
Metagenomic Analysis Reveals A Gut Microbiota Structure and Function Alteration between Healthy and Diarrheic Juvenile Yaks


 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Microbiome Sample Processing and Sequencing
2.3. Bioinformatics and Statistical Analysis
2.4. Functional Profile Analysis of the Intestinal Bacterial Community via PICRUSt
2.5. Statistical Analysis
3. Results
3.1. Sequence Analyses
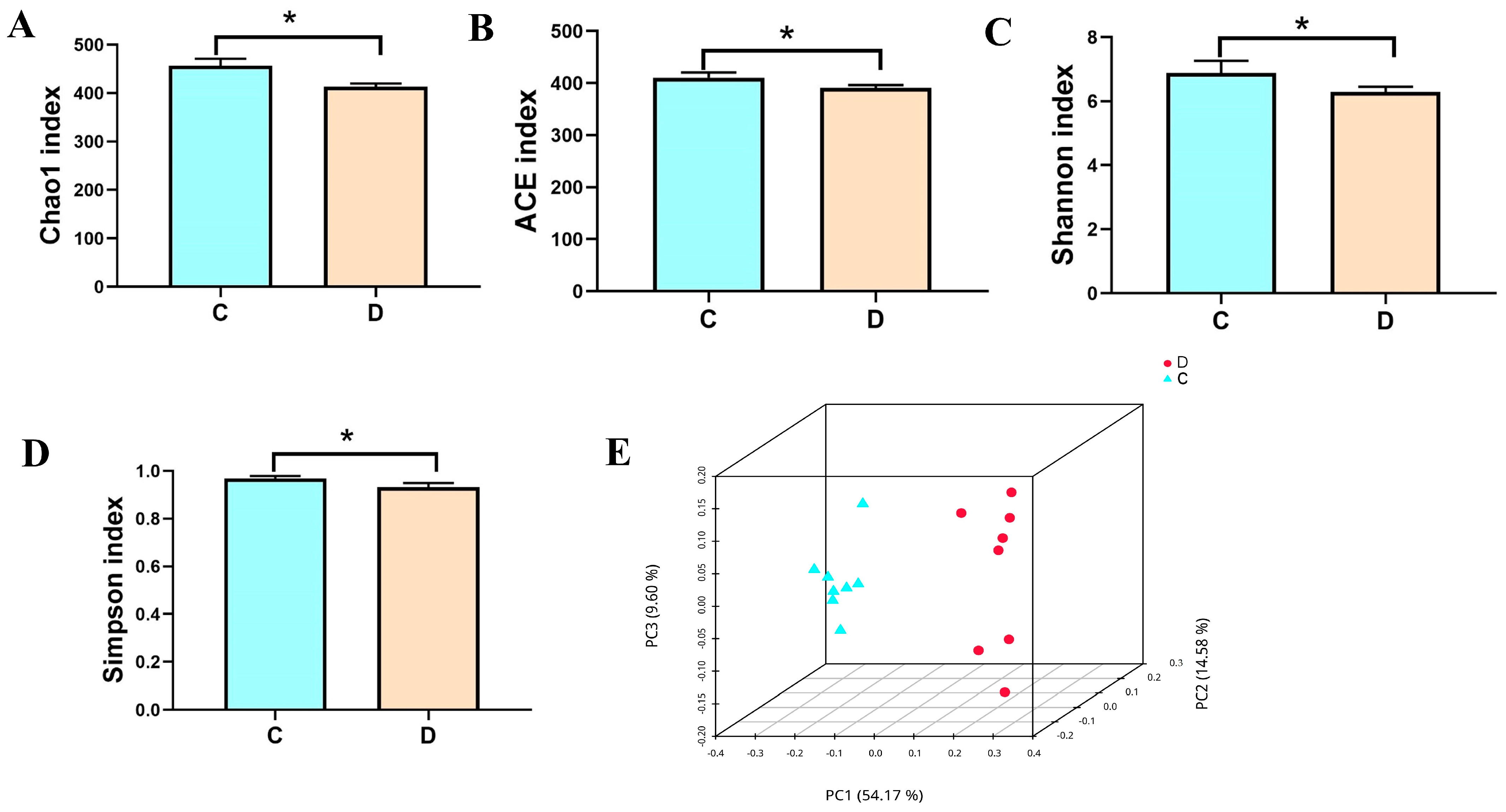
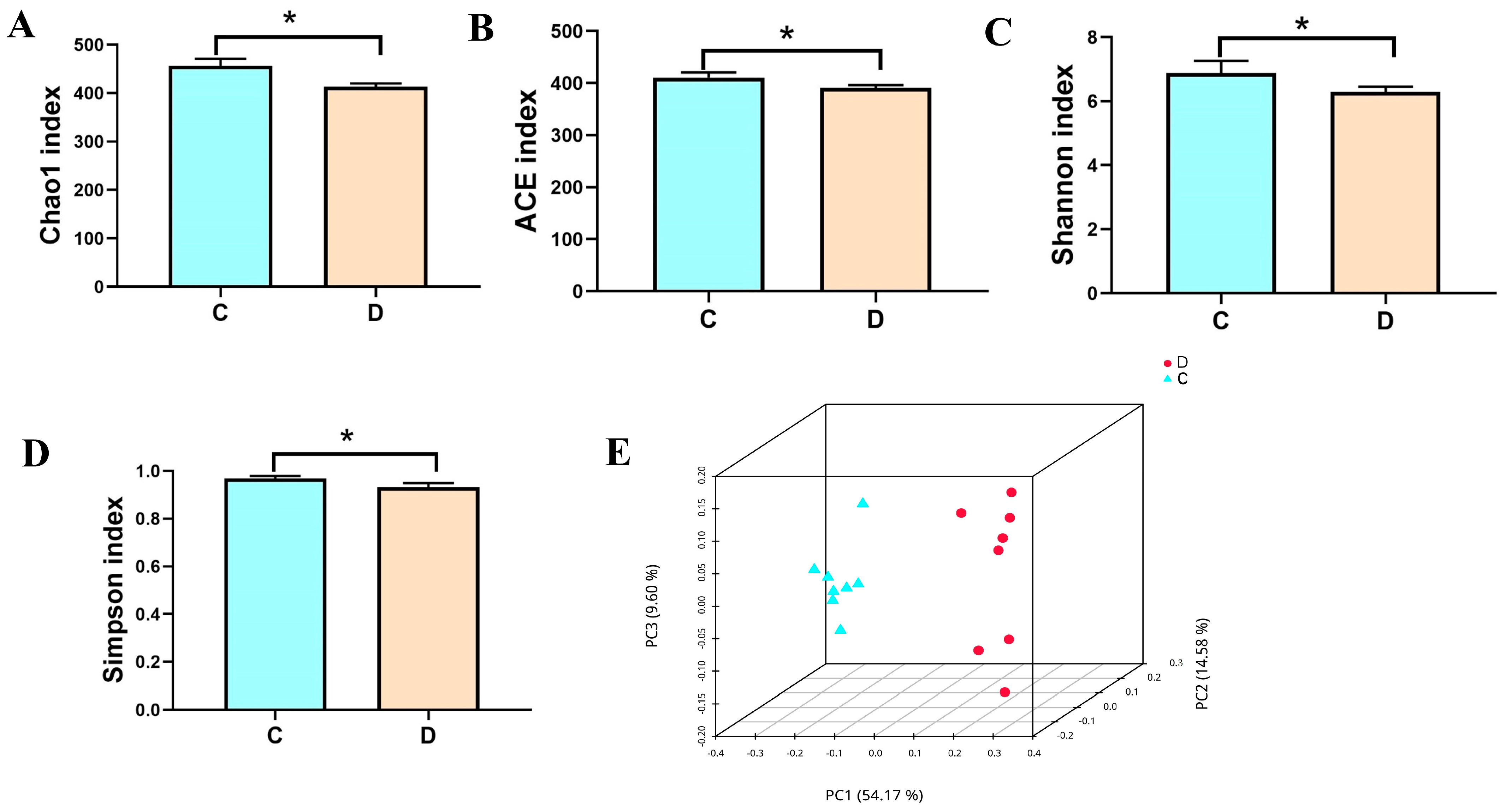
3.2. Analysis of the Microbial Diversity in the Healthy and Diarrheic Yaks
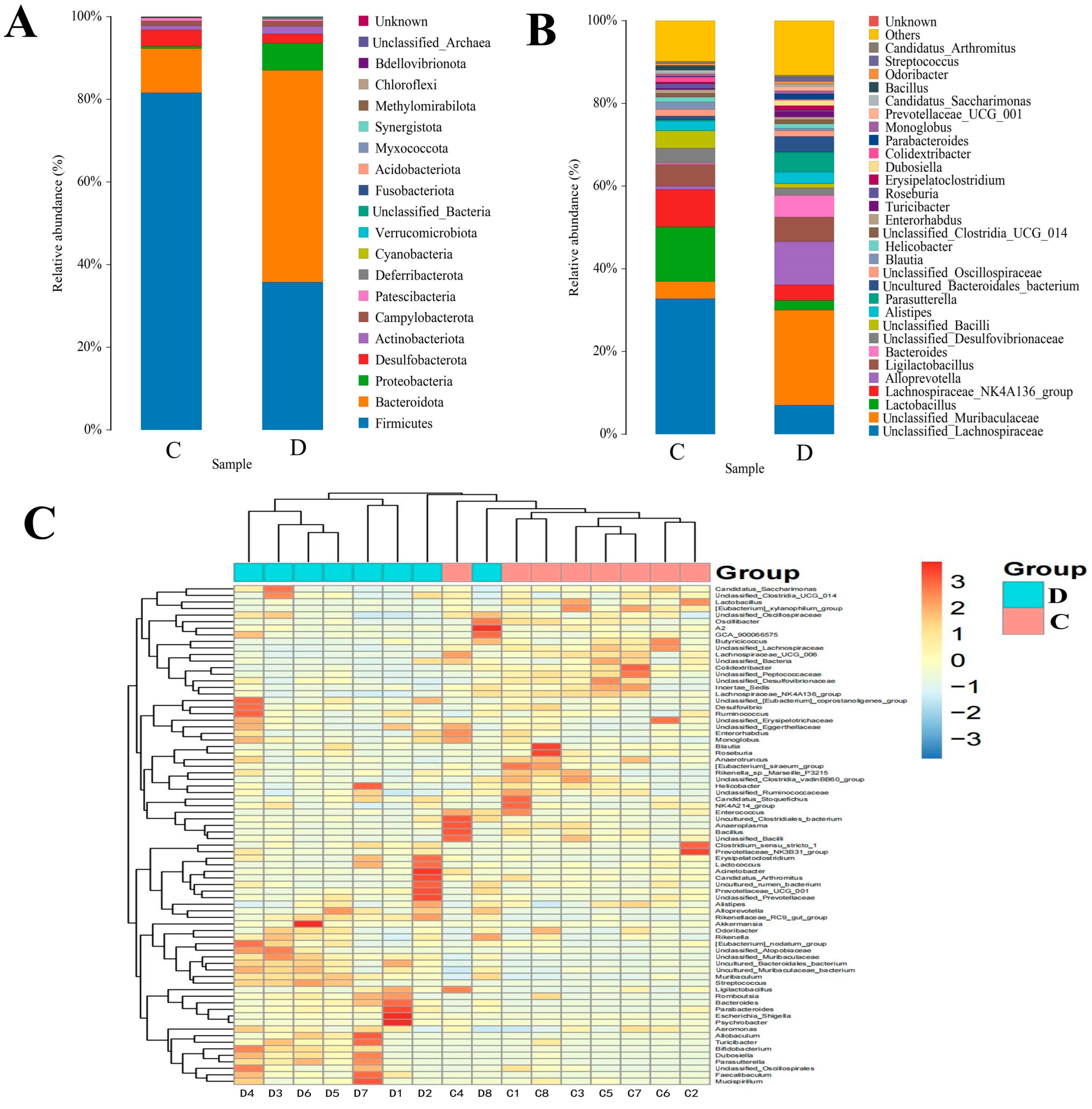
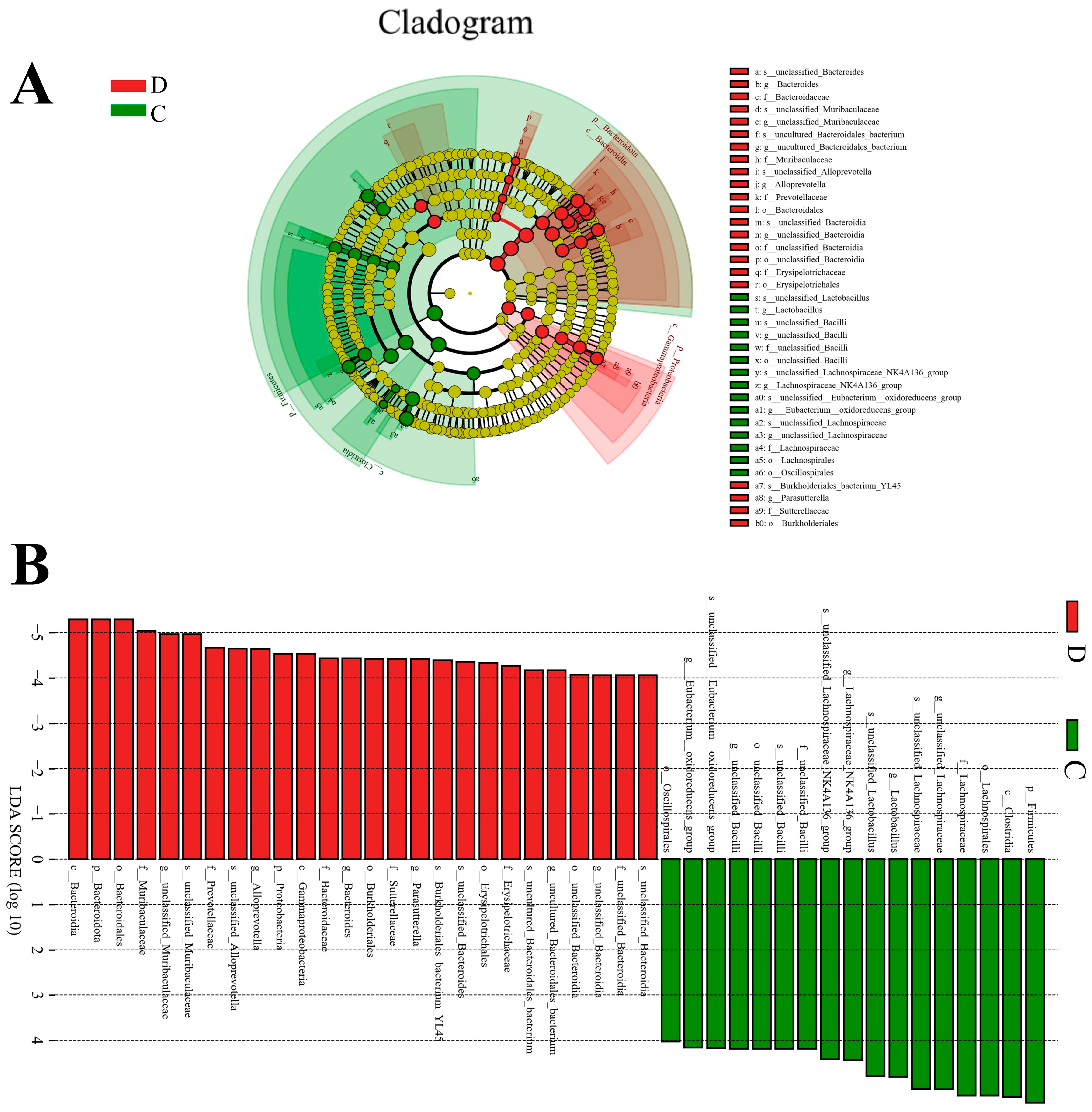
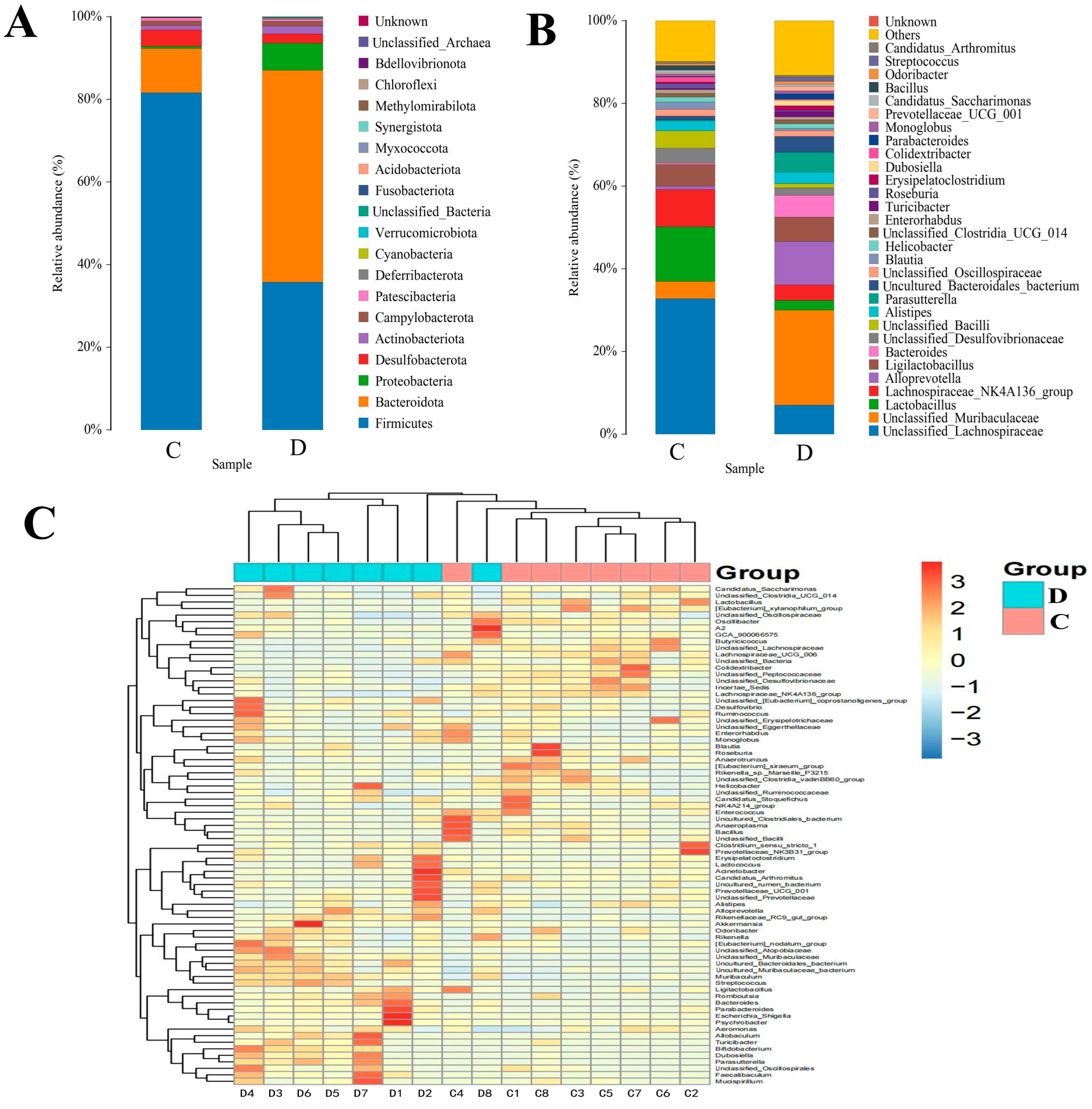
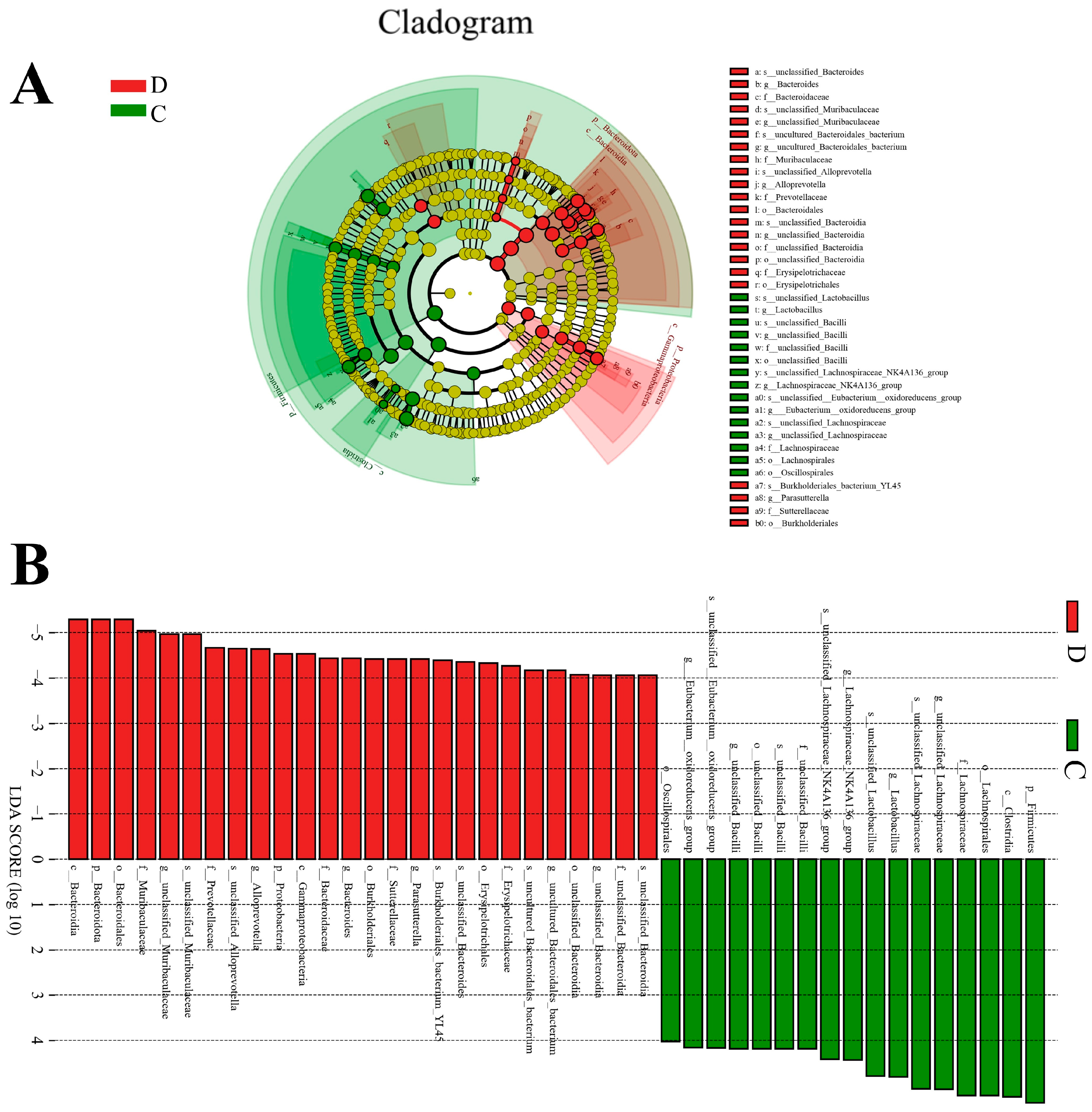
3.3. Composition Analysis of the Gut Microbial Community in the Healthy and Diarrheic Yaks
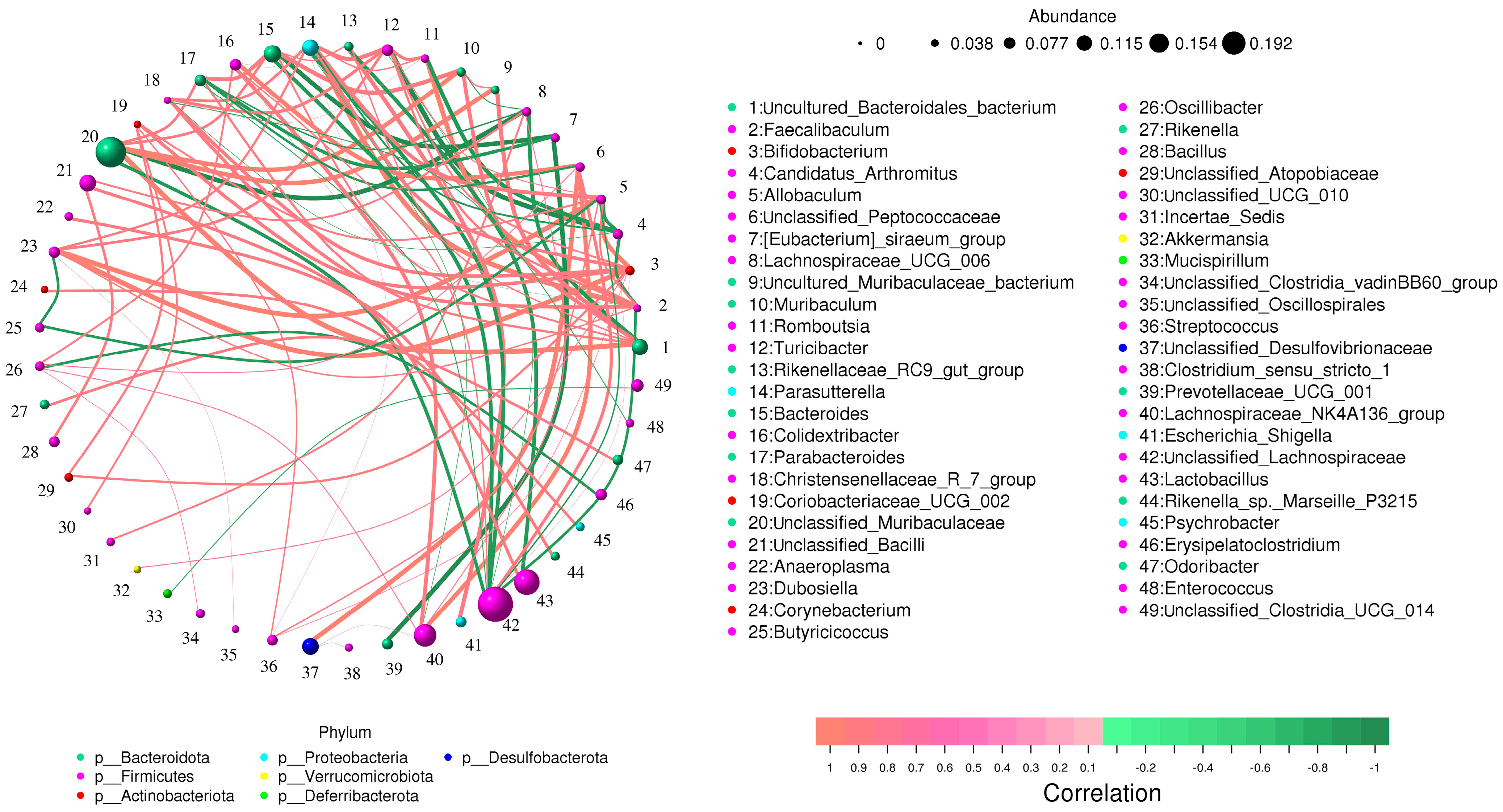
3.4. Correlation Network Analysis
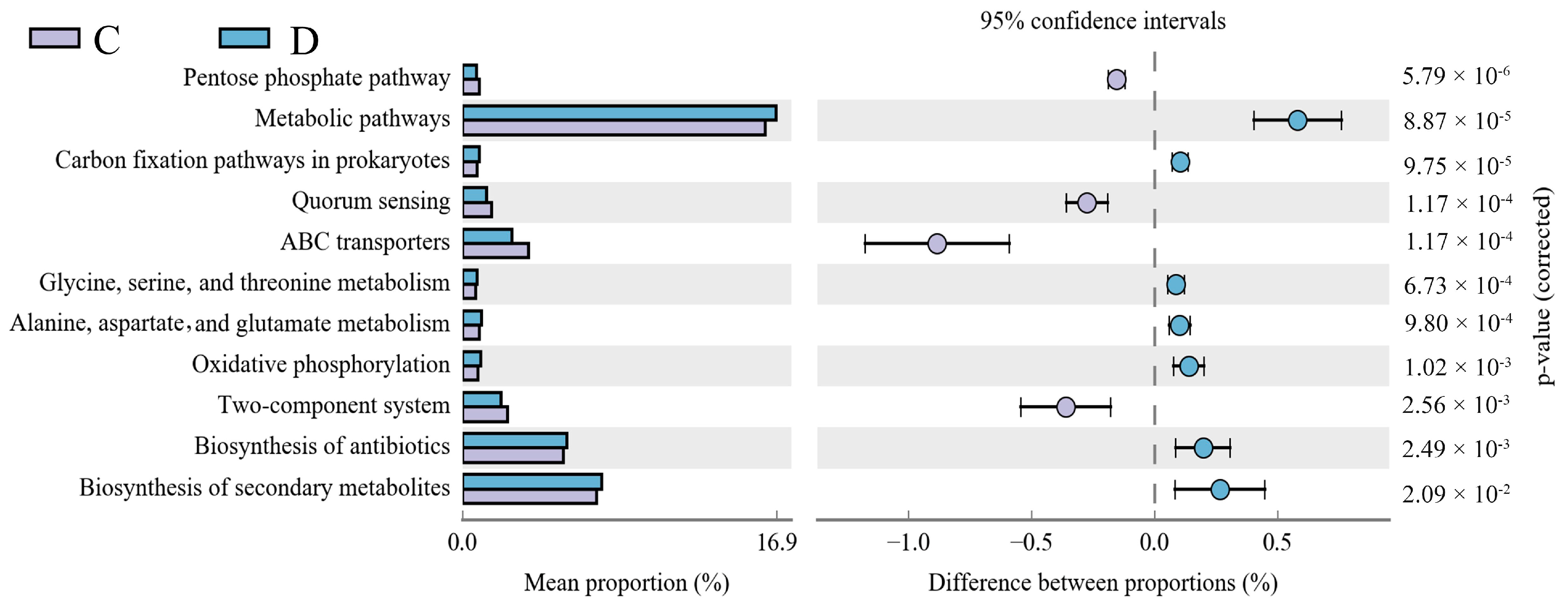
3.5. Functional Predictions of the Intestinal Microbes in the Diarrheic Group and Healthy Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adak, A.; Khan, M.R. An Insight Into Gut Microbiota and Its Functionalities. Cell. Mol. Life Sci. 2018, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, S.; Jiang, X.; Feng, C.; Gong, S.; Ma, J.; Fang, Z.; Yin, J.; Yin, Y. Gut Microbiota and Diarrhea: An Updated Review. Front. Cell. Infect. Microbiol. 2021, 11, 625210. [Google Scholar] [CrossRef]
- Régnier, M.; van Hul, M.; Knauf, C.; Cani, P.D. Gut Microbiome, Endocrine Control of Gut Barrier Function and Metabolic Diseases. J. Endocrinol. 2021, 248, R67–R82. [Google Scholar] [CrossRef] [PubMed]
- Choden, T.; Cohen, N.A. The Gut Microbiome and the Immune System. Explor. Med. 2022, 3, 219–233. [Google Scholar] [CrossRef]
- Du, Y.; Gao, Y.; Hu, M.; Hou, J.; Yang, L.; Wang, X.; Du, W.; Liu, J.; Xu, Q. Colonization and Development of the Gut Microbiome in Calves. J. Anim. Sci. Biotechnol. 2023, 14, 46. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; Lawrence, A.; et al. In the Metabolic Function of the Gut Microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Ma, J.; Piao, X.; Mahfuz, S.; Long, S.; Wang, J. The Interaction among Gut Microbes, the Intestinal Barrier and Short Chain Fatty Acids. Anim. Nutr. 2021, 9, 159–174. [Google Scholar] [CrossRef]
- Kong, Q.; Liu, S.; Li, A.; Wang, Y.; Zhang, L.; Iqbal, M.; Jamil, T.; Shang, Z.; Suo, L.-S.; Li, J. Characterization of Fungal Microbial Diversity in Healthy and Diarrheal Tibetan Piglets. BMC Microbiol. 2021, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wang, Y.; Hao, J.; Wang, L.; Quan, L.; Duan, K.; Kulyar, M.F.-E.; Ullah, K.; Zhang, J.; Wu, Y.; et al. Long-Term Hexavalent Chromium Exposure Disturbs the Gut Microbial Homeostasis of Chickens. Ecotoxicol. Environ. Saf. 2022, 237, 113532. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Neculae, E.; Costea, C.F.; Ciocoiu, M.; Hurjui, L.L.; Tarniceriu, C.C.; Maranduca, M.A.; Lacatusu, C.M.; Floria, M.; et al. Role of Gut Microbiota on Onset and Progression of Microvascular Complications of Type 2 Diabetes (T2DM). Nutrients 2020, 12, 3719. [Google Scholar] [CrossRef]
- Qi, M.; Cao, Z.; Shang, P.; Zhang, H.; Hussain, R.; Mehmood, K.; Chang, Z.; Wu, Q.; Dong, H. Comparative Analysis of Fecal Microbiota Composition Diversity in Tibetan Piglets Suffering from Diarrheagenic Escherichia coli (DEC). Microb. Pathog. 2021, 158, 105106. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Wang, X.; Hu, M.; Hou, J.; Du, Y.; Si, W.; Yang, L.; Xu, L.; Xu, Q. Modulating Gastrointestinal Microbiota to Alleviate Diarrhea in Calves. Front. Microbiol. 2023, 14, 1181545. [Google Scholar] [CrossRef] [PubMed]
- Gryaznova, M.V.; Dvoretskaya, Y.D.; Syromyatnikov, M.Y.; Shabunin, S.V.; Parshin, P.A.; Mikhaylov, E.V.; Strelnikov, N.A.; Popov, V.N. Changes in the Microbiome Profile in Different Parts of the Intestine in Piglets with Diarrhea. Animals 2022, 12, 320. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, K.; Shahzad, M.; Zhang, H.; Luo, H.; Qiu, G.; Lan, Y.; Wang, X.; Mehmood, K.; Li, J. Analysis of the Intestinal Microbial Community in Healthy and Diarrheal Perinatal Yaks by High-Throughput Sequencing. Microb. Pathog. 2017, 111, 60–70. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Zhu, L.; Xu, Y.; Liu, N.; Sun, X.; Hu, L.; Huang, H.; Wei, K.; Zhu, R. Dynamic Distribution of Gut Microbiota in Goats at Different Ages and Health States. Front. Microbiol. 2018, 9, 2509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, A.; Liu, J.; Mehmood, K.; Wangdui, B.; Shi, H.; Luo, X.; Zhang, H.; Li, J. L. pseudomesenteroides and L. johnsonii isolated from Yaks in Tibet Modulate Gut Microbiota in Mice to Ameliorate Enteroinvasive Escherichia coli-Induced Diarrhea. Microb. Pathog. 2019, 132, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, A.; Zhang, L.; Waqas, M.; Mehmood, K.; Iqbal, M.; Muyou, C.; Li, Z.; Lian, Y.; Sizhu, S.; et al. Probiotic Potential of Lactobacillus on the Intestinal Microflora against Escherichia coli Induced Mice Model through High-Throughput Sequencing. Microb. Pathog. 2019, 137, 103760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, B.; Liang, Q.; Jin, X.; Tang, L.; Ding, Q.; Wang, Z.; Wei, Z. Porcine Deltacoronavirus Infection Alters Bacterial Communities in the Colon and Feces of Neonatal Piglets. Microbiologyopen 2020, 9, e1036. [Google Scholar] [CrossRef]
- Burrello, C.; Garavaglia, F.; Cribiù, F.M.; Ercoli, G.; Lopez, G.; Troisi, J.; Colucci, A.; Guglietta, S.; Carloni, S.; Guglielmetti, S.; et al. Therapeutic Faecal Microbiota Transplantation Controls Intestinal Inflammation through IL10 Secretion by Immune Cells. Nat. Commun. 2018, 9, 5184. [Google Scholar] [CrossRef]
- Li, X.-Y.; He, C.; Zhu, Y.; Lu, N.-H. Role of Gut Microbiota on Intestinal Barrier Function in Acute Pancreatitis. World J. Gastroenterol. 2020, 26, 2187–2193. [Google Scholar] [CrossRef]
- Gomez, D.; Arroyo, L.; Costa, M.; Viel, L.; Weese, J. Characterization of the Fecal Bacterial Microbiota of Healthy and Diarrheic Dairy Calves. J. Veter-Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, B.; Li, F.; He, Y.; Wang, L.; Kulyar, M.F.-E.; Li, H.; Fu, Y.; Zhu, H.; Wang, Y.; et al. Integrated Bacterial and Fungal Diversity Analysis Reveals the Gut Microbial Alterations in Diarrheic Giraffes. Front. Microbiol. 2021, 12, 712092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, X.; Li, A.; Waqas, M.; Gao, X.; Li, K.; Xie, G.; Zhang, J.; Mehmood, K.; Zhao, S.; et al. Characterization of the Microbial Community Structure in Intestinal Segments of Yak (Bos grunniens). Anaerobe 2019, 61, 102115. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Liang, C.; Guo, X.; Wu, X.; Wang, H.; Johnson, K.; Yan, P. Physiological Insight into The High-Altitude Adaptations in Domesticated Yaks (Bos grunniens) along the Qinghai-Tibetan Plateau Altitudinal Gradient. Livest. Sci. 2014, 162, 233–239. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, B.; Yue, H.; Wang, Y.; Zhou, F.; Zhang, Q.; Tang, C. A Novel Astrovirus Species in the Gut of Yaks with Diarrhoea in the Qinghai–Tibetan Plateau, 2013. J. Gen. Virol. 2015, 96, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Mehmood, K.; Zhang, H.; Jiang, X.; Shahzad, M.; Dong, X.; Li, J. Characterization of Fungus Microbial Diversity in Healthy and Diarrheal Yaks in Gannan Region of Tibet Autonomous Prefecture. Acta Trop. 2018, 182, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Zhang, W.; Kulyar, M.F.-E.; Ullah, K.; Han, Z.; Qin, J.; Bi, C.; Wang, Y.; Li, K. Comparative Analysis Of Gut Microbiota In Healthy and Diarrheic Yaks. Microb. Cell Fact. 2022, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; An, S. Identifying the Biogeographic Patterns of Rare and Abundant Bacterial Communities Using Different Primer Sets on the Loess Plateau. Microorganisms 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Rehman, M.U.; Zhang, H.; Iqbal, M.K.; Mehmood, K.; Huang, S.; Nabi, F.; Luo, H.; Lan, Y.; Li, J. Antibiotic Resistance, Serogroups, Virulence Genes, and Phylogenetic Groups of Escherichia coli Isolated from Yaks with Diarrhea in Qinghai Plateau, China. Gut Pathog. 2017, 9, 24. [Google Scholar] [CrossRef]
- Lu, S.; Zou, W.; Chen, X.; Sun, G.; Cidan, Y.; Almutairi, M.H.; Dunzhu, L.; Nazar, M.; Mehmood, K.; Zhu, Y.; et al. Effects of Cryptosporidium Parvum Infection on Intestinal Fungal Microbiota in Yaks (Bos grunniens). Microb. Pathog. 2023, 183, 106322. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yi, X.; Wu, B.; Li, X.; Ye, B.; Deng, Z.; Hu, S.; Li, D.; Wu, H.; Zhou, Z.; et al. Neonatal Calf Diarrhea Is Associated with Decreased Bacterial Diversity and Altered Gut Microbiome Profiles. Fermentation 2023, 9, 827. [Google Scholar] [CrossRef]
- Dias, J.; Marcondes, M.I.; de Souza, S.M.; Silva, B.C.d.M.e.; Noronha, M.F.; Resende, R.T.; Machado, F.S.; Mantovani, H.C.; Dill-McFarland, K.A.; Suen, G. Bacterial Community Dynamics across the Gastrointestinal Tracts of Dairy Calves during Preweaning Development. Appl. Environ. Microbiol. 2018, 84, e02675-17. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-S.; Jo, H.E.; Lee, J.; Choi, K.-S.; Yu, D.; Oh, Y.-S.; Park, J.; Choi, H.-J. Alteration of the Gut Microbiota in Post-Weaned Calves Following Recovery from Bovine Coronavirus-Mediated Diarrhea. J. Anim. Sci. Technol. 2021, 63, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ren, Y.; Wen, X.; Yue, S.; Wang, Z.; Wang, L.; Peng, Q.; Hu, R.; Zou, H.; Jiang, Y.; et al. Comparison of Coated and Uncoated Trace Elements on Growth Performance, Apparent Digestibility, Intestinal Development and Microbial Diversity in Growing Sheep. Front. Microbiol. 2022, 13, 1080182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Z.; Kao, R.D.; Han, J.; Du, M.; Ahmad, A.A.; Wang, S.; Salekdeh, G.H.; Long, R.; Yan, P.; et al. Maternal Fecal Microbes Contribute to Shaping the Early Life Assembly of the Intestinal Microbiota of Co-inhabiting Yak and Cattle Calves. Front. Microbiol. 2022, 13, 916735. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yang, Y.; Zhang, Y.; Lv, S.; Jin, T.; Li, K.; Han, Z.; Li, Y. Microbiome Analysis Reveals the Alterations In Gut Microbiota in Different Intestinal Segments of Yimeng Black Goats. Microb. Pathog. 2021, 155, 104900. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Tremblay, D.M.; Moineau, S. Characterization of 1706, a Virulent Phage from Lactococcus lactis with Similarities to Prophages from Other Firmicutes. Virology 2008, 373, 298–309. [Google Scholar] [CrossRef]
- Li, A.; Yang, Y.; Qin, S.; Lv, S.; Jin, T.; Li, K.; Han, Z.; Li, Y. Microbiome Analysis Reveals Gut Microbiota Alteration of Early-Weaned Yimeng Black Goats with the Effect of Milk Replacer and Age. Microb. Cell Fact. 2021, 20, 78. [Google Scholar] [CrossRef]
- Mahowald, M.A.; Rey, F.E.; Seedorf, H.; Turnbaugh, P.J.; Fulton, R.S.; Wollam, A.; Shah, N.; Wang, C.; Magrini, V.; Wilson, R.K.; et al. Characterizing a Model Human Gut Microbiota Composed of Members of Its Two Dominant Bacterial Phyla. Proc. Natl. Acad. Sci. USA 2009, 106, 5859–5864. [Google Scholar] [CrossRef]
- Yang, H.; Xiao, Y.; Gui, G.; Li, J.; Wang, J.; Li, D. Microbial Community and Short-Chain Fatty Acid Profile in Gastrointestinal tract of Goose. Poult. Sci. 2018, 97, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Kraus, V.B. Does Lipopolysaccharide-Mediated Inflammation Have a Role in OA? Nat. Rev. Rheumatol. 2016, 12, 123–129. [Google Scholar] [CrossRef]
- Idicula, D.V.; Parappilly, S.J.; Joy, N.; Balan, J.; George, S.M. Salutary Attributes of Probiotic Human Gut Lactobacilli for Gut Health. Lett. Appl. Microbiol. 2023, 76, ovad011. [Google Scholar] [CrossRef]
- Chee, W.J.Y.; Chew, S.Y.; Than, L.T.L. Vaginal Microbiota and the Potential of Lactobacillus derivatives in Maintaining Vaginal Health. Microb. Cell Fact. 2020, 19, 203. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, B.; Lu, S.; Wang, S.; Ren, X.; Liu, R.; Dong, H.; Li, K.; Fouad, D.; Ataya, F.S.; et al. Metagenomic Analysis for Exploring the Potential of Lactobacillus Yoelii FYL1 to Mitigate Bacterial Diarrhea and Changes in the Gut Microbiota of Juvenile Yaks. Microb. Pathog. 2024, 186, 106496. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ni, Y.; Wang, Z.; Tu, W.; Ni, L.; Zhuge, F.; Zheng, A.; Hu, L.; Zhao, Y.; Zheng, L.; et al. Spermidine Improves Gut Barrier Integrity and Gut Microbiota Function in Diet-Induced Obese Mice. Gut Microbes 2020, 12, 1–19. [Google Scholar] [CrossRef]
- The, H.C.; Le, S.-N.H. Dynamic of the Human Gut Microbiome under Infectious Diarrhea. Curr. Opin. Microbiol. 2022, 66, 79–85. [Google Scholar] [CrossRef]
- Wu, M.-R.; Chou, T.-S.; Huang, C.-Y.; Hsiao, J.-K. A Potential Probiotic- Lachnospiraceae NK4A136 Group: Evidence from the Restoration of the Dietary Pattern from a High-Fat Diet. Angew. Chem. Int. Ed. 2021, 6, 951–952. [Google Scholar]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A New Functional Genus with Potential Probiotic Properties? Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef]
- Mao, B.; Guo, W.; Liu, X.; Cui, S.; Zhang, Q.; Zhao, J.; Tang, X.; Zhang, H. Potential Probiotic Properties of Blautia producta Against Lipopolysaccharide-Induced Acute Liver Injury. Probiotics Antimicrob. Proteins 2023, 15, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ge, B.; Zhang, X.; Liu, W.; Duan, J.; Fu, D. Effect of Fu Brick Tea on Improving Metabolic Disorders in Type 2 Diabetes Mice. J. Tea Sci. 2022, 42, 63–75. [Google Scholar]
- Cheng, H.; Zhang, D.; Wu, J.; Liu, J.; Tan, Y.; Feng, W.; Peng, C. Atractylodes macrocephala Koidz. Volatile Oil Relieves Acute Ulcerative Colitis Via Regulating Gut Microbiota and Gut Microbiota Metabolism. Front. Immunol. 2023, 14, 1127785. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Chen, Y.; Ma, Z.; Zhang, X.; Shi, D.; Khan, J.A.; Liu, H. Gut Microbiota-Derived Short Chain Fatty Acids Are Potential Mediators in Gut Inflammation. Anim. Nutr. 2021, 8, 350–360. [Google Scholar] [CrossRef]
- Kim, B.; Kwon, J.; Kim, M.-S.; Park, H.; Ji, Y.; Holzapfel, W.; Hyun, C.-K. Protective Effects of Bacillus Probiotics against High-Fat Diet-Induced Metabolic Disorders in Mice. PLoS ONE 2018, 13, e0210120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ding, T.; Chen, Y.; Yang, W.; Rao, J.; Liu, D.; Yi, B. Arecoline Aggravates Acute Ulcerative Colitis in Mice by Affecting Intestinal Microbiota and Serum Metabolites. Front. Immunol. 2023, 14, 1197922. [Google Scholar] [CrossRef] [PubMed]
- Rabizadeh, S.; Rhee, K.-J.; Wu, S.; Huso, D.; Gan, C.M.; Golub, J.E.; Wu, X.; Zhang, M.; Sears, C.L. Enterotoxigenic Bacteroides fragilis: A Potential Instigator of Colitis. Inflamm. Bowel Dis. 2007, 13, 1475–1483. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Shao, S.; Yu, H.; Wang, D.; Li, C.; Yuan, Q.; Liu, W.; Cao, J.; Wang, X.; et al. Rabdosia Serra Alleviates Dextran Sulfate Sodium Salt-Induced Colitis in Mice through Anti-Inflammation, Regulating Th17/Treg Balance, Maintaining Intestinal Barrier Integrity, and Modulating Gut Microbiota. J. Pharm. Anal. 2022, 12, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, J.; Wang, J.; Zhou, H.; Lu, Y.; Lou, L.; Zheng, J.; Tian, L.; Wang, X.; Cao, Z.; et al. Dysbiosis of Intestinal Microbiota and Decreased Antimicrobial Peptide Level in Paneth Cells during Hypertriglyceridemia-Related Acute Necrotizing Pancreatitis in Rats. Front. Microbiol. 2017, 8, 776. [Google Scholar] [CrossRef]
- Li, Z.; Shen, Y.; Xin, J.; Xu, X.; Ding, Q.; Chen, W.; Wang, J.; Lv, Y.; Wei, X.; Wei, Y.; et al. Cryptotanshinone Alleviates Radiation-Induced Lung Fibrosis via Modulation of Gut Microbiota and Bile Acid Metabolism. Phytother. Res. 2023, 37, 4557–4571. [Google Scholar] [CrossRef]
- Liu, C.-E.; Pan, Y.-M.; Du, Z.-L.; Wu, C.; Hong, X.-Y.; Sun, Y.-H.; Li, H.-F.; Liu, J. Composition Characteristics of the Gut Microbiota in Infants and Young Children of under 6 Years old between Beijing and Japan. Transl. Pediatr. 2021, 10, 790–806. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Im, W.-T.; Kim, M.; Kim, D.; Seo, Y.H.; Yong, D.; Jeong, S.H.; Lee, K. Parabacteroides chongii sp. nov., Isolated from Blood of a Patient with Peritonitis. J. Microbiol. 2018, 56, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, D.; Sun, H. Herba Origani Alleviated DSS-Induced Ulcerative Colitis in Mice through Remolding Gut Microbiota to Regulate Bile Acid and Short-Chain Fatty Acid Metabolisms. Biomed. Pharmacother. 2023, 161, 114409. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.O.; Towarnicki, S.G. Mitochondria, the Gut Microbiome and ROS. Cell. Signal. 2020, 75, 109737. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y. Potential for Therapeutic Use of Hydrogen Sulfide in Oxidative Stress-Induced Neurodegenerative Diseases. Int. J. Med Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Clean Reads | Effective Reads | AvgLen(bp) | Effective (%) |
|---|---|---|---|---|---|
| C1 | 79,966 | 79,834 | 78,775 | 414 | 98.51 |
| C2 | 79,069 | 78,924 | 78,034 | 413 | 98.69 |
| C3 | 79,930 | 79,798 | 78,106 | 412 | 97.71 |
| C4 | 80,082 | 79,932 | 78,858 | 413 | 98.47 |
| C5 | 80,162 | 79,980 | 77086 | 415 | 96.16 |
| C6 | 69,034 | 68,913 | 67,730 | 416 | 98.11 |
| C7 | 57,481 | 57,336 | 56,588 | 415 | 98.45 |
| C8 | 79,998 | 79,865 | 78,867 | 418 | 98.59 |
| D1 | 79,966 | 79,834 | 787,75 | 415 | 98.51 |
| D2 | 80,097 | 79,944 | 79,196 | 414 | 98.87 |
| D3 | 79,971 | 79,827 | 78,281 | 415 | 97.89 |
| D4 | 79,836 | 79,689 | 78,560 | 414 | 98.40 |
| D5 | 79,979 | 79,825 | 78,529 | 413 | 98.19 |
| D6 | 80,029 | 79,874 | 78,653 | 415 | 98.28 |
| D7 | 64,354 | 64,235 | 63,175 | 416 | 98.17 |
| D8 | 78,158 | 78,008 | 76,781 | 415 | 98.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Mo, Q.; Kulyar, M.F.-e.-A.; Guan, J.; Zhang, X.; Luo, X.; Li, J. Metagenomic Analysis Reveals A Gut Microbiota Structure and Function Alteration between Healthy and Diarrheic Juvenile Yaks. Animals 2024, 14, 1181. https://doi.org/10.3390/ani14081181
Zhao H, Mo Q, Kulyar MF-e-A, Guan J, Zhang X, Luo X, Li J. Metagenomic Analysis Reveals A Gut Microbiota Structure and Function Alteration between Healthy and Diarrheic Juvenile Yaks. Animals. 2024; 14(8):1181. https://doi.org/10.3390/ani14081181
Chicago/Turabian StyleZhao, Hongwen, Quan Mo, Muhammad Fakhar-e-Alam Kulyar, Jiuqiang Guan, Xiangfei Zhang, Xiaolin Luo, and Jiakui Li. 2024. "Metagenomic Analysis Reveals A Gut Microbiota Structure and Function Alteration between Healthy and Diarrheic Juvenile Yaks" Animals 14, no. 8: 1181. https://doi.org/10.3390/ani14081181