Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology
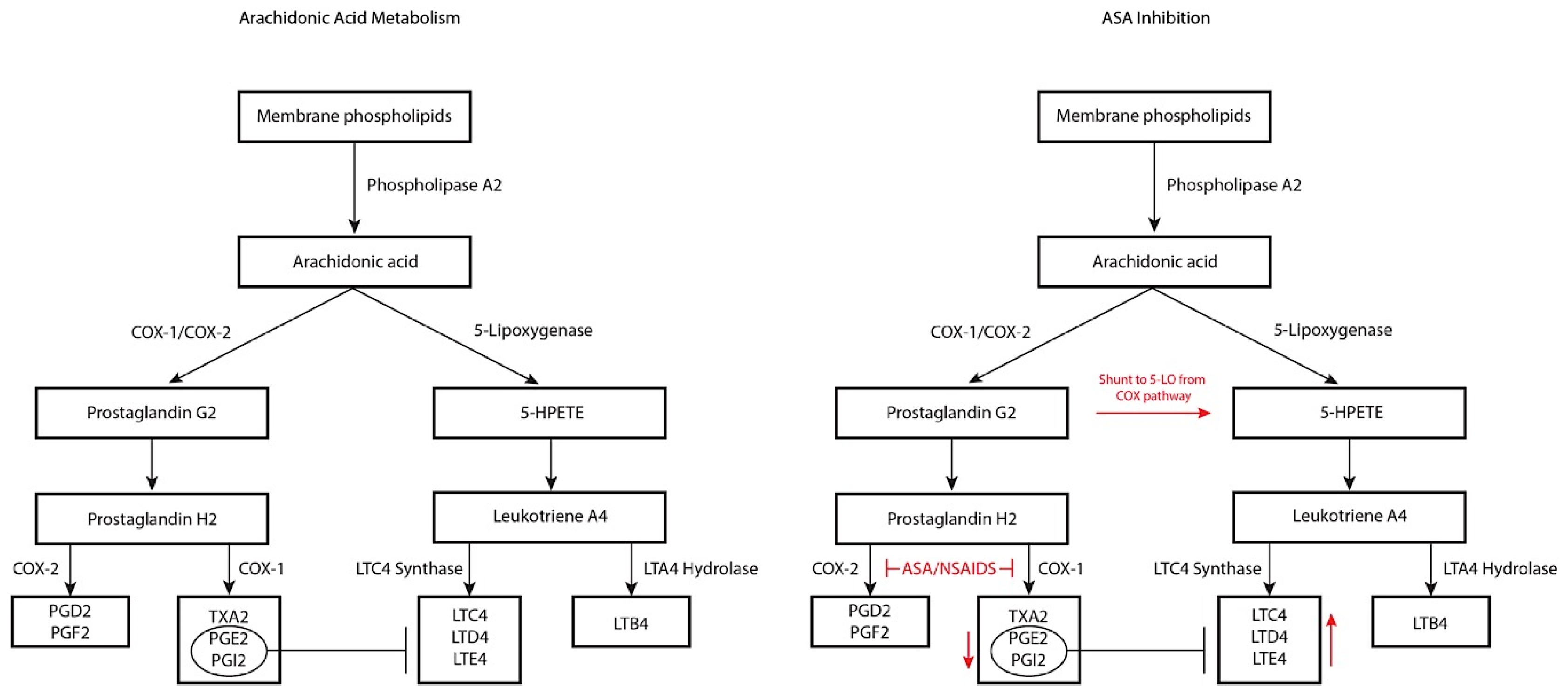
3. Pathophysiology
4. Diagnostic Workup
5. Medical Treatment
5.1. Corticosteroids
5.2. Leukotriene Modifiers
5.3. Aspirin Desensitization
5.4. Monoclonal Antibodies
6. Surgical Procedures and Outcomes
6.1. Functional Endoscopic Sinus Surgery
6.2. Endoscopic Modified Lothrop Procedure/Draf 3
6.3. Complete Total Ethmoidectomy with Mucosal Stripping
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Widal, F.; Abrami, P.; Lermoyez, J. First complete description of the aspirin idiosyncrasy-asthma-nasal polyposis syndrome (plus urticaria)--1922 (with a note on aspirin desensitization). J. Asthma 1987, 24, 297–300. [Google Scholar] [PubMed]
- Samter, M.; Beers, R.F., Jr. Intolerance to aspirin. Clinical studies and consideration of its pathogenesis. Ann. Intern. Med. 1968, 68, 975–983. [Google Scholar] [CrossRef]
- Sakalar, E.G.; Muluk, N.B.; Kar, M.; Cingi, C. Aspirin-exacerbated respiratory disease and current treatment modalities. Eur. Arch. Otorhinolaryngol. 2017, 274, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Cho, K.S. Samter’s Triad: State of the Art. Clin. Exp. Otorhinolaryngol. 2018, 11, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, A.; Nizankowska, E.; Duplaga, M. Natural history of aspirin-induced asthma. AIANE Investigators. European Network on Aspirin-Induced Asthma. Eur. Respir. J. 2000, 16, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, A.; Stevenson, D.D. Aspirin-induced asthma: Advances in pathogenesis, diagnosis, and management. J. Allergy Clin. Immunol. 2003, 111, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Gudziol, V.; Michel, M.; Sonnefeld, C.; Koschel, D.; Hummel, T. Olfaction and sinonasal symptoms in patients with CRSwNP and AERD and without AERD: A cross-sectional and longitudinal study. Eur. Arch. Otorhinolaryngol. 2017, 274, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Lumry, W.R.; Curd, J.G.; Zeiger, R.S.; Pleskow, W.W.; Stevenson, D.D. Aspirin-sensitive rhinosinusitis: The clinical syndrome and effects of aspirin administration. J. Allergy Clin. Immunol. 1983, 71, 580–587. [Google Scholar] [CrossRef]
- Stevenson, D.; Szczeklik, A. Clinical and pathologic perspectives on aspirin sensitivity and asthma. J. Allergy Clin. Immunol. 2006, 118, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Rajan, J.P.; Wineinger, N.E.; Stevenson, D.D.; White, A.A. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: A meta-analysis of the literature. J. Allergy Clin. Immunol. 2015, 135, 676–681.e1. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Costello, J.; Hodge, L. Systematic review of prevalence of aspirin induced asthma and its implications for clinical practice. BMJ 2004, 328, 434. [Google Scholar] [CrossRef] [PubMed]
- Berges-Gimeno, M.P.; Simon, R.A.; Stevenson, D.D. The natural history and clinical characteristics of aspirin-exacerbated respiratory disease. Ann. Allergy Asthma Immunol. 2002, 89, 474–478. [Google Scholar] [CrossRef]
- White, A.A.; Stevenson, D.D. Aspirin-exacerbated respiratory disease: Update on pathogenesis and desensitization. Semin. Respir. Crit. Care Med. 2012, 33, 588–594. [Google Scholar] [CrossRef]
- Kidon, M.I.; Kang, L.W.; Chin, C.W.; Hoon, L.S.; See, Y.; Goh, A.; Lin, J.T.; Chay, O.M. Early presentation with angioedema and urticaria in cross-reactive hypersensitivity to nonsteroidal antiinflammatory drugs among young, Asian, atopic children. Pediatrics 2005, 116, e675–e680. [Google Scholar] [CrossRef] [PubMed]
- White, A.A.; Stevenson, D.D. Aspirin-Exacerbated Respiratory Disease. N. Engl. J. Med. 2018, 379, 1060–1070. [Google Scholar] [CrossRef]
- Peters-Golden, M.; Gleason, M.M.; Togias, A. Cysteinyl leukotrienes: Multi-functional mediators in allergic rhinitis. Clin. Exp. Allergy 2006, 36, 689–703. [Google Scholar] [CrossRef]
- Steinke, J.W.; Borish, L. Factors driving the aspirin exacerbated respiratory disease phenotype. Am. J. Rhinol. Allergy 2015, 29, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Steinke, J.W.; Liu, L.; Huyett, P.; Negri, J.; Payne, S.C.; Borish, L. Prominent role of IFN-gamma in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2013, 132, 856–865.e3. [Google Scholar] [CrossRef]
- Milanovic, M.; Terszowski, G.; Struck, D.; Liesenfeld, O.; Carstanjen, D. IFN consensus sequence binding protein (Icsbp) is critical for eosinophil development. J. Immunol. 2008, 181, 5045–5053. [Google Scholar] [CrossRef]
- Mellor, E.A.; Austen, K.F.; Boyce, J.A. Cysteinyl leukotrienes and uridine diphosphate induce cytokine generation by human mast cells through an interleukin 4-regulated pathway that is inhibited by leukotriene receptor antagonists. J. Exp. Med. 2002, 195, 583–592. [Google Scholar] [CrossRef]
- Johns, C.B.; Laidlaw, T.M. Elevated total serum IgE in nonatopic patients with aspirin-exacerbated respiratory disease. Am. J. Rhinol. Allergy 2014, 28, 287–289. [Google Scholar] [CrossRef]
- Cahill, K.N.; Murphy, K.; Singer, J.; Israel, E.; Boyce, J.A.; Laidlaw, T.M. Plasma tryptase elevation during aspirin-induced reactions in aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2019, 143, 799–803.e2. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.N.; Bensko, J.C.; Boyce, J.A.; Laidlaw, T.M. Prostaglandin D(2): A dominant mediator of aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2015, 135, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Tanaka, K.; Yoshie, O.; Ogawa, K.; Kenmotsu, K.; Takamori, Y.; Ichimasa, M.; Sugamura, K.; Nakamura, M.; Takano, S.; et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001, 193, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Cavagnero, K.; Doherty, T.A. Cytokine and Lipid Mediator Regulation of Group 2 Innate Lymphoid Cells (ILC2s) in Human Allergic Airway Disease. J. Cytokine Biol. 2017, 2. [Google Scholar] [CrossRef]
- Eastman, J.J.; Cavagnero, K.J.; Deconde, A.S.; Kim, A.S.; Karta, M.R.; Broide, D.H.; Zuraw, B.L.; White, A.A.; Christiansen, S.C.; Doherty, T.A. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2017, 140, 101–108.e3. [Google Scholar] [CrossRef]
- Buchheit, K.M.; Cahill, K.N.; Katz, H.R.; Murphy, K.C.; Feng, C.; Lee-Sarwar, K.; Lai, J.; Bhattacharyya, N.; Israel, E.; Boyce, J.A.; et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2016, 137, 1566–1576.e5. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Jerico, D.-R.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, R.J.; Gregory, L.G.; Peiro, T.; Akthar, S.; Campbell, G.A.; Walker, S.A.; Lloyd, C.M. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J. Allergy Clin. Immunol. 2014, 134, 583–592.e6. [Google Scholar] [CrossRef]
- Hung, L.Y.; Lewkowich, I.P.; Dawson, L.A.; Downey, J.; Yang, Y.; Smith, D.E.; Herbert, D.R. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc. Natl. Acad. Sci. USA 2013, 110, 282–287. [Google Scholar] [CrossRef]
- Liu, T.; Kanaoka, Y.; Barrett, N.A.; Feng, C.; Garofalo, D.; Lai, J.; Buchheit, K.; Bhattacharya, N.; Laidlaw, T.M.; Katz, H.R.; et al. Aspirin-Exacerbated Respiratory Disease Involves a Cysteinyl Leukotriene-Driven IL-33-Mediated Mast Cell Activation Pathway. J. Immunol. 2015, 195, 3537–3545. [Google Scholar] [CrossRef]
- Austen, K.F.; Maekawa, A.; Kanaoka, Y.; Boyce, J.A. The leukotriene E4 puzzle: Finding the missing pieces and revealing the pathobiologic implications. J. Allergy Clin. Immunol. 2009, 124, 406–414; quiz 415–416. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Gyles, S.L.; Wettey, F.R.; Gazi, L.; Townsend, E.; Hunter, M.G.; Pettipher, R. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J. Immunol. 2005, 175, 6531–6536. [Google Scholar] [CrossRef]
- Paruchuri, S.; Jiang, Y.; Feng, C.; Francis, S.A.; Plutzky, J.; Boyce, J.A. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J. Biol. Chem. 2008, 283, 16477–16487. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Kidder, M.S.; Bhattacharyya, N.; Xing, W.; Shen, S.; Milne, G.L.; Castells, M.C.; Chhay, H.; Boyce, J.A. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood 2012, 119, 3790–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoine, C.; Murphy, R.C.; Henson, P.M.; Maclouf, J. Time-dependent utilization of platelet arachidonic acid by the neutrophil in formation of 5-lipoxygenase products in platelet-neutrophil co-incubations. Biochim. Biophys. Acta 1992, 1128, 139–146. [Google Scholar] [CrossRef]
- Raiden, S.; Schettini, J.; Salamone, G.; Trevani, A.; Vermeulen, M.; Gamberale, R.; Giordano, M.; Geffner, J. Human platelets produce granulocyte-macrophage colony-stimulating factor and delay eosinophil apoptosis. Lab. Investig. 2003, 83, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A.; Resendiz-Hernandez, J.M.; Falfan-Valencia, R.; Teran, L.M. Biochemical pathogenesis of aspirin exacerbated respiratory disease (AERD). Clin. Biochem. 2013, 46, 566–578. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Boyce, J.A. Pathogenesis of aspirin-exacerbated respiratory disease and reactions. Immunol. Allergy Clin. N. Am. 2013, 33, 195–210. [Google Scholar] [CrossRef]
- Human Nose. Available online: http://pngimg.com/uploads/nose/nose_PNG2.png (accessed on 12 March 2019).
- Mast Cell. Hematopoiesis (Human) Diagram en. Available online: https://commons.wikimedia.org/wiki/File:Mast_cell.svg (accessed on 12 March 2019).
- Art, S.M. Vasodilation. Available online: https://smart.servier.com/smart_image/vasodilation/ (accessed on 12 March 2019).
- Physiology, A. 1907 Granular Leukocytes. Connexions Web Site. Available online: http://cnx.org/content/col11496/1.6/ (accessed on 12 March 2019).
- Szczeklik, A.; Nizankowska, E. Clinical features and diagnosis of aspirin induced asthma. Thorax 2000, 55 (Suppl. 2), S42–S44. [Google Scholar] [CrossRef] [Green Version]
- Nizankowska-Mogilnicka, E.; Bochenek, G.; Mastalerz, L.; Swierczynska, M.; Picado, C.; Scadding, G.; Kowalski, M.L.; Setkowicz, M.; Ring, J.; Brockow, K.; et al. EAACI/GA2LEN guideline: Aspirin provocation tests for diagnosis of aspirin hypersensitivity. Allergy 2007, 62, 1111–1118. [Google Scholar] [CrossRef]
- Hope, A.P.; Woessner, K.A.; Simon, R.A.; Stevenson, D.D. Rational approach to aspirin dosing during oral challenges and desensitization of patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2009, 123, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Nizankowska, E.; Bestynska-Krypel, A.; Cmiel, A.; Szczeklik, A. Oral and bronchial provocation tests with aspirin for diagnosis of aspirin-induced asthma. Eur. Respir. J. 2000, 15, 863–869. [Google Scholar] [CrossRef] [Green Version]
- Nasser, S.M.; Patel, M.; Bell, G.S.; Lee, T.H. The effect of aspirin desensitization on urinary leukotriene E4 concentrations in aspirin-sensitive asthma. Am. J. Respir. Crit. Care Med. 1995, 151, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, G.; Stachura, T.; Szafraniec, K.; Plutecka, H.; Sanak, M.; Nizankowska-Mogilnicka, E.; Sladek, K. Diagnostic Accuracy of Urinary LTE4 Measurement to Predict Aspirin-Exacerbated Respiratory Disease in Patients with Asthma. J. Allergy Clin. Immunol. Pract. 2018, 6, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.U.; White, A.A.; Ding, D.; Dursun, A.B.; Woessner, K.M.; Simon, R.A.; Stevenson, D.D. Use of intranasal ketorolac and modified oral aspirin challenge for desensitization of aspirin-exacerbated respiratory disease. Ann. Allergy Asthma Immunol. 2010, 105, 130–135. [Google Scholar] [CrossRef]
- Scott, D.R.; White, A.A. Approach to desensitization in aspirin-exacerbated respiratory disease. Ann. Allergy Asthma Immunol. 2014, 112, 13–17. [Google Scholar] [CrossRef]
- DeGregorio, G.A.; Singer, J.; Cahill, K.N.; Laidlaw, T. A 1-Day, 90-Minute Aspirin Challenge and Desensitization Protocol in Aspirin-Exacerbated Respiratory Disease. J. Allergy Clin. Immunol. Pract. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mastalerz, L.; Milewski, M.; Duplaga, M.; Nizankowska, E.; Szczeklik, A. Intranasal fluticasone propionate for chronic eosinophilic rhinitis in patients with aspirin-induced asthma. Allergy 1997, 52, 895–900. [Google Scholar] [CrossRef]
- Sarnes, E.; Crofford, L.; Watson, M.; Dennis, G.; Kan, H.; Bass, D. Incidence and US costs of corticosteroid-associated adverse events: A systematic literature review. Clin. Ther. 2011, 33, 1413–1432. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, S.E.; Malmstrom, K.; Nizankowska, E.; Dahlen, B.; Kuna, P.; Kowalski, M.; Lumry, W.R.; Picado, C.; Stevenson, D.D.; Bousquet, J.; et al. Improvement of aspirin-intolerant asthma by montelukast, a leukotriene antagonist: A randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 9–14. [Google Scholar] [CrossRef] [PubMed]
- De Lepeleire, I.; Reiss, T.F.; Rochette, F.; Botto, A.; Zhang, J.; Kundu, S.; Decramer, M. Montelukast causes prolonged, potent leukotriene D4-receptor antagonism in the airways of patients with asthma. Clin. Pharmacol. Ther. 1997, 61, 83–92. [Google Scholar] [CrossRef]
- Dahlen, B.; Nizankowska, E.; Szczeklik, A.; Zetterstrom, O.; Bochenek, G.; Kumlin, M.; Mastalerz, L.; Pinis, G.; Swanson, L.J.; Boodhoo, T.I.; et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am. J. Respir. Crit. Care Med. 1998, 157, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Ta, V.; White, A.A. Survey-Defined Patient Experiences With Aspirin-Exacerbated Respiratory Disease. J. Allergy Clin. Immunol. Pract. 2015, 3, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, T.M.; Fuentes, D.J.; Wang, Y. Efficacy of Zileuton in Patients with Asthma and History of Aspirin Sensitivity: A Retrospective Analysis of Data from Two Phase 3 Studies. J. Allergy Clin. Immunol. 2017, 139, AB384. [Google Scholar] [CrossRef]
- White, A.; Ludington, E.; Mehra, P.; Stevenson, D.D.; Simon, R.A. Effect of leukotriene modifier drugs on the safety of oral aspirin challenges. Ann. Allergy Asthma Immunol. 2006, 97, 688–693. [Google Scholar] [CrossRef]
- Heise, C.E.; O’Dowd, B.F.; Figueroa, D.J.; Sawyer, N.; Nguyen, T.; Im, D.S.; Stocco, R.; Bellefeuille, J.N.; Abramovitz, M.; Cheng, R.; et al. Characterization of the human cysteinyl leukotriene 2 receptor. J. Biol. Chem. 2000, 275, 30531–30536. [Google Scholar] [CrossRef]
- Adelman, J.; McLean, C.; Shaigany, K.; Krouse, J.H. The Role of Surgery in Management of Samter’s Triad: A Systematic Review. Otolaryngol. Head Neck Surg. 2016, 155, 220–237. [Google Scholar] [CrossRef]
- Xu, J.J.; Sowerby, L.; Rotenberg, B.W. Aspirin desensitization for aspirin-exacerbated respiratory disease (Samter’s Triad): A systematic review of the literature. Int. Forum Allergy Rhinol. 2013, 3, 915–920. [Google Scholar] [CrossRef]
- Bobolea, I.; Del Pozo, V.; Sanz, V.; Cabanas, R.; Fiandor, A.; Alfonso-Carrillo, C.; Salcedo, M.A.; Heredia Revuelto, R.; Quirce, S. Aspirin desensitization in aspirin-exacerbated respiratory disease: New insights into the molecular mechanisms. Respir. Med. 2018, 143, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Arm, J.P.; O’Hickey, S.P.; Spur, B.W.; Lee, T.H. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am. Rev. Respir. Dis. 1989, 140, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.D.; Simon, R.A. Selection of patients for aspirin desensitization treatment. J. Allergy Clin. Immunol. 2006, 118, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.M.; Aronica, M.A.; Maierson, E.S.; Wang, X.F.; Vasas, D.C.; Hazen, S.L. Omalizumab can inhibit respiratory reaction during aspirin desensitization. Ann. Allergy Asthma Immunol. 2018, 121, 98–104. [Google Scholar] [CrossRef]
- Hayashi, H.; Mitsui, C.; Nakatani, E.; Fukutomi, Y.; Kajiwara, K.; Watai, K.; Sekiya, K.; Tsuburai, T.; Akiyama, K.; Hasegawa, Y.; et al. Omalizumab reduces cysteinyl leukotriene and 9alpha,11beta-prostaglandin F2 overproduction in aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2016, 137, 1585–1587.e4. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, P.; Calus, L.; Van Zele, T.; Blomme, K.; De Ruyck, N.; Bauters, W.; Hellings, P.; Brusselle, G.; De Bacquer, D.; van Cauwenberge, P.; et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J. Allergy Clin. Immunol. 2013, 131, 110–116.e1. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, D.P.; Bodtger, U.; Sidenius, K.; Maltbaek, N.; Pedersen, L.; Madsen, H.; Andersson, E.A.; Norgaard, O.; Madsen, L.K.; Chawes, B.L. Efficacy, adverse events, and inter-drug comparison of mepolizumab and reslizumab anti-IL-5 treatments of severe asthma - a systematic review and meta-analysis. Eur. Clin. Respir. J. 2018, 5, 1536097. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, P.; Van Bruaene, N.; Cattaert, T.; Van Steen, K.; Van Zele, T.; Acke, F.; De Ruyck, N.; Blomme, K.; Sousa, A.R.; Marshall, R.P.; et al. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J. Allergy Clin. Immunol. 2011, 128, 989–995.e8. [Google Scholar] [CrossRef]
- Chupp, G.L.; Bradford, E.S.; Albers, F.C.; Bratton, D.J.; Wang-Jairaj, J.; Nelsen, L.M.; Trevor, J.L.; Magnan, A.; Ten Brinke, A. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): A randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir. Med. 2017, 5, 390–400. [Google Scholar] [CrossRef]
- Bachert, C.; Sousa, A.R.; Lund, V.J.; Scadding, G.K.; Gevaert, P.; Nasser, S.; Durham, S.R.; Cornet, M.E.; Kariyawasam, H.H.; Gilbert, J.; et al. Reduced need for surgery in severe nasal polyposis with mepolizumab: Randomized trial. J. Allergy Clin. Immunol. 2017, 140, 1024–1031.e14. [Google Scholar] [CrossRef]
- Tuttle, K.L.; Buchheit, K.M.; Laidlaw, T.M.; Cahill, K.N. A retrospective analysis of mepolizumab in subjects with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. Pract. 2018, 6, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Mannent, L.; Naclerio, R.M.; Mullol, J.; Ferguson, B.J.; Gevaert, P.; Hellings, P.; Jiao, L.; Wang, L.; Evans, R.R.; et al. Effect of Subcutaneous Dupilumab on Nasal Polyp Burden in Patients With Chronic Sinusitis and Nasal Polyposis: A Randomized Clinical Trial. JAMA 2016, 315, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Castro, M.; Corren, J.; Maspero, J.; Wang, L.; Zhang, B.; Pirozzi, G.; Sutherland, E.R.; Evans, R.R.; Joish, V.N.; et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting beta2 agonist: A randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet 2016, 388, 31–44. [Google Scholar] [CrossRef]
- Mullol, J.; Laidlaw, T.M.; Dong, Q. Dupilumab improves nasal polyp burden and asthma control in patients with CRSwNP and NSAID-ERD. Allergy Asthma Immunol. Res. 2018, 73, 203. [Google Scholar]
- McFadden, E.A.; Kany, R.J.; Fink, J.N.; Toohill, R.J. Surgery for sinusitis and aspirin triad. Laryngoscope 1990, 100, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Abouali, O.; Keshavarzian, E.; Farhadi, G.; Faramarzi, A.; Ahmadi, G.; Bagheri, M. Micro and nanoparticle deposition in human nasal passage pre and post virtual maxillary sinus endoscopic surgery. Respir. Physiol. Neurobiol. 2012, 181, 335–345. [Google Scholar] [CrossRef]
- Kumar, H.; Jain, R. Review: The role of computational simulation in understanding the postoperative sinonasal environment. Clin. Biomech. 2018. [Google Scholar] [CrossRef]
- Jerschow, E.; Edin, M.; Chi, Y.; Hurst, B.; Abuzeid, W.; Akbar, N.; Gibber, M.; Fried, M.; Han, W.; Pelletier, T.; et al. Sinus surgery is associated with a decrease in aspirin-induced reaction severity in AERD patients. J. Allergy Clin. Immunol. Pract. 2018. [Google Scholar] [CrossRef]
- Mendelsohn, D.; Jeremic, G.; Wright, E.; Rotenberg, B. Revision rates after endoscopic sinus surgery: A recurrence analysis. Ann. Otol. Rhinol. Laryngol. 2011, 120, 162–166. [Google Scholar] [CrossRef]
- Lee, R.; Stevenson, D. Aspirin-Exacerbated Respiratory Disease: Evaluation and Management. Allergy Asthma Immunol. Res. 2011, 3, 3–10. [Google Scholar] [CrossRef]
- Cho, K.S.; Soudry, E.; Psaltis, A.J.; Nadeau, K.C.; McGhee, S.A.; Nayak, J.V.; Hwang, P.H. Long-term sinonasal outcomes of aspirin desensitization in aspirin exacerbated respiratory disease. Otolaryngol. Head Neck Surg. 2014, 151, 575–581. [Google Scholar] [CrossRef]
- Havel, M.; Ertl, L.; Braunschweig, F.; Markmann, S.; Leunig, A.; Gamarra, F.; Kramer, M.F. Sinonasal outcome under aspirin desensitization following functional endoscopic sinus surgery in patients with aspirin triad. Eur. Arch. Otorhinolaryngol. 2013, 270, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.M.; Waldram, J.D.; Woessner, K.M.; White, A.A. Long-term Clinical Outcomes of Aspirin Desensitization With Continuous Daily Aspirin Therapy in Aspirin-exacerbated Respiratory Disease. Am. J. Rhinol. Allergy 2018, 32, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.D.; Hankammer, M.A.; Mathison, D.A.; Christiansen, S.C.; Simon, R.A. Aspirin desensitization treatment of aspirin-sensitive patients with rhinosinusitis-asthma: Long-term outcomes. J. Allergy Clin. Immunol. 1996, 98, 751–758. [Google Scholar] [CrossRef]
- Adappa, N.D.; Ranasinghe, V.J.; Trope, M.; Brooks, S.G.; Glicksman, J.T.; Parasher, A.K.; Palmer, J.N.; Bosso, J.V. Outcomes after complete endoscopic sinus surgery and aspirin desensitization in aspirin-exacerbated respiratory disease. Int. Forum Allergy Rhinol. 2018, 8, 49–53. [Google Scholar] [CrossRef]
- Stevens, W.; Peters, A.; Hirsch, A.; Nordberg, C.; Schwartz, B.; Mercer, D.; Mahdavinia, M.; Grammer, L.; Hulse, K.; Kern, R.; et al. Clinical Characteristics of Patients with Chronic Rhinosinusitis with Nasal Polyps, Asthma, and Aspirin-Exacerbated Respiratory Disease. J. Allergy Clin. Immunol. Pract. 2017, 5, 1061–1070. [Google Scholar] [CrossRef]
- Morrissey, D.K.; Bassiouni, A.; Psaltis, A.J.; Naidoo, Y.; Wormald, P.J. Outcomes of revision endoscopic modified Lothrop procedure. Int. Forum Allergy Rhinol. 2016, 6, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ragab, S.M.; Lund, V.J.; Scadding, G. Evaluation of the medical and surgical treatment of chronic rhinosinusitis: A prospective, randomised, controlled trial. Laryngoscope 2004, 114, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.A.; Marchiano, E.; Vazquez, A. Extended Endoscopic and Open Sinus Surgery for Refractory Chronic Rhinosinusitis. Otolaryngol. Clin. N. Am. 2017, 50, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Metson, R.; Sindwani, R. Endoscopic surgery for frontal sinusitis–a graduated approach. Otolaryngol. Clin. N. Am. 2004, 37, 411–422. [Google Scholar] [CrossRef]
- Abuzeid, W.M.; Vakil, M.; Lin, J.; Fastenberg, J.; Akbar, N.A.; Fried, M.P.; Fang, C.H. Endoscopic modified Lothrop procedure after failure of primary endoscopic sinus surgery: A meta-analysis. Int. Forum Allergy Rhinol. 2018, 8, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, Y.; Bassiouni, A.; Keen, M.; Wormald, P.J. Long-term outcomes for the endoscopic modified Lothrop/Draf III procedure: A 10-year review. Laryngoscope 2014, 124, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Stammberger, H.; Posawetz, W. Functional endoscopic sinus surgery. Concept, indications and results of the Messerklinger technique. Eur. Arch. Otorhinolaryngol. 1990, 247, 63–76. [Google Scholar] [CrossRef]
- DeConde, A.; Suh, J.; Mace, J.; Alt, J.; Smith, T. Outcomes of complete vs targeted approaches to endoscopic sinus surgery. Int. Forum Allergy Rhinol. 2015, 5, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuzeid, W.M.; Mace, J.C.; Costa, M.L.; Rudmik, L.; Soler, Z.M.; Kim, G.S.; Smith, T.L.; Hwang, P.H. Outcomes of chronic frontal sinusitis treated with ethmoidectomy: A prospective study. Int. Forum Allergy Rhinol. 2016, 6, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.A.; Vazquez, A.; Liu, J.K.; Baredes, S. Endoscopic Approaches to the Frontal Sinus: Modifications of the Existing Techniques and Proposed Classification. Otolaryngol. Clin. N. Am. 2016, 49, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Craig, J.; Cohen, N.; Adappa, N.; Khalili, S.; Palmer, J. Sinus irrigations before and after surgery-Visualization through computational fluid dynamics simulations. Laryngoscope 2017, 126, E90–E96. [Google Scholar] [CrossRef]
- Anderson, P.; Sindwani, R. Safety and efficacy of the endoscopic modified Lothrop procedure: A systematic review and meta-analysis. Laryngoscope 2009, 119, 1828–1833. [Google Scholar] [CrossRef]
- Jankowski, R.; Pigret, D.; Decroocq, F.; Blum, A.; Gillet, P. Comparison of radical (nasalisation) and functional ethmoidectomy in patients with severe sinonasal polyposis. A retrospective study. Rev. Laryngol. Otol. Rhinol. (Bord) 2006, 127, 131–140. [Google Scholar]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.L.; Lee, A.Y.; Abuzeid, W.M. Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management. Med. Sci. 2019, 7, 45. https://doi.org/10.3390/medsci7030045
Li KL, Lee AY, Abuzeid WM. Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management. Medical Sciences. 2019; 7(3):45. https://doi.org/10.3390/medsci7030045
Chicago/Turabian StyleLi, Kevin L., Andrew Y. Lee, and Waleed M. Abuzeid. 2019. "Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management" Medical Sciences 7, no. 3: 45. https://doi.org/10.3390/medsci7030045
APA StyleLi, K. L., Lee, A. Y., & Abuzeid, W. M. (2019). Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management. Medical Sciences, 7(3), 45. https://doi.org/10.3390/medsci7030045