Catalytic Conversion of α-Pinene to High-Density Fuel Candidates Over Stannic Chloride Molten Salt Hydrates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. General Procedure for Catalytic Conversion of α-pinene by Stannic Chloride Molten Salt Hydrates
3. Results and Discussion
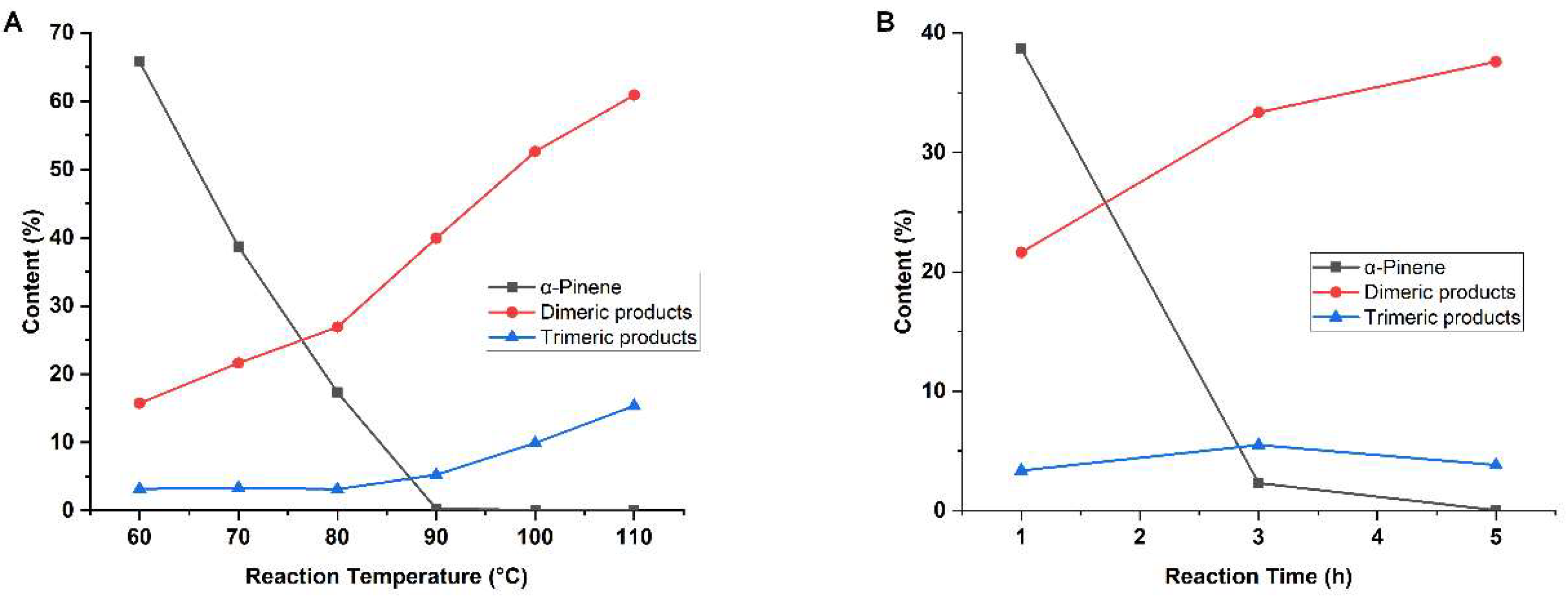
3.1. Catalytic Conversion of α-Pinene over Stannic Chloride Molten Salt Hydrates
3.2. Rationalization of the Formation of Monoterpene Isomers and Dimeric Hydrocarbons
3.3. Rationalization of the Formation of Heteroatom-Containing Dimeric Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xu, J.; Li, N.; Yang, X.; Li, G.; Wang, A.; Cong, Y.; Wang, X.; Zhang, T. Synthesis of diesel and jet fuel range alkanes with furfural and angelica lactone. ACS Catal. 2017, 7, 5880–5886. [Google Scholar] [CrossRef]
- Xia, Q.; Xia, Y.; Xi, J.; Liu, X.; Zhang, Y.; Guo, Y.; Wang, Y. Selective One-Pot Production of High-Grade Diesel-Range Alkanes from Furfural and 2-Methylfuran over Pd/NbOPO4. ChemSusChem 2017, 10, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Otsuki, A.L.; Mascal, M. Production of cellulosic gasoline via levulinic ester self-condensation. Green Chem. 2018, 20, 3804–3808. [Google Scholar] [CrossRef]
- Garcia, D.; Villa Holguín, A.L.; Lapuerta, M.; Bustamante, F.; Alarcón, E. Oxyfunctionalization of turpentine for fuel applications. Energy Fuels 2020, 34, 579–586. [Google Scholar] [CrossRef]
- Cho, S.M.; Kim, J.H.; Kim, S.H.; Park, S.Y.; Kim, J.C.; Choi, I.G. A comparative study on the fuel properties of biodiesel from woody essential oil depending on terpene composition. Fuel 2018, 218, 375–384. [Google Scholar] [CrossRef]
- Babu, A.M.; Saravanan, C.; Vikneswaran, M.; Jeo, V.E.; Sasikala, J. Visualization of in-cylinder combustion using endoscope in spark ignition engine fueled with pine oil blended gasoline. Fuel 2020, 263, 116707. [Google Scholar] [CrossRef]
- Ashok, B.; Raj, R.T.K.; Nanthagopal, K.; Krishnan, R.; Subbarao, R. Lemon peel oil–A novel renewable alternative energy source for diesel engine. Energy Convers. Manag. 2017, 139, 110–121. [Google Scholar] [CrossRef]
- Cho, S.M.; Hong, C.Y.; Park, S.Y.; Lee, D.S.; Choi, J.H.; Koo, B.; Choi, I.G. Application of Sulfated Tin (IV) Oxide Solid Superacid Catalyst to Partial Coupling Reaction of α-Pinene to Produce Less Viscous High-Density Fuel. Energies 2019, 12, 1905. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Wang, Z.; Yue, X.; Yu, F.; Xie, C.; Yu, S. Biomass high energy density fuel transformed from α-pinene catalyzed by Brönsted-Lewis acidic heteropoly inorganic-organic salt. Renew. Energy 2018, 123, 218–226. [Google Scholar] [CrossRef]
- Meylemans, H.A.; Baldwin, L.C.; Harvey, B.G. Low-temperature properties of renewable high-density fuel blends. Energy Fuels 2013, 27, 883–888. [Google Scholar] [CrossRef]
- Deng, W.; Kennedy, J.R.; Tsilomelekis, G.; Zheng, W.; Nikolakis, V. Cellulose hydrolysis in acidified LiBr molten salt hydrate media. Ind. Eng. Chem. Res. 2015, 54, 5226–5236. [Google Scholar] [CrossRef]
- Yang, X.; Li, N.; Lin, X.; Pan, X.; Zhou, Y. Selective cleavage of the aryl ether bonds in lignin for depolymerization by acidic lithium bromide molten salt hydrate under mild conditions. J. Agric. Food Chem. 2016, 64, 8379–8387. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.G.; Zhang, S.; Pan, X. Effective conversion of biomass into bromomethylfurfural, furfural, and depolymerized lignin in lithium bromide molten salt hydrate of a biphasic system. RSC Adv. 2017, 7, 300–308. [Google Scholar] [CrossRef] [Green Version]
- de Almeida, R.M.; Li, J.; Nederlof, C.; O’Connor, P.; Makkee, M.; Moulijn, J.A. Cellulose conversion to isosorbide in molten salt hydrate media. ChemSusChem 2010, 3, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Pedersen, C.M.; Wang, Y.; Hou, X. NMR insights on the properties of ZnCl2 molten salt hydrate medium through its interaction with SnCl4 and fructose. ACS Sustain. Chem. Eng. 2014, 2, 2576–2581. [Google Scholar] [CrossRef]
- Wang, Y.; Pedersen, C.M.; Qiao, Y.; Deng, T.; Shi, J.; Hou, X. In Situ NMR spectroscopy: Inulin biomass conversion in ZnCl2 molten salt hydrate medium—SnCl4 addition controls product distribution. Carbohydr. Polym. 2015, 115, 439–443. [Google Scholar] [CrossRef]
- Hu, S.; Zhang, Z.; Song, J.; Zhou, Y.; Han, B. Efficient conversion of glucose into 5-hydroxymethylfurfural catalyzed by a common Lewis acid SnCl4 in an ionic liquid. Green Chem. 2009, 11, 1746–1749. [Google Scholar] [CrossRef]
- Omari, K.W.; Besaw, J.E.; Kerton, F.M. Hydrolysis of chitosan to yield levulinic acid and 5-hydroxymethylfurfural in water under microwave irradiation. Green Chem. 2012, 14, 1480–1487. [Google Scholar] [CrossRef] [Green Version]
- Kumazawa, K.; Ishihara, K.; Yamamoto, H. Tin (IV) chloride-chiral pyrogallol derivatives as new Lewis acid-assisted chiral Brønsted acids for enantioselective polyene cyclization. Org. Lett. 2004, 6, 2551–2554. [Google Scholar] [CrossRef]
- Nakamura, S.; Ishihara, K.; Yamamoto, H. Enantioselective biomimetic cyclization of isoprenoids using lewis acid-assisted chiral brønsted acids: Abnormal claisen rearrangements and successive cyclizations. J. Am. Chem. Soc. 2000, 122, 8131–8140. [Google Scholar] [CrossRef]
- Ishihara, K.; Kaneeda, M.; Yamamoto, H. Lewis Acid assisted chiral Bronsted acid for enantioselective protonation of silyl enol ethers and ketene bis (trialkylsilyl) acetals. J. Am. Chem. Soc. 1994, 116, 11179–11180. [Google Scholar] [CrossRef]
- Ishihara, K.; Nakashima, D.; Hiraiwa, Y.; Yamamoto, H. The crystallographic structure of a Lewis acid-assisted chiral Brønsted acid as an enantioselective protonation reagent for silyl enol ethers. J. Am. Chem. Soc. 2003, 125, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Zou, J.-J.; Feng, R.; Zhang, X.; Wang, L. HPW/MCM-41 catalyzed isomerization and dimerization of pure pinene and crude turpentine. Catal. Today 2014, 234, 271–277. [Google Scholar] [CrossRef]
- Lana, E.J.L.; da Silva Rocha, K.A.; Kozhevnikov, I.V.; Gusevskaya, E.V. One-pot synthesis of diisobornyl ether from camphene using heteropoly acid catalysts. J. Mol. Catal. A Chem. 2006, 243, 258–263. [Google Scholar] [CrossRef]
- Sidorenko, A.Y.; Aho, A.; Ganbaatar, J.; Batsuren, D.; Utenkova, D.; Sen’kov, G.; Wärnå, J.; Murzin, D.Y.; Agabekov, V. Catalytic isomerization of α-pinene and 3-carene in the presence of modified layered aluminosilicates. Mol. Catal. 2017, 443, 193–202. [Google Scholar] [CrossRef]
- Shihada, A.F.; Abushamleh, A.S.; Weller, F. Crystal Structures and Raman Spectra of cis-[SnCl4 (H2O)2]·2H2O, cis-[SnCl4 (H2O)2]·3H2O,[Sn2Cl6 (OH)2 (H2O)2]·4H2O, and [HL][SnCl5(H2O)]·2.5H2O (L = 3-acetyl-5-benzyl-1-phenyl-4, 5-dihydro-1, 2, 4-triazine-6-one oxime, C18H18N4O2). Z. Anorg. Allg. Chem. 2004, 630, 841–847. [Google Scholar] [CrossRef]
- Zhao, P.Q.; Xu, L.W.; Xia, C.G. Transition metal-based Lewis acid catalyzed ring opening of epoxides using amines under solvent-free conditions. Synlett 2004, 2004, 0846–0850. [Google Scholar] [CrossRef]
- Wang, M.; Song, Z.; Liang, Y. SnCl4·5H2O-Catalyzed Synthesis of Β-Amino Carbonyl Compounds Via a Direct Mannich-Type Reaction. Prep. Biochem. Biotechnol. 2010, 41, 1–6. [Google Scholar] [CrossRef]
- Chen, D.; Wang, D.; Wu, W.; Xiao, L. SnCl4·5H2O: A Highly Efficient Catalyst for Hydration of Alkyne. Appl. Sci. 2015, 5, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Jasti, R.; Anderson, C.D.; Rychnovsky, S.D. Utilization of an oxonia-cope rearrangement as a mechanistic probe for prins cyclizations. J. Am. Chem. Soc. 2005, 127, 9939–9945. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.-M.; Choi, J.-H.; Kim, J.-H.; Koo, B.; Choi, I.-G. Catalytic Conversion of α-Pinene to High-Density Fuel Candidates Over Stannic Chloride Molten Salt Hydrates. Appl. Sci. 2020, 10, 7517. https://doi.org/10.3390/app10217517
Cho S-M, Choi J-H, Kim J-H, Koo B, Choi I-G. Catalytic Conversion of α-Pinene to High-Density Fuel Candidates Over Stannic Chloride Molten Salt Hydrates. Applied Sciences. 2020; 10(21):7517. https://doi.org/10.3390/app10217517
Chicago/Turabian StyleCho, Seong-Min, June-Ho Choi, Jong-Hwa Kim, Bonwook Koo, and In-Gyu Choi. 2020. "Catalytic Conversion of α-Pinene to High-Density Fuel Candidates Over Stannic Chloride Molten Salt Hydrates" Applied Sciences 10, no. 21: 7517. https://doi.org/10.3390/app10217517