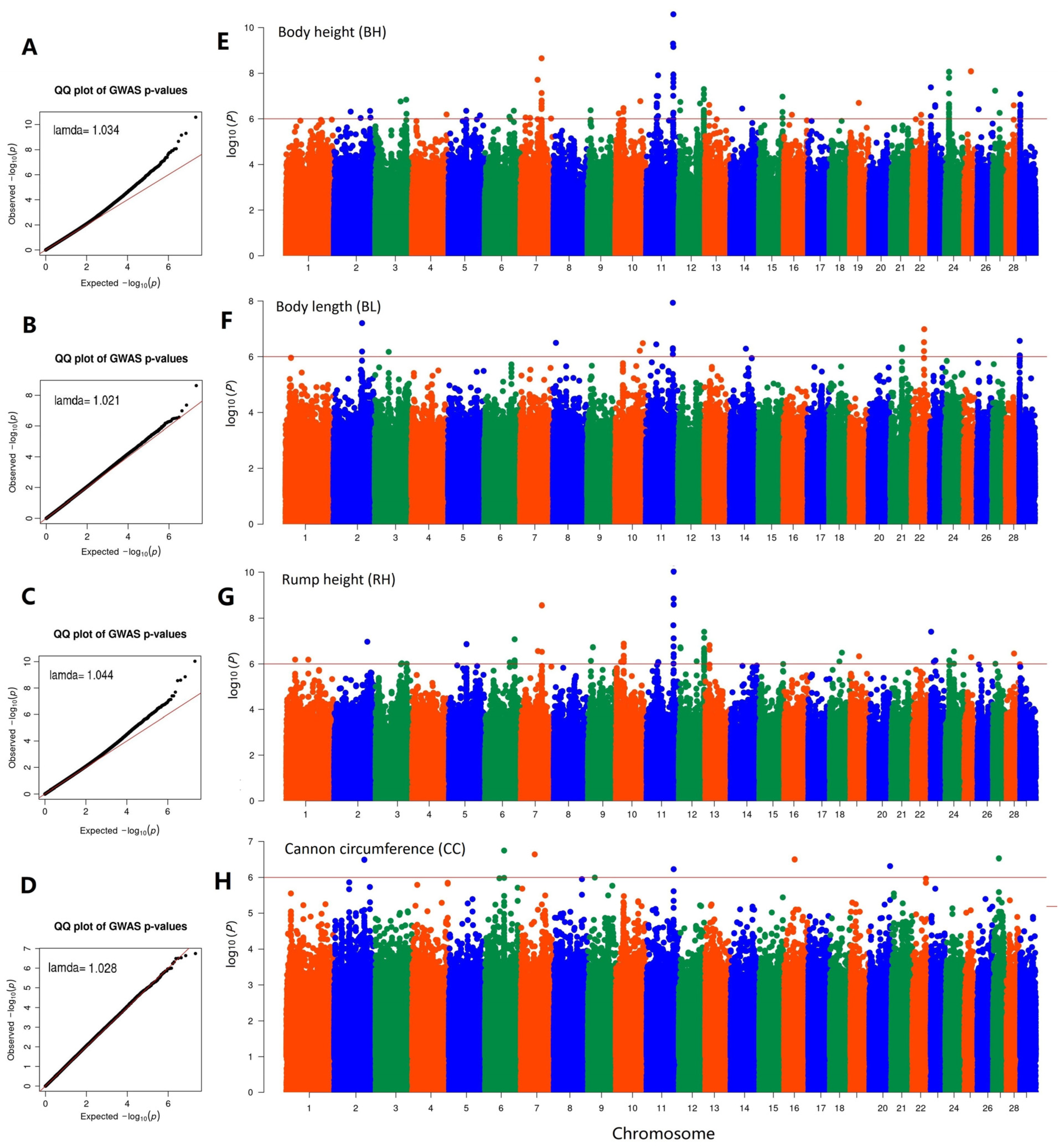
2.4. Genome-Wide Association Studies
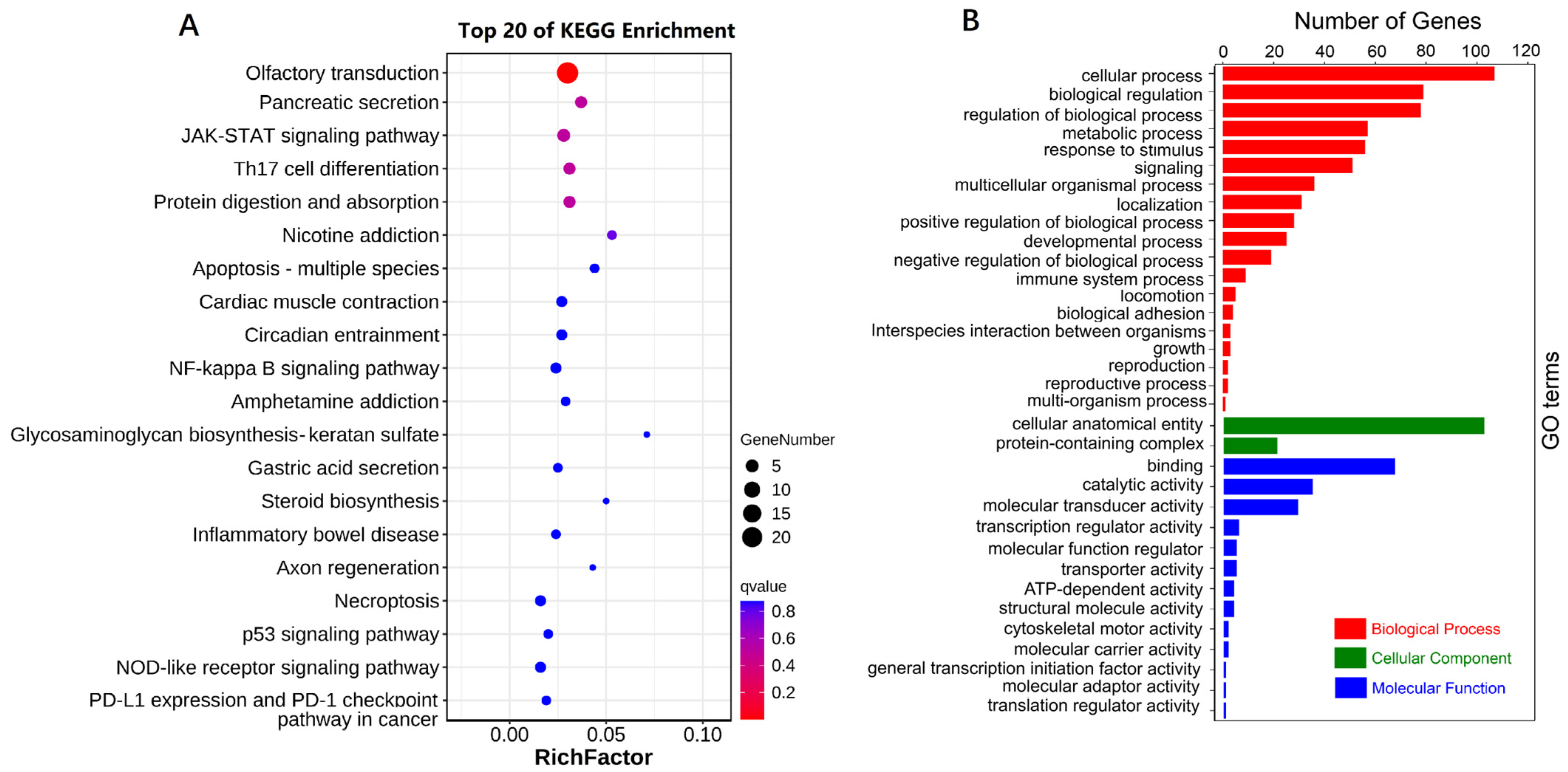
2.5. Functional Enrichment Analysis
3. Results
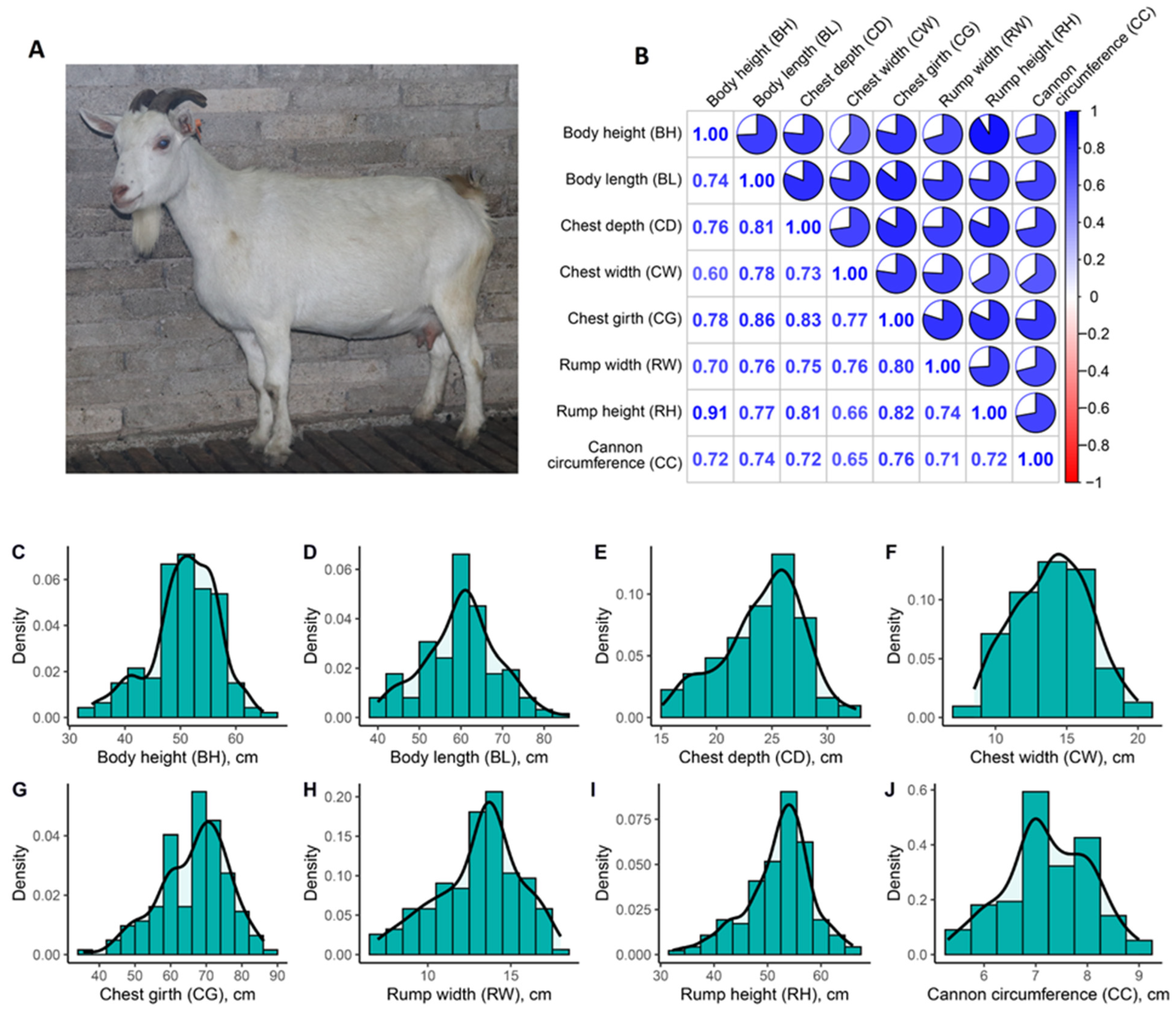
3.1. Phenotypic Analysis
3.2. Whole-Genome Sequencing and Variant Calling
3.3. Population Structure Analysis
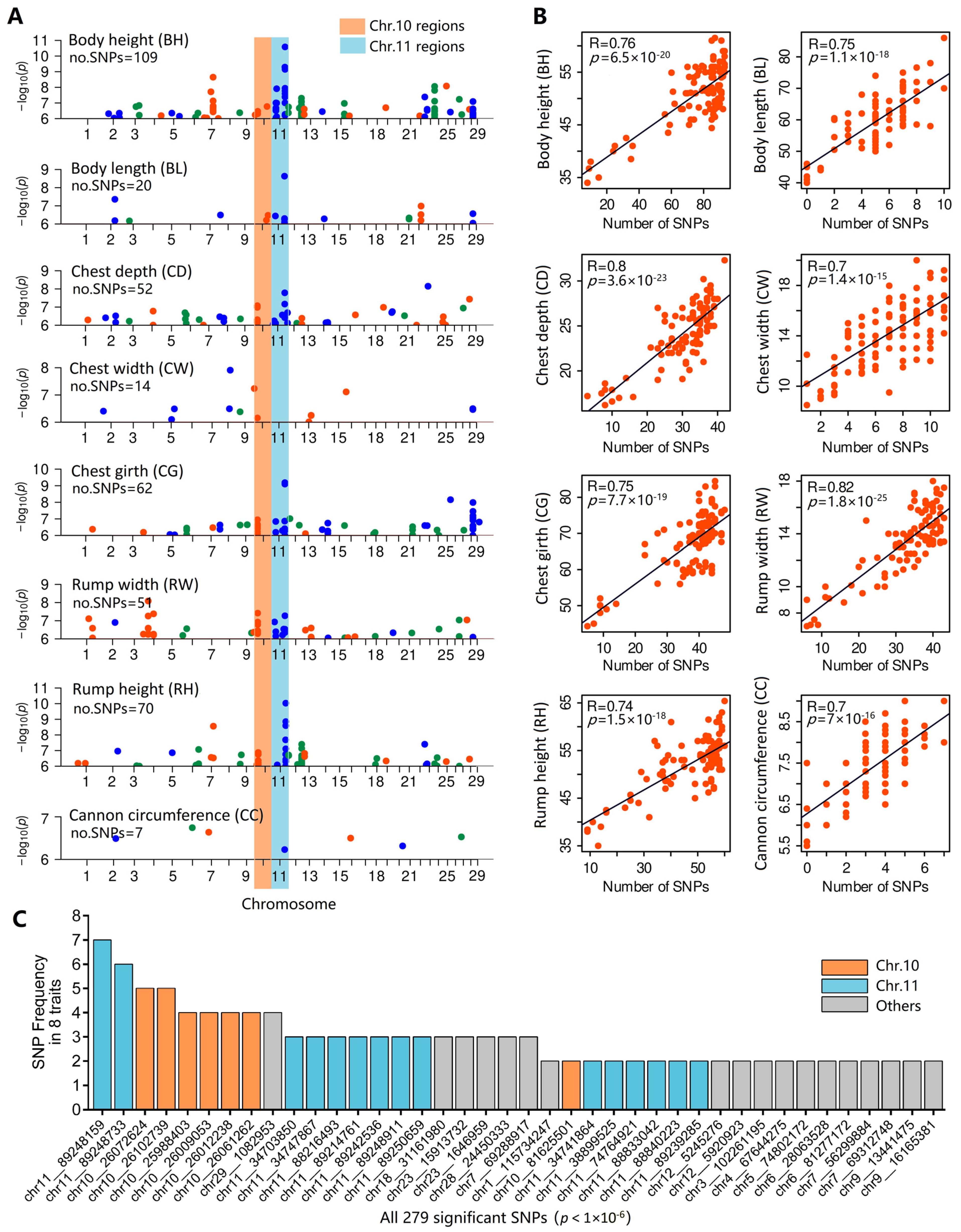
3.4. Genome-Wide Association Study
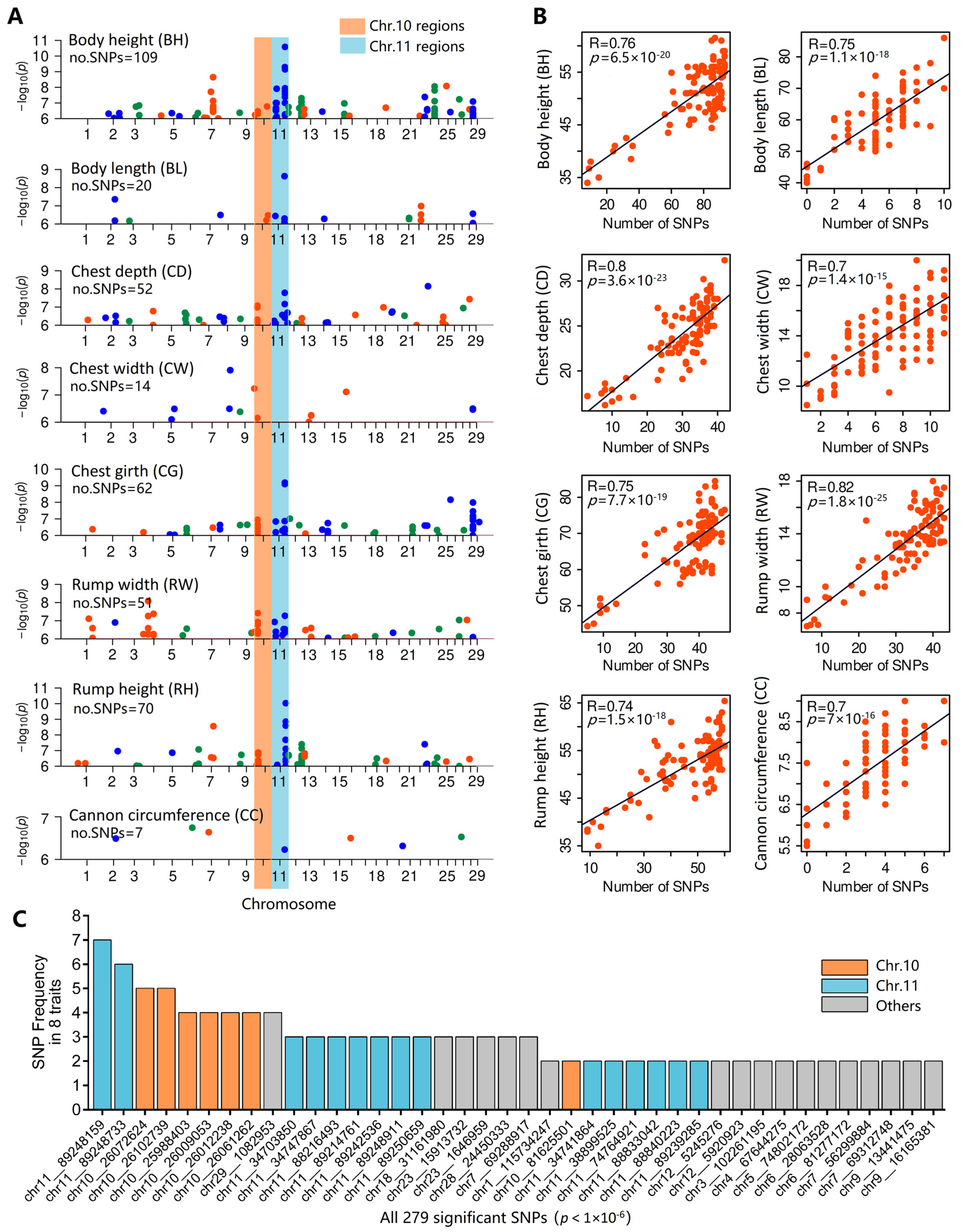
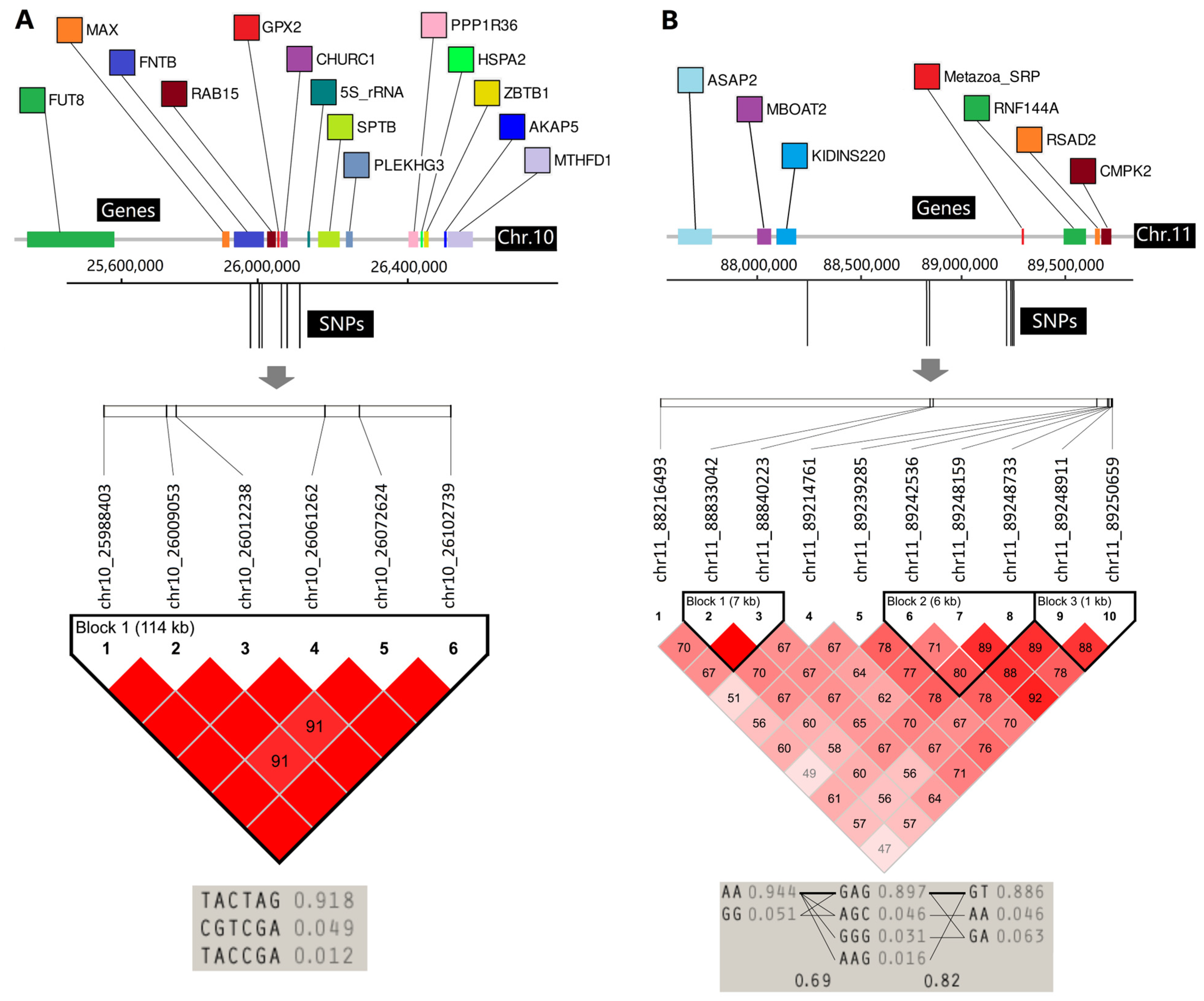
3.5. Identification of Core SNPs
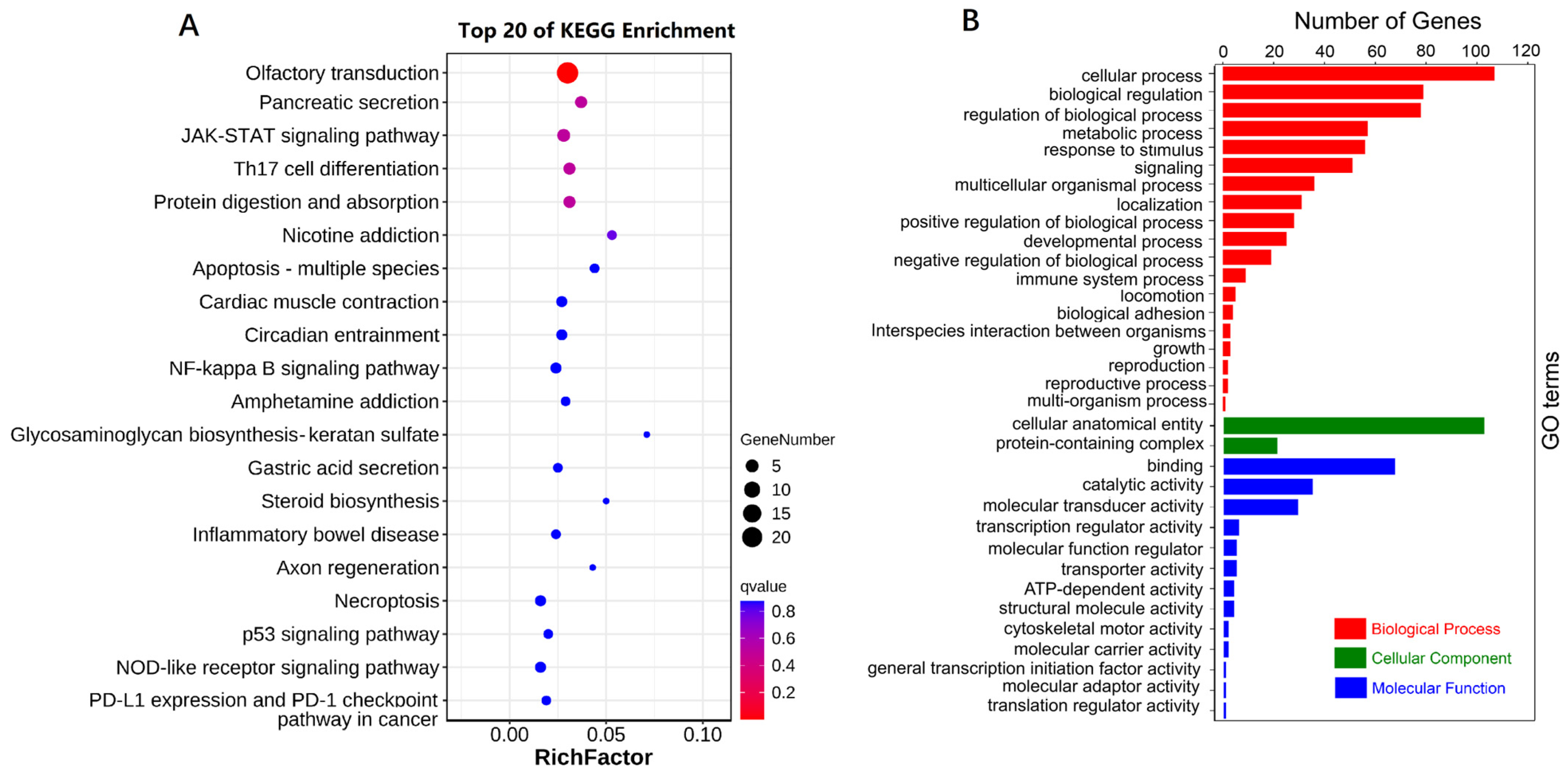
3.6. SNP Annotations and GO and KEGG Enrichment
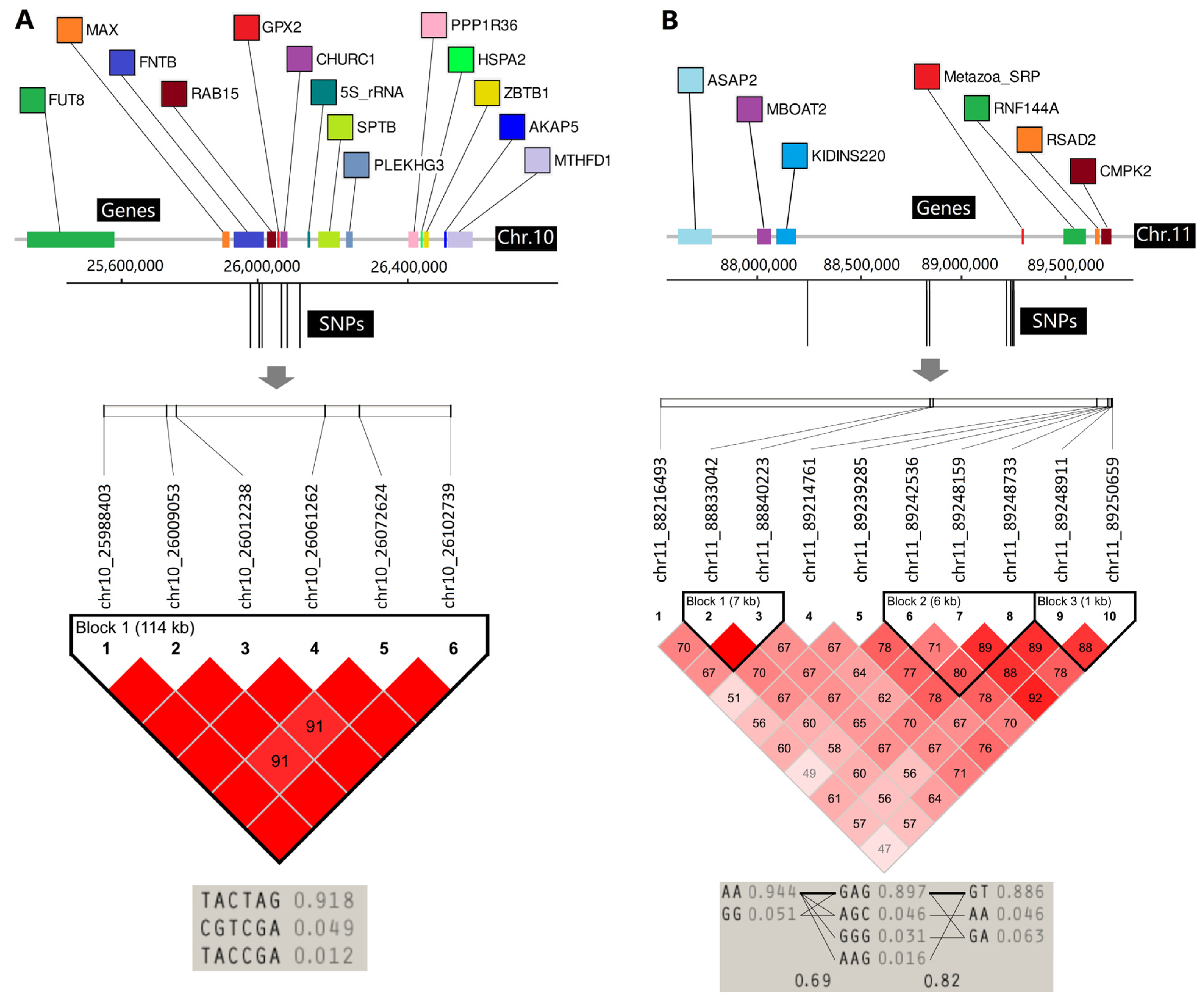
3.7. LD Analyses of Two Important Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naderi, S.; Rezaei, H.R.; Pompanon, F.; Blum, M.G.; Negrini, R.; Naghash, H.R.; Balkiz, O.; Mashkour, M.; Gaggiotti, O.E.; Ajmone-Marsan, P.; et al. The goat domestication process inferred from large-scale mitochondrial DNA analysis of wild and domestic individuals. Proc. Natl. Acad. Sci. USA 2008, 105, 17659–17664. [Google Scholar] [CrossRef]
- Mohlatlole, R.P.; Dzomba, E.F.; Muchadeyi, F.C. Addressing production challenges in goat production systems of South Africa: The genomics approach. Small Rumin. Res. 2015, 131, 43–49. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Alharbi, W.S.; Rashid, M. A review of deep learning applications in human genomics using next-generation sequencing data. Hum. Genom. 2022, 16, 26. [Google Scholar] [CrossRef]
- Haldar, A.; Pal, P.; Datta, M.; Paul, R.; Pal, S.K.; Majumdar, D.; Biswas, C.K.; Pan, S. Prolificacy and Its Relationship with Age, Body Weight, Parity, Previous Litter Size and Body Linear Type Traits in Meat-type Goats. Asian-Australas J. Anim. Sci. 2014, 27, 628–634. [Google Scholar] [CrossRef]
- Ncube, K.T.; Dzomba, E.F.; Hadebe, K.; Soma, P.; Frylinck, L.; Muchadeyi, F.C. Carcass Quality Profiles and Associated Genomic Regions of South African Goat Populations Investigated Using Goat SNP50K Genotypes. Animals 2022, 12, 364. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.P.; Fortes, M.R.S.; Bresolin, T.; Mota, L.F.M.; Albuquerque, L.G.; Carvalheiro, R. Multitrait meta-analysis identified genomic regions associated with sexual precocity in tropical beef cattle. J. Anim. Sci. 2018, 96, 4087–4099. [Google Scholar] [CrossRef]
- Luigi-Sierra, M.G.; Landi, V.; Guan, D.; Delgado, J.V.; Castello, A.; Cabrera, B.; Marmol-Sanchez, E.; Alvarez, J.F.; Gomez-Carpio, M.; Martinez, A.; et al. A genome-wide association analysis for body, udder, and leg conformation traits recorded in Murciano-Granadina goats. J. Dairy. Sci. 2020, 103, 11605–11617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, C.; Guo, Y.; She, S.; Wang, B.; Jiang, Y.; Bai, Y.; Song, X.; Li, L.; Shi, L.; et al. Screening of Deletion Variants within the Goat PRDM6 Gene and Its Effects on Growth Traits. Animals 2020, 10, 208. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, H.; Lei, C.; Pan, C.; Chen, H.; Lin, Q.; Lan, X. Effects of genetic variations within goat PITX2 gene on growth traits and mRNA expression. Anim. Biotechnol. 2020, 31, 107–114. [Google Scholar] [CrossRef]
- Liu, X.; Ma, L.; Wang, M.; Wang, K.; Li, J.; Yan, H.; Zhu, H.; Lan, X. Two indel variants of prolactin receptor (PRLR) gene are associated with growth traits in goat. Anim. Biotechnol. 2020, 31, 314–323. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.A.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Sutera, A.M.; Di Gerlando, R.; Mastrangelo, S.; Sardina, M.T.; D’Alessandro, E.; Portolano, B.; Tolone, M. Genome-wide association study for milk production traits in an economically important local dairy sheep breed. Ital. J. Anim. Sci. 2021, 20, 1500–1505. [Google Scholar] [CrossRef]
- Tilahun, Y.; Gipson, T.A.; Alexander, T.; McCallum, M.L.; Hoyt, P.R. Genome-Wide Association Study towards Genomic Predictive Power for High Production and Quality of Milk in American Alpine Goats. Int. J. Genom. 2020, 2020, 6035694. [Google Scholar] [CrossRef]
- Salgado Pardo, J.I.; Delgado Bermejo, J.V.; Gonzalez Ariza, A.; Leon Jurado, J.M.; Marin Navas, C.; Iglesias Pastrana, C.; Martinez Martinez, M.d.A.; Navas Gonzalez, F.J. Candidate Genes and Their Expressions Involved in the Regulation of Milk and Meat Production and Quality in Goats (Capra hircus). Animals 2022, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Woodward-Greene, J.; Kang, X.; Pan, M.G.; Rosen, B.; Van Tassell, C.P.; Chen, H.; Liu, G.E. Genome-wide CNV analysis revealed variants associated with growth traits in African indigenous goats. Genomics 2020, 112, 1477–1480. [Google Scholar] [CrossRef]
- Moaeen-ud-Din, M.; Muner, R.D.; Khan, M.S. Genome wide association study identifies novel candidate genes for growth and body conformation traits in goats. Sci. Rep. 2022, 12, 9891. [Google Scholar] [CrossRef]
- Easa, A.A.; Selionova, M.; Aibazov, M.; Mamontova, T.; Sermyagin, A.; Belous, A.; Abdelmanova, A.; Deniskova, T.; Zinovieva, N. Identification of Genomic Regions and Candidate Genes Associated with Body Weight and Body Conformation Traits in Karachai Goats. Genes 2022, 13, 1773. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Sun, R.; Fang, X.; Zhang, J.; Zhao, Z.; Huang, D.; Zhao, Y.; Zhao, Y. Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats. Animals 2022, 12, 548. [Google Scholar] [CrossRef]
- Ofori, S.A.; Hagan, J.K.; Kyei, F. Morphometric characterization and differentiation of West African Dwarf goat populations in Ghana. Trop. Anim. Health Prod. 2021, 53, 69. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Freed, D.A.R.; Weber, J.A.; Edwards, J.S. The Sentieon Genomics Tools—A fast and accurate solution to variant calling from next-generation sequence data. bioRxiv, 1157. [Google Scholar]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821-U136. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.A.; Liu, K.Z.; Liu-Fang, G.; Nakka, P.; Ramachandran, S. Pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics 2016, 32, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, N.; Brien, C.; Oakey, H.; Berger, B.; Saade, S.; Ho, Y.S.; Schmöckel, S.M.; Tester, M.; Negrao, S. Salinity tolerance loci revealed in rice using high-throughput non-invasive phenotyping. Nat. Commun. 2016, 7, 13342. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. Qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, J.N.B.; Fahmy, M.H. Breeding goats for meat production: 3. Selection and breeding strategies. Small Rumin. Res. 2007, 67, 113–125. [Google Scholar] [CrossRef]
- Kuthu, Z.H.; Javed, K.; Babar, M.E.; Sattar, A.; Abdullah, M. Environmental effects on growth traits of teddy goats. J. Anim. Plant Sci. 2013, 23, 692–698. [Google Scholar]
- Ayele, S.; Urge, M.; Animut, G.; Yusuf, M. Comparative slaughter performance and carcass quality of three Ethiopian fat-tailed hair sheep breeds supplemented with two levels of concentrate. Trop. Anim. Health Prod. 2019, 51, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Budimir, K.; Trombetta, M.F.; Francioni, M.; Toderi, M.; D’Ottavio, P. Slaughter performance and carcass and meat quality of Bergamasca light lambs according to slaughter age. Small Rumin. Res. 2018, 164, 1–7. [Google Scholar] [CrossRef]
- Tuomi, J. Mammalian reproductive strategies: A generalized relation of litter size to body size. Oecologia 1980, 45, 39–44. [Google Scholar] [CrossRef]
- Xiao, T.; Li, Y.; Yue, L.; Lu, Z.; Yuan, C.; Song, Y.; Yang, B.; Liu, J.; Guo, T. Correlation of 20 Single-Nucleotide Polymorphisms with Weight and Wool Traits in Alpine Merino Sheep. Animals 2024, 14, 127. [Google Scholar] [CrossRef]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef]
- Abo-Ismail, M.K.; Brito, L.F.; Miller, S.P.; Sargolzaei, M.; Grossi, D.A.; Moore, S.S.; Plastow, G.; Stothard, P.; Nayeri, S.; Schenkel, F.S. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet. Sel. Evol. 2017, 49, 82. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; He, X.Y.; Pan, L.X.; Wang, J.W.; Gan, S.Q.; Chu, M.X. Genome-wide association study of body weight and conformation traits in neonatal sheep. Anim. Genet. 2020, 51, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cao, Y.; Shan, H.; Wu, J.; Song, X.; Jiang, Y. The GWAS Analysis of Body Size and Population Verification of Related SNPs in Hu Sheep. Front. Genet. 2021, 12, 642552. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Song, H.L.; Jiang, Y.F.; Jiang, Y.; Zhang, F.X.; Liu, Y.B.; Shi, Y.; Ding, X.D.; Wang, C.D. A Single-Step Genome Wide Association Study on Body Size Traits Using Imputation-Based Whole-Genome Sequence Data in Yorkshire Pigs. Front. Genet. 2021, 12, 629049. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fang, M.; Liu, L.; Wang, S.; Liu, J.; Ding, X.; Zhang, S.; Zhang, Q.; Zhang, Y.; Qiao, L.; et al. Genome wide association studies for body conformation traits in the Chinese Holstein cattle population. Bmc Genom. 2013, 14, 897. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Qiu, Y.; Zhuang, Z.; Wu, J.; Li, X.; Ruan, D.; Xu, C.; Zheng, E.; Yang, M.; Cai, G.; et al. Genome-Wide Association Study of Body Conformation Traits in a Three-Way Crossbred Commercial Pig Population. Animals 2023, 13, 2414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhuang, Z.; Yang, M.; Ding, R.; Quan, J.; Zhou, S.; Gu, T.; Xu, Z.; Zheng, E.; Cai, G.; et al. Genome-Wide Detection of Genetic Loci and Candidate Genes for Body Conformation Traits in Duroc x Landrace x Yorkshire Crossbred Pigs. Front. Genet. 2021, 12, 664343. [Google Scholar] [CrossRef] [PubMed]
- Manca, E.; Cesarani, A.; Gaspa, G.; Sorbolini, S.; Macciotta, N.P.P.; Dimauro, C. Use of the Multivariate Discriminant Analysis for Genome-Wide Association Studies in Cattle. Animals 2020, 10, 1300. [Google Scholar] [CrossRef]
- Ham, T.-h.; Ja-Hong, L.; Seong-gyu, J.; Kim, M.; Zhang, H.; Kim, N.-E.; Kwon, S.-W.; Lee, J. Genome-Wide Association Study of Blast Resistant in Korean Rice (Oryza sativa L.) Breed Lines. Plant Breed. Biotechnol. 2022, 10, 139–144. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Shaw, R.K.; Zhang, X.; Li, J.; Li, L.; Li, S.; Adnan, M.; Jiang, F.; Bi, Y.; et al. Genome-Wide Association Study and Prediction of Tassel Weight of Tropical Maize Germplasm in Multi-Parent Population. Int. J. Mol. Sci. 2024, 25, 1756. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Tarsani, E.; Hager-Theodorides, A.L.; Mastranestasis, I.; Gkelia, D.; Hadjigeorgiou, I. Genetic differentiation of mainland-island sheep of Greece: Implications for identifying candidate genes for long-term local adaptation. PLoS ONE 2021, 16, e0257461. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, H.S.; Jung, D.; Link, T.; Arnold, A.; Kantelhardt, E.; Thomssen, C.; Wimberger, P.; Vetter, M.; Kuhlmann, J.D. FNTB Promoter Polymorphisms Are Independent Predictors of Survival in Patients with Triple Negative Breast Cancer. Cancers 2022, 14, 468. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chen, Y.X.; Chen, Y.; Mou, Y.; Sun, X.T.; Dai, D.P.; Zhao, C.Z.; Yang, J.; Hu, S.J.; Guo, X. Overexpression of FNTB and the activation of Ras induce hypertrophy and promote apoptosis and autophagic cell death in cardiomyocytes. J. Cell. Mol. Med. 2020, 24, 8998–9011. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Xu, H.; Mao, L.; Xu, B.; Fu, X.; Cheng, J.; Zhao, R.; Cheng, Z.; Liu, X.; Xu, J.; et al. A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes. Cancers 2022, 14, 5309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Cao, J.-L.; Zhang, Y.; Liao, L.; Deng, L.; Yang, S.-Y.; Hu, S.-Y.; Ning, Y.; Zhang, F.-L.; Li, D.-Q. RNF144A exerts tumor suppressor function in breast cancer through targeting YY1 for proteasomal degradation to downregulate GMFG expression. Med. Oncol. 2022, 39, 48. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Wang, F.; Chen, W.; Gao, C.; Wang, J. RNF144A suppresses ovarian cancer stem cell properties and tumor progression through regulation of LIN28B degradation via the ubiquitin-proteasome pathway. Cell Biol. Toxicol. 2022, 38, 809–824. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Body Height | Body Length | Chest Depth | Chest Width | Chest Girth | Rump Width | Rump Height | Cannon Bone Circumference |
|---|---|---|---|---|---|---|---|---|
| (BH) | (BL) | (CD) | (CW) | (CG) | (RW) | (RH) | (CC) | |
| Mean | 50.73 | 60.09 | 24.20 | 13.99 | 66.60 | 13.09 | 52.14 | 7.22 |
| Std | 6.08 | 8.84 | 3.65 | 2.62 | 9.57 | 2.49 | 6.17 | 0.82 |
| Max | 65.00 | 86.00 | 32.60 | 20.00 | 86.20 | 18.00 | 66.00 | 9.00 |
| Min | 34.00 | 40.00 | 15.10 | 8.50 | 35.50 | 7.00 | 32.20 | 5.30 |
| CV | 12.0% | 14.7% | 15.1% | 18.7% | 14.4% | 19.0% | 11.8% | 11.4% |
| Interval No. | Interval Length | SNP Maker | Frequency | Chromosome | Position | Mean of PVE | Genes (±500 kb) |
|---|---|---|---|---|---|---|---|
| 1 | 114 Kb | chr10_25988403 | 4 | chr10 | 25,988,403 | 22.41% | FUT8, MAX, FNTB, RAB15, GPX2, CHURC1, 5S_rRNA, SPTB, PLEKHG3, PPP1R36, HSPA2, ZBTB1, AKAP5, MTHFD1 |
| chr10_26009053 | 4 | chr10 | 26,009,053 | 21.64% | |||
| chr10_26012238 | 4 | chr10 | 26,012,238 | 25.21% | |||
| chr10_26061262 | 4 | chr10 | 26,061,262 | 22.73% | |||
| chr10_26072624 | 5 | chr10 | 26,072,624 | 24.78% | |||
| chr10_26102739 | 5 | chr10 | 26,102,739 | 26.30% | |||
| 2 | 1.03 Mb | chr11_88216493 | 3 | chr11 | 88,216,493 | 20.81% | ASAP2, MBOAT2, KIDINS220, Metazoa_SRP, RNF144A, RSAD2, CMPK |
| chr11_88833042 | 2 | chr11 | 88,833,042 | 20.54% | |||
| chr11_88840223 | 2 | chr11 | 88,840,223 | 20.77% | |||
| chr11_89214761 | 3 | chr11 | 89,214,761 | 20.94% | |||
| chr11_89239285 | 2 | chr11 | 89,239,285 | 20.81% | |||
| chr11_89242536 | 3 | chr11 | 89,242,536 | 21.35% | |||
| chr11_89248159 | 7 | chr11 | 89,248,159 | 15.45% | |||
| chr11_89248733 | 6 | chr11 | 89,248,733 | 28.40% | |||
| chr11_89248911 | 3 | chr11 | 89,248,911 | 20.18% | |||
| chr11_89250659 | 3 | chr11 | 89,250,659 | 8.65% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Zhou, D.; Tan, X.; Zhao, Z.; Lv, Y.; Tian, X.; Ren, L.; Wang, Y.; Li, J.; Zhao, Y.; et al. Genome-Wide Association Study of Body Conformation Traits in Tashi Goats (Capra hircus). Animals 2024, 14, 1145. https://doi.org/10.3390/ani14081145
Yang R, Zhou D, Tan X, Zhao Z, Lv Y, Tian X, Ren L, Wang Y, Li J, Zhao Y, et al. Genome-Wide Association Study of Body Conformation Traits in Tashi Goats (Capra hircus). Animals. 2024; 14(8):1145. https://doi.org/10.3390/ani14081145
Chicago/Turabian StyleYang, Rong, Di Zhou, Xiaoshan Tan, Zhonghai Zhao, Yanli Lv, Xingzhou Tian, Liqun Ren, Yan Wang, Jun Li, Yongju Zhao, and et al. 2024. "Genome-Wide Association Study of Body Conformation Traits in Tashi Goats (Capra hircus)" Animals 14, no. 8: 1145. https://doi.org/10.3390/ani14081145