1. Introduction
Arsenic is considered as one of the most toxic pollutants due to its mutagenic and carcinogenic effect on human beings. The continuous industrialization of developing countries will probably lead to an increase in arsenic release in the environment, which will cause potentially serious problems throughout the world [
1]. The presence of arsenic in water is due to the dissolution of minerals from subterranean strata or from anthropogenic activities such as the leaching of manmade arsenic compounds deriving from smelting of metal ores, agricultural pesticides, desiccants and wood preservatives production [
2]. In particular, the presence of arsenic in water supplies has been indicated as responsible for arsenical dermatitis, enlargement of liver, neurological effects, heart disease and several forms of cancer [
3].
Arsenic is sensitive to mobilization at pH values typical of natural waters (pH 6.5–8.5) under both oxidizing and reducing conditions; while it can occur in the environment in several oxidation states (−3, 0, +3 and +5), in natural waters it is mostly found in inorganic forms as oxyanions of trivalent arsenite [As(III)] or pentavalent arsenate [As(V)] [
4]. According to the EPA (Environmental Protection Agency) the acceptable level of As in drinking water is fixed at 0.010 mg/L [
5].
The oxidation-reduction conditions affect the speciation of arsenic: in oxygen-rich environments, where aerobic conditions persist, arsenate [As(V)] is prevalent and exists as a monovalent (H
2AsO
4−) or divalent (HAsO
42−) anion, whereas arsenite [As(III)] exists as acid (H
3AsO
3) and anionic (H
2AsO
3−) species in a moderately reducing environment, where anoxic conditions persist [
6,
7]. Between the two forms, As(III) is more “mobile” and at the same time much more toxic than As(V) which is easily fixed onto soil particles [
8].
Several techniques have been developed to remove arsenic from water: coagulation, reverse osmosis, adsorption and anion exchange processes are some of the commonly used [
9,
10]. Due to both their low cost and a relatively operational ease, there is now a raising interest on sorption methods that seem to be the most promising way to remove arsenic from water sources [
11]. Several materials have already been studied for the adsorption of arsenic from water, including metal-loaded coral limestone [
12,
13] hematite and feldspar, sandy soils [
14], activated carbon [
15,
16,
17,
18], activated alumina [
19,
20], lanthanum-impregnated silica gel [
21], hydrous zirconium oxide [
22] and hydrous iron oxide [
23] and zero valent iron [
24,
25]. Ohki et al. [
13] found an adsorption capability of 150 µg/g As(V) on coral limestone, while Xu et al. [
14] investigated the adsorption capacity of drinking water treatment solids (DWTS) in As(V) removal processes: at its best, DWTS adsorption capability was found to be 170 mg/g, although heavily affected by pH conditions.
Recently, activated carbons (both commercial and synthetized at lab scale) were heavily tested to remove arsenic from water sources: Pattanayak et al. [
26] found that char carbon can remove 89 mg/g of As(III) and 34.46 mg/g of As(V) from water under strong acidic conditions (pH = 2). Coconut husk carbon showed also high performances: this material was tested by Manju et al. [
27] on arsenic removal from industrial wastewater, and they found that it was able to adsorb 146.3 mg/g of As(III).
In recent years, other materials with anion exchange properties gained attention as potentially good candidates to remove arsenic from water sources, since their anion exchange capacity can be useful in the removal process. Among the possible candidates, zeolites seem to be quite promising. Zeolites are inorganic cation-exchangers of natural and synthetic origin. Many deposits of natural zeolites of sedimentary origin, commonly referred to as zeolitc tuffs, are diffused all over the globe [
28].The documented characteristics of affinity of natural zeolites toward several heavy metal cations and their modest cost, due to the wide availability, make the ion-exchange an advantageous procedure, suitable for pollution removal [
29,
30].
In order to make a zeolite able to exchange anions, the cations found on the raw zeolite surface can be replaced by long chained cationic surfactants such as hexadecyltrimethylammonium (HDTMA), by an ion exchange process; due to the large dimensions of such molecules, the exchange takes place on the zeolite grain surface only. If the formation of a double layer (micelle formation) of organic molecules is induced by ion-exchange and tail–tail interaction among the surfactant hydrophobic chains, the charge on the zeolite surface changes from negative to positive [
31,
32]. The positive surface charge provides sites for the exchange of anions such as chromate and arsenate. In this way, surfactant-modified zeolites (SMZ) can act as both cation and anion inorganic exchangers.
Figure 1 shows the mechanism that leads to the formation first of the monolayer by surface ion exchange and later of the patchy or complete bilayers by interaction of the hydrophobic tails of the surfactant molecules.
Recently, natural zeolites, particularly clinoptilolite, modified with cationic surfactant, were used to remove different types of contaminants from water: nitrates [
9], chromates [
33,
34], cesium, lead, zinc [
34], perchloroethylene [
35], sulfates [
36], NSAIDs [
37] and antibiotics [
38].
Regarding the As(V) removal, some researchers examined the behavior of both HDTMA-modified, mordenite and clinoptilolite rich tuff, in the elimination of this polluting oxyanion from aqueous solutions [
39]. The authors, although basing on the results of thermodynamic runs, showed that the two samples removed arsenic with different kinetics. Moreover, in agreement with an earlier study, they established that surfactant surface coverage plays a key role in sorption; in particular, when the HDTMA loading level on zeolite exceeds monolayer coverage, a substantial increase in arsenate sorption capacity could be attained.
Li et al. [
40] also carried out a comparison between a surface-modified clinoptilolite and kaolinite minerals in the arsenic removal process. For this aim, the authors performed some equilibrium runs by contacting the modified systems with arsenate or arsenite solution at 0.1–2.0 mM in tubes, under continuous stirring for 24 h. The outcomes exhibited a notable increase in arsenate sorption capacity when the loading level of HDTMA on zeolite and kaolinite surfaces overrun monolayer coverage up to the bilayer formation (200% of the ECEC).
Furthermore, Mendoza-Barrón et al. [
41] emphasized the effect of pH on the As(V) removal process through a clinoptilolite sample modified with HDTMA-Br. In aforementioned work, after the surface modification of a clinoptilolite-rich rock to improve its anionic capacity, adsorption equilibrium runs of As(V) from water solutions were carried out. Based on the results, it was proved that the affinity of the modified zeolite for As(V) was extremely dependent on the form of arsenic species implicated in the solution at different pH. The adsorption capacity increased with solution pH, from pH 5 to pH 7. This implied that, among the different As forms, the affinity of the HAsO
42− toward the SMZ was higher than that of the H
2AsO
4−.
However, most of the published research has mainly considered the study of the As removal mechanism through the execution of thermodynamic test. On the contrary, few are the data concerning kinetic analysis in batch or in dynamic conditions to better simulate the effects and the feasibility of a more realistic As abatement process such as the pump and treat column. Moreover, clinoptilolite was the main zeolite sample investigated.
On this basis, the aim of the present research is to verify a possible use of two natural zeolites—a phillipsite-rich tuff (PHI) from Marano (Naples, Italy) and a clinoptilolite-rich tuff (CLI) from Eskişehir (Turkey)—modified by exchange with cationic surfactants HDTMA bromide and HDTMA chloride, as alternative adsorbents for removing arsenate from water solutions.
The ability of these surfactant-modified natural zeolites (SMNZs) to remove As(V) from water was studied at first by kinetic and thermodynamic experiments in batch, to verify the efficiency and the selectivity of modified zeolites towards As(V). Later, dynamic runs were performed by eluting fixed-bed columns containing the selected SMNZs with As(V) solutions to verify their ability to remove the anion under more realistic conditions.
3. Results and Discussion
3.1. Sample Characterization
The XRPD patterns of CLI and PHI samples are reported in
Figure 2A,B. The CLI sample contains clinoptilolite as the only exchanger phase, while the PHI sample is more heterogeneous, containing at least three zeolitic exchangers, mainly phillipsite with minor amounts of chabazite and analcime.
Quantitative X-ray analysis carried out using the RIR/Rietveld method supplied the following mineralogical composition: 79% clinoptilolite, 5% feldspar, 1% quartz, 15% clays minerals for CLI and 44% phillipsite, 4% chabazite, 10% analcime, 32% feldspar, 20% clays minerals for PHI, respectively. Concerning the clinoptilolite sample, XRPD analysis confirms that the sample consists of clinoptilolite as the only phase with exchange properties. Therefore, the CLI sample can be considered as a single exchanger containing only 21% of non-exchanging phase.
Concerning the PHI sample, the analysis underlined its heterogeneity, although phillipsite is the zeolite present in the maximum concentration. The second zeolite in order of abundance, the analcime, has a much denser microstructure than the other two (phillipsite and chabazite), which makes its contribution to the exchange process negligible, at least up to the end of our experiments. The last zeolitic phase, chabazite, is present in a very low concentration. This makes it possible to describe the PHI sample as a single exchanger phase with a Si/Al ratio of 2.38.
Table 1 reports the chemical analysis after acid digestion of the selected samples. Although it is a bulk analysis, it is possible to highlight how the CLI sample is mainly a potassic/calcic tuff, while PHI is a potassic/sodic/calcic tuff. This is perfectly in agreement with the genesis and origins of the two selected tuffs.
The CEC of the two samples was 1.97 meq g−1 and 2.03 meq g−1 for CLI and PHI, respectively. These relatively close values arise from the higher cation content of the phillipsite in the PHI sample that compensates for its amount, which is lower than that of clinoptilolite in the CLI sample. The ECEC values were 0.142 meq g−1 and 0.130 meq g−1, for CLI and PHI, respectively. Such values are for both materials roughly 1 out of 10 of the total exchange capacity, which accounts for the surface/bulk ratio of the tuff grains.
Finally, the AEC values obtained for the four samples are: CLI-Br = 0.142 meq g−1; CLI-Cl = 0.071 meq g−1; PHI-Br = 0.115 meq g−1; PHI-Cl = 0.058 meq g−1.
The data show the following evidence:
The AEC value of both HDTMA-Br modified samples is about equal to the ECEC values: this indicates that a double layered micelle is formed on the zeolite surface;
On the contrary, the AEC value of both HDTMA-Cl modified samples is lower than the ECEC values: in this case, a patchy bilayer is formed on the zeolite surface.
This demonstrates that the counterion affects the micelle formation more than the zeolite present in the sample: even if the genesis and structure of the two tuffs are different, the morphology of the zeolites present is almost the same, characterized by a rigid framework with internal channels oriented along two preferential directions (planar-like structure). This type of morphology, together with the high Si/Al ratio typical of these natural zeolites, leaves many free surfaces available to interact with large surfactant molecules, which, however, cannot access the most internal cationic sites of the zeolitic framework, but are able to micellize on the surface of the same tuffs. The obtained results are confirmed by previous experiments in which a clinoptilolite-rich sample coming from New Mexico was also used [
31,
42]. Lastly, through the HPLC analysis of the liquid phase, no release of HDTMA by SMNZs was found, at least under the conditions in which the AEC evaluation was performed. This indicates a fair stability of the surfactant zeolite system both in the case of patchy and complete bilayer formation. Moreover, already in previous works [
37], it was pointed out that a loss in surfactant could occur in extreme stirring or washing conditions—a situation not applied in this research.
3.2. Ion Exchange Runs
3.2.1. Preliminary Runs
As previously pointed out, the effect of the pH on the removal process was checked, prior to further investigating the behavior of the SMNZs, by performing some preliminary tests at different pH values. In fact, it is well known that as the pH changes, both the surface charge of the SMNZ and the As speciation will change. The effect of pH on the As species uptake is reported in
Figure 3 for CLI-Br and PHI-Br samples.
From the comparison of the equilibrium values obtained at different pH, it can be observed that the higher uptake occurs at pH = 7 for both SMNZs. These results, supported by literature data [
41], seem to indicate that the SMZ is selective towards HAsO
42− against H
2AsO
4−, because the concentration of the former increases with the pH, while the positive charge (and consequently its ability to remove anions) of the SMZ decreases with the pH (actually, as previously reported [
57], the point of zero charge (PZC) of the modified zeolites is around 8). The most favorable pH condition is then met when (at pH around 7) the higher amount of HAsO
42− (the species with the greatest affinity towards the exchanger) is associated with the positively charged surface of the exchanger [
41]. Concerning the CLI-Cl and PHI-Cl samples, similar results have been obtained (here not reported for the sake of brevity).
3.2.2. Kinetics of the Anion Exchange
Figure 4 shows the kinetics of the anion exchange of As species on CLI-Br, CLI-Cl (
Figure 4A), PHI-Br and PHI-Cl (
Figure 4B) samples. All the kinetics appear reasonably fast: the equilibrium conditions had already been reached after just 50 min. For each tuff, the HDTMA-Br modified sample is always more effective than the HDTMA-Cl modified one: for PHI-based SMNZ samples, the maximum loading of As for PHI-Br is 4.21 mg g
−1, while for PHI-Cl is 1.20 mg g
−1.
The same thing holds true for the clinoptilolite-based SMNZ samples: the maximum loading value for CLI-Br is 8.14 mg g−1, while the value for CLI-Cl is 4.52 mg g−1. These results confirm once again how the surfactant that has Br− as a counterion generates a much more complete and compact micellar structure on the surface of the zeolite compared to the surfactant that has Cl− as a counterion, which can generate only a patchy bilayer. This results in the greater AEC of both Br-based SMNZs, which is practically equal to the ECEC of the two samples, while the surface AEC of the samples modified with HDTMA-Cl is equal to half of the ECEC.
Moreover, clinoptilolite-based SMNZs can always remove more As than phillipsite-based ones. This could be justified by the ECEC of the former that is greater due to a higher amount of zeolite in the sample: the mineralogical analysis showed that while the clinoptilolitic tuff has a zeolite content of 79%, the phillipsitic tuff reaches a maximum of 58% (even if the analcime contribution to the exchange is very low). However, both the modified samples were not able, in any of the experiments, to exploit the totality of their AEC, as reported in
Table 2, in which the AEC, for practical purposes, is reported as mg g
−1 of exchangeable arsenic.
This evidence is once again justified by the steric hindrance and shape complexity of the As anions compared to the “simple” Br
− and Cl
−. Such complexity forces a rearrangement of the micelle (either bilayer or patchy bilayer) to allow the entrance of the As species into its structure. This micellar rearrangement prevents the complete exploitation of its anionic exchange sites, because, as already proven [
32], the micellar structure is not perfectly flexible and has limited capacity to accommodate complex anions.
Kinetic exchange data were modeled with the pseudo-first and pseudo-second order equations (see
Section 2.4.1). The results are shown in
Figure 5 and
Table 3.
From the characteristic parameters of the models, as well as from the trend of the model curves reported in
Figure 5, it is clear how the kinetics of anion exchange are closer to a pseudo-second order trend, as also confirmed by the obtained values of R
2.
3.2.3. Thermodynamics of Anion Exchange
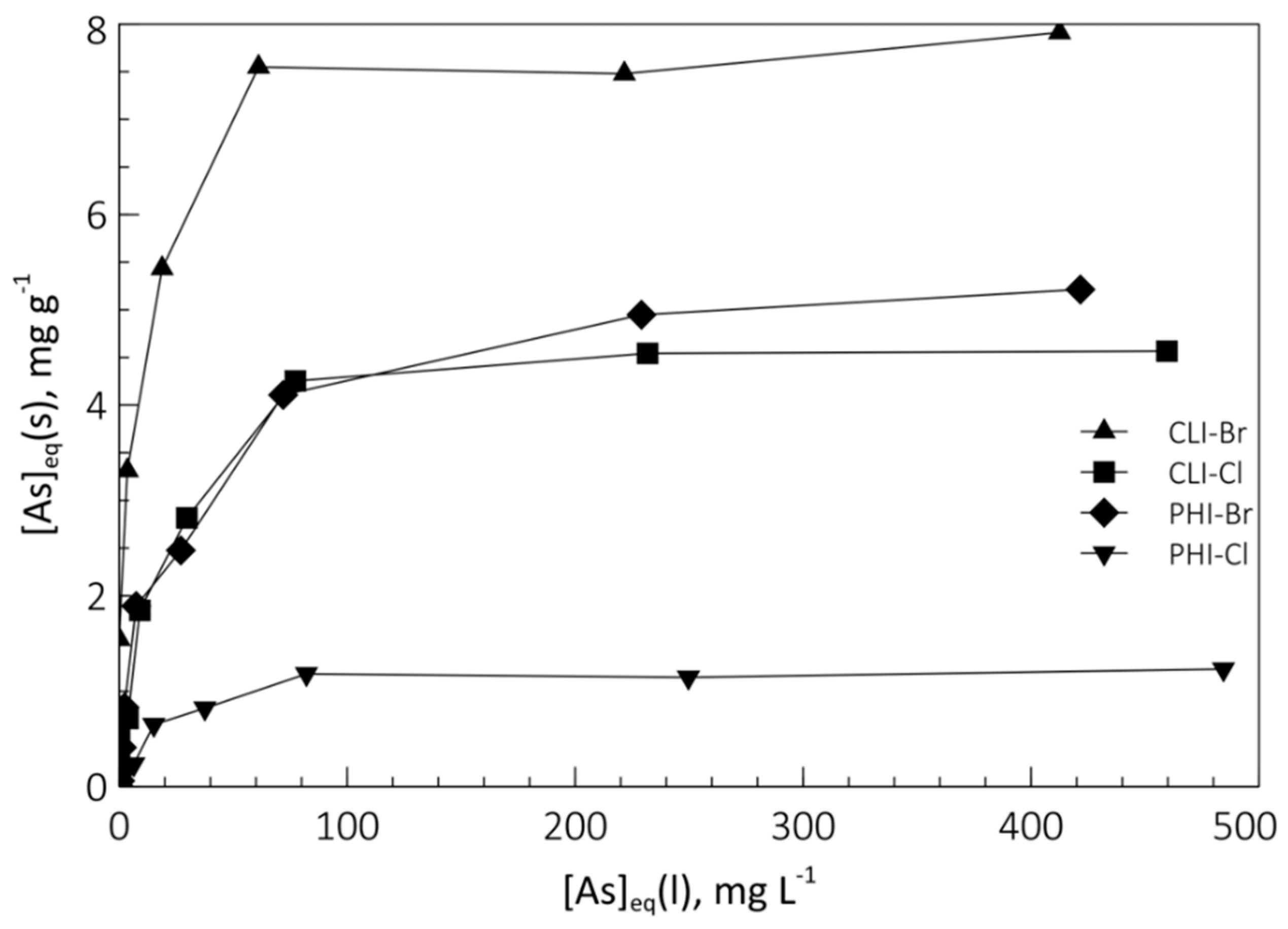
Figure 6 shows the As species exchange isotherms in SMNZ-Br and SMNZ-Cl, obtained at pH = 7. The graphs show the equilibrium concentration of the ingoing cation in the solid phase (mg g
−1) as a function of its equilibrium concentration in the liquid phase (mg L
−1).
Isotherms data allow us to state that:
Samples obtained with the use of HDTMA-Br perform better than those obtained with HDTMA-Cl: concerning the phillipsite-rich tuff, maximum exchange is of 5.2 mg g−1 for PHI-Br and 1.2 mg g−1 for PHI-Cl, while for clinoptilolite-rich tuff, maximum exchange is 7.9 mg g−1 for CLT-Br and 4.6 mg g−1 for CLT-Cl. This is once again justified by what was previously verified about the micelle formation on the zeolitic surface: if the counterion that balances the positive charge of the nitrogenous head of HDTMA+ is Br−, the micellar structure that is obtained on the zeolite surface is complete and compact (the bilayer has an anion exchange capacity, AEC, equal to its ECEC). If Cl− is the counterion, the micellar structure that is generated is like a patchy bilayer (partial micelle structure), which has obviously less exchangeable anions with respect to the previous case.
Clinoptilolite modified samples perform better than phillipsite modified samples. This can be due to the different density of HDTMA in the micelle, that in turn depends on the Al on the surface of the zeolite: the higher (4.90) Si/Al ratio of clinoptilolite will produce a micelle sparser than that produced on the phillipsite surface (Si/Al ratio = 2.38). This allows a higher amount of complex anions such as HAsO42− and H2AsO4− to enter the micellar structure, as the micelle can better rearrange itself to accommodate them.
The As anion can only partially exploit the surface anionic exchange capacity of both SMNZs investigated. This is a direct consequence of what was stated earlier: the uptake of a complex anion forces a rearrangement of the micellar structure, which probably displaces some exchange sites and makes them unavailable for further exchange.
The equilibrium data were modeled with Langmuir, Sips and Toth equations—already described in the Materials and Method section (see
Section 2.4.2).
Figure 7 shows the results of these modelings, while
Table 4 shows the significant parameters determined from each best fitting curve.
The modeling results show that the Toth model seems to be the one that made the experimental thermodynamic data dense; this is also confirmed by the values of R2, which are always higher for the Toth model. In any case, since the Langmuir isotherm is a two-parameter model that assumes adsorption on a defined number of sites that have the same affinity for the adsorbate, which forms a monolayer, and considering that the exchange of ions should not give multilayer formation, we will consider the Langmuir isotherm model as the most appropriate for our system.
Lastly, the data in the table confirm the maximum uptake values recorded by the thermodynamic tests in which the maximum value of removable As was 5.34 and 1.29 mg g−1 for the PHI-Br and PHI-Cl samples, respectively, and 7.86 and 4.87 mg g−1 for CLI-Br and CLI-Cl samples, respectively.
3.3. Dynamic Runs
Figure 8 reports the breakthrough curves for all investigated SMNZs. From the inspection of the plotted data, it is evident that all samples have a similar performance up to breakthrough, being able to treat about 20–30 bed volumes. In particular, the best and worst results were obtained for CLI-Br (30 bed volumes, 610 mL) and CLI-Cl (22 bed volumes, 440 mL), respectively, while PHI-based samples were quite insensitive to their anionic form (about 25 bed volumes, corresponding to about 490 mL).
Besides the treated volume up to breakthrough, the curves show different slopes, which indicate different mass transfer zones. Furthermore, in some case, a change in the curve slope is clear, especially in the case of PHI-based samples: such strange behavior can be due to another active phase (chabazite, analcime) which exchanges As, but at a slower rate.
To obtain a better estimation of the mass transfer zone, the calculation was performed on the curve data ranging from the breakthrough point up to the change of slope. Results are reported in
Table 5: the PHI-based samples showed the shortest MTZ, while CLI-based the longest one.
Such evidence could be due to a faster diffusion of the As ions on the particles surface, allowed by a more dense micelle structure, as previously seen. Furthermore, the different amount of As (V) removed, if compared with the batch kinetic tests, could be due to the high elution rate selected for the present runs, higher than the diffusion rate and the exchange kinetics; thus not giving time to fully exploit the AEC of the tested SMNZs. The obtained values of the Thomas model k constant (see
Table 6) are consistent with this hypothesis, being higher for PHI-based samples.
Among the four samples, PHI-Cl showed the best performances in terms of selectivity (0.24) and efficiency (0.15,
Table 5): such values, although quite low, were obtained under severe conditions, and should be considered only for the sake of comparison among the four samples. The q values obtained from the Thomas model are also in very good agreement with the M-AEC obtained with the Michaels approach, except for the PHI-Cl sample, which suffers from a noticeable change in slope.
4. Conclusions
The present paper reports the results of an investigation of the possible use of surface-modified phillipsite- and clinoptilolite-rich tuffs as alternative ion exchangers for removing As from wastewater. Several surfactant-modified natural zeolite (SMNZ) exchangers were prepared by contacting tuff samples with HDTMA-Br or HDTMA-Cl solutions under continuous flow. On the obtained samples, the estimation of the anionic exchange capacity together with a kinetic and thermodynamic characterization was performed in batch conditions, while a fixed-bed test was also performed in order to characterize their behaviour under severe dynamic conditions.
The influence of pH on As removal was preliminarily investigated, showing that the best performances were obtained for neutral pH values. For each tuff, modification with HDTMA-Br produced samples with an exchange capacity higher than those modified with HDTMA-Cl, suggesting the formation of a complete and compact micellar structure, opposite to the patchy bilayer obtained on the samples modified with HDTMA-Cl. Kinetic tests demonstrated that As removal is in all cases reasonably fast: it reached 80% in 1 h and 100% in 2 h. Data modeling results suggested that the ion exchange process follows a pseudo-second order kinetic. The thermodynamic tests were satisfactorily modeled with the Langmuir equation, confirming the better performance of the Br-modified samples over the Cl-modified ones: the maximum amount of removable As was 7.86 mg g−1 for CLI-Br sample.
Moreover, the dynamic tests showed that every sample was able to treat at least 22 bed volumes: the CLI-Br sample showed the best performance (30 bed volumes), even if the PHI-Cl sample showed slightly higher efficiency and selectivity. Such results suggest a possible use of SMNZs as ion exchangers for the removal of As from wastewaters.
Lastly, as also verified in previous papers [
32], there is a different selectivity on SMNZs for various anions present in waters. Therefore, it is important to study the effects of these possible interfering cations. However, in this first study, it was intended to verify only the removal efficiency of the As (V) by our modified zeolite supports (mainly as regards phillipsite rich-tuff). Future research, which have already begun, will be directed at verifying (with appropriate kinetic tests) the effect of the presence of other anionic species; this will certainly give us more complete information on the removal efficiency of our SMNZs.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}