Ab Initio Study of the Effect of Mono-Vacancies on the Metastability of Ga-Stabilized δ-Pu


Abstract
:1. Introduction
2. Materials and Methods
3. Results
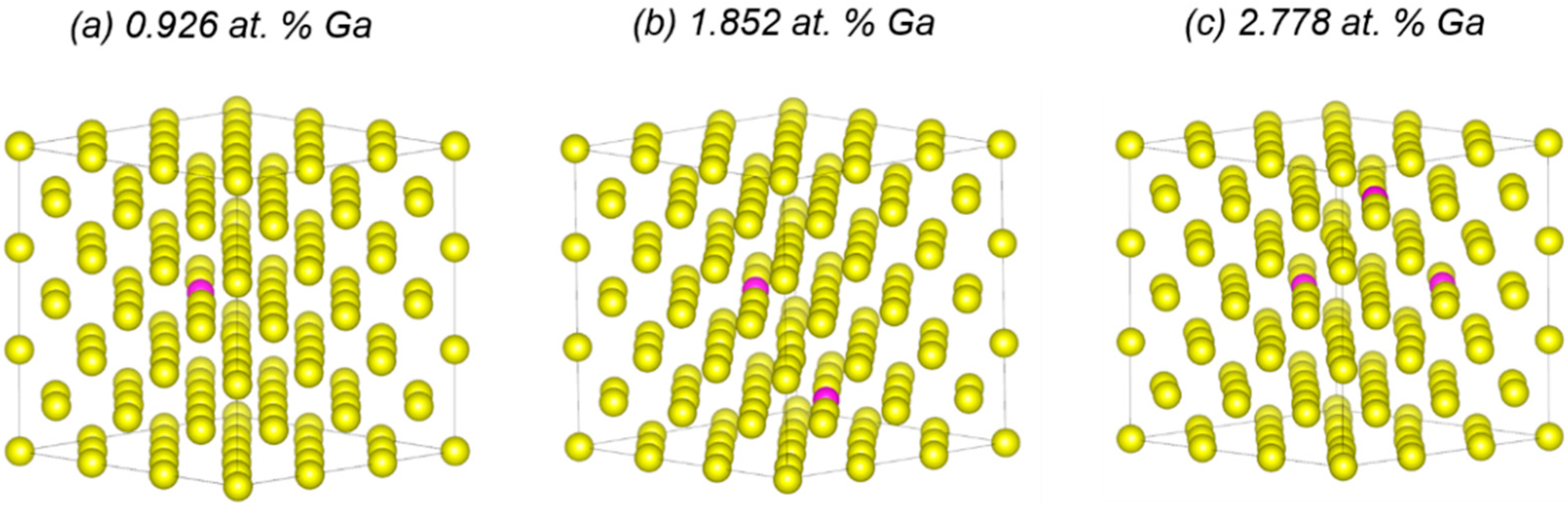
3.1. Structural Effects
3.2. Electronic Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hecker, S.S. Plutonium and its alloys. Los Alamos Sci. 2000, 26, 290. [Google Scholar]
- Hecker, S. Plutonium—An element at odds with itself. Los Alamos Sci. 2000, 26, 16–23. [Google Scholar]
- Ellinger, F.H.; Land, C.C.; Struebing, V.O. The plutonium-gallium system. J. Nucl. Mater. 1964, 12, 226–236. [Google Scholar] [CrossRef]
- Lawson, A.C.; Roberts, J.A.; Martinez, B.; Richardson, J.W. Invar effect in Pu-Ga alloys. Philos. Mag. B 2002, 82, 1837–1845. [Google Scholar] [CrossRef]
- Adler, P.; Olson, G.B. Thermodynamics and kinetics of δ→ α martensitic transformation in Pu alloys. Metall. Trans. A 1988, 19, 2705–2711. [Google Scholar] [CrossRef]
- Hirth, J.P.; Mitchell, J.N.; Schwartz, D.S.; Mitchell, T.E. On the fcc→monoclinic martensite transformation in a Pu–1.7 at.% Ga alloy. Acta Mater. 2006, 54, 1917–1925. [Google Scholar] [CrossRef]
- Clark, D.L.; Hecker, S.S.; Jarvinen, G.D.; Neu, M.P. Plutonium Chapter 7: The Chemistry of the Actinide and Transactinide and Elements; Morss, L.R., Edelstein, N.M., Fuger, J., Eds.; Springer: Dordrecht, The Netherlands, 2006; p. 813. [Google Scholar]
- Hecker, S.; Harbur, D.; Zocco, T. Phase stability and phase transformations in Pu–Ga alloys. Prog. Mater. Sci. 2004, 49, 429–485. [Google Scholar] [CrossRef]
- Hecker, S.S. Plutonium—An element never at equilibrium. Metall. Mater. Trans. A 2008, 39, 1585–1592. [Google Scholar] [CrossRef]
- Wolfer, W.G. Radiation effects in plutonium. Los Alamos Sci. 2000, 26, 274. [Google Scholar]
- Mitchell, J.N.; Freibert, F.J.; Schwartz, D.S.; Bange, M.E. Unconventional δ-phase stabilization in Pu–Ga alloys. J. Nucl. Mater. 2009, 385, 95–98. [Google Scholar] [CrossRef]
- Wolfer, W.G.; Kubota, A.; Söderlind, P.; Landa, A.I.; Oudot, B.; Sadigh, B.; Sturgeon, J.B.; Surh, M.P. Density changes in Ga-stabilized δ-Pu, and what they mean. J. Alloys Compd. 2007, 444–445, 72–79. [Google Scholar] [CrossRef]
- Chebotarev, N.T.; Utkina, O.N. Relationship Between Structure and Some Properties of δ-Pu and γ-U Alloys Plutonium and Other Actinides. In Proceedings of the Fifth International Conference on Plutonium and Other Actinides, Baden Baden, Germany, 10–13 September 1975; Blank, H., Linder, R., Eds.; North-Holland Publishing Company: Amsterdam, The Netherlands, 1975; p. 559. [Google Scholar]
- Ravat, B.; Oudot, B.; Baclet, N. Study by XRD of the lattice swelling of PuGa alloys induced by self-irradiation. J. Nucl. Mater. 2007, 366, 288–296. [Google Scholar] [CrossRef]
- Olive, D.T.; Wang, D.L.; Booth, C.H.; Bauer, E.D.; Pugmire, A.L.; Freibert, F.J.; McCall, S.K.; Wall, M.A.; Allen, P.G. Isochronal annealing effects on local structure, crystalline fraction, and undamaged region size of radiation damage in Ga-stabilized δ-Pu. J. Appl. Phys. 2016, 120, 035103. [Google Scholar] [CrossRef] [Green Version]
- Hecker, S.S.; Timofeeva, L.F. A tale of two diagrams. Los Alamos Sci. 2000, 26, 244–251. [Google Scholar]
- Massalski, T.B.; Schwartz, A.J. Connections between the Pu–Ga phase diagram in the Pu-rich region and the low temperature phase transformations. J. Alloys Compd. 2007, 444–445, 98–103. [Google Scholar] [CrossRef]
- Turchi, P.; Kaufman, L.; Zhou, S.; Liu, Z.-K. Thermostatics and kinetics of transformations in Pu-based alloys. J. Alloys Compd. 2007, 444, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, S.C.; Schwartz, D.S.; Taylor, C.D.; Ray, A.K. Ab initio study of gallium stabilized δ-plutonium alloys and hydrogen-vacancy complexes. J. Phys. Condens. Matter 2014, 26, 235502. [Google Scholar] [CrossRef]
- Sadigh, B.; Wolfer, W.G. Gallium stabilization of δ−Pu: Density-functional calculations. Phys. Rev. B 2005, 72, 205122. [Google Scholar] [CrossRef]
- Cox, L.E.; Martinez, R.; Nickel, J.H.; Conradson, S.D.; Allen, P.G. Short-range atomic structure of 1 wt. % Ga δ-stabilized plutonium by x-ray-absorption fine-structure spectroscopy. Phys. Rev. B 1995, 51, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Faure, P.; Deslandes, B.; Bazin, D.; Tailland, C.; Doukhan, R.; Fournier, J.M.; Falanga, A. Lattice collapse around gallium in PuGa alloys as revealed by X-ray absorption spectroscopy. J. Alloys Compd. 1996, 244, 131–139. [Google Scholar] [CrossRef]
- Söderlind, P.; Sadigh, B. Density-Functional Calculations of α, β, γ, δ, δ′, and ϵ Plutonium. Phys. Rev. Lett. 2004, 92, 185702. [Google Scholar] [CrossRef] [PubMed]
- Söderlind, P.; Landa, A.; Sadigh, B. Density-functional theory for plutonium. Adv. Phys. 2019, 68, 1–47. [Google Scholar] [CrossRef]
- Lashley, J.C.; Lawson, A.; McQueeney, R.J.; Lander, G.H. Absence of magnetic moments in plutonium. Phys. Rev. B 2005, 72, 054416. [Google Scholar] [CrossRef] [Green Version]
- Savrasov, S.Y.; Kotliar, G. Ground State Theory of δ−Pu. Phys. Rev. Lett. 2000, 84, 3670. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-X.; Albers, R.; Haule, K.; Kotliar, G.; Wills, J. Site-selective electronic correlation in α-plutonium metal. Nat. Commun. 2013, 4, 2644. [Google Scholar] [PubMed] [Green Version]
- Amadon, B. First-principles DFT+DMFT calculations of structural properties of actinides: Role of Hund’s exchange, spin-orbit coupling, and crystal structure. Phys. Rev. B 2016, 94, 115148. [Google Scholar]
- Bouchet, J.; Siberchicot, B.; Jollet, F.; Pasturel, A. Equilibrium properties of delta-Pu: LDA+Ucalculations (LDAequiv local density approximation). J. Phys. Condens. Matter 2000, 12, 1723–1733. [Google Scholar]
- Chen, S.P. Correlation-induced anomalies and extreme sensitivity in fcc Pu. Philos. Mag. 2009, 89, 1813–1822. [Google Scholar]
- Moore, K.T.; Söderlind, P.; Schwartz, A.J.; Laughlin, D.E. Symmetry and Stability of δ Plutonium: The Influence of Electronic Structure. Phys. Rev. Lett. 2006, 96, 206402. [Google Scholar]
- Hernandez, S.C.; Freibert, F.J.; Hoagland, R.G.; Uberuaga, B.P.; Wills, J.M. Spin density stabilization of local distortions induced by a monovacancy in δ-Pu. Phys. Rev. Mater. 2018, 2, 085005. [Google Scholar]
- Landa, A.; Söderlind, P. First-principles calculations of stability of δ-Pu–Am alloys. J. Alloys Compd. 2004, 376, 62–67. [Google Scholar]
- Landa, A.; Söderlind, P.; Ruban, A. Monte Carlo simulations of the stability of δ-Pu. J. Phys. Condens. Matter 2003, 15, L371–L376. [Google Scholar]
- Landa, A.; Söderlind, P. Stability of δ-Pu alloys from first-principles theory. J. Alloys Compd. 2003, 354, 99–103. [Google Scholar]
- Robert, G.; Pasturel, A.; Siberchicot, B. Vacancy formation in δ-plutonium: A density-functional study in the generalized gradient approximation. EPL (Europhys. Lett.) 2005, 71, 412. [Google Scholar]
- Söderlind, P. Quantifying the importance of orbital over spin correlations in δ-Pu within density-functional theory. Phys. Rev. B 2008, 77, 085101. [Google Scholar] [CrossRef]
- Islam, M.F.; Ray, A.K. On the Interplay Between Spin Polarization, Orbital Polarization and Spin-Orbit Coupling in Actinides from Pa to Cm. J. Comput. Theor. Nanosci. 2009, 6, 1458–1467. [Google Scholar] [CrossRef]
- McCall, S.K.; Fluss, M.J.; Chung, B.W.; McElfresh, M.W.; Jackson, D.D.; Chapline, G.F. Emergent magnetic moments produced by self-damage in plutonium. Proc. Natl. Acad. Sci. USA 2006, 103, 17179–17183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Rose, R.L.; Robbins, J.L.; Massalski, T.B. Heat content and heat capacity of a Pu/1 wt % Ga delta-stabilized alloy at elevated temperatures. J. Nucl. Mater. 1970, 36, 99–107. [Google Scholar] [CrossRef]
- Robert, G.; Pasturel, A.; Siberchicot, B. Thermodynamic, alloying and defect properties of plutonium: Density-functional calculations. J. Alloys Compd. 2007, 444–445, 191–196. [Google Scholar] [CrossRef]
- Richard, N.; Faure, P.; Rofidal, P.; Truffier, J.L.; Bazin, D. PuGa alloys: An X-ray absorption study. J. Alloys Compd. 1998, 271–273, 879–881. [Google Scholar] [CrossRef]
- van Ek, J.; Sterne, P.A.; Gonis, A. Phase stability of plutonium. Phys. Rev. B 1993, 48, 16280–16289. [Google Scholar] [CrossRef]
- Jullien, R.; d’Agliano, E.G.; Coqblin, B. Hybridized Nondegenerate 6d and 5f Virtual-Bound-States Model for Actinides Metals. Phys. Rev. B 1972, 6, 2139–2155. [Google Scholar]
- Jullien, R.; d’Agliano, E.G.; Coqblin, B. A simple model for the study of magnetism in transition-based alloys with actinide impurities. J. Low Temp. Phys. 1973, 10, 685–705. [Google Scholar] [CrossRef]
- Hernandez, S.C.; Richmond, S.; Venhaus, T.J.; Huda, M.N. The Influence of Ga on H Reactivity with Ga-Stabilized δ-Pu Alloys. J. Phys. Chem. C 2017, 121, 19162–19172. [Google Scholar] [CrossRef]
- Hernandez, S.C.; Venhaus, T.J.; Huda, M.N. Atomic oxygen adsorption on 3.125 at.% Ga stabilized δ-Pu (111) surface. J. Alloys Compd. 2015, 643, 253–262. [Google Scholar]
- Conradson, S.D.; Bock, N.; Castro, J.M.; Conradson, D.R.; Cox, L.E.; Dmowski, W.; Dooley, D.E.; Egami, T.; Espinosa-Faller, F.J.; Freibert, F.J.; et al. Nanoscale heterogeneity, premartensitic nucleation, and a new plutonium structure in metastable δ fcc Pu-Ga alloys. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 89, 224102. [Google Scholar]
- Freibert, F.J.; Hernandez, S.C.; Migliori, A. Chapter 9: Thermophysical Properties of Plutonium Metal and Its Alloys. In Plutonium Handbook, 2nd ed.; Clark, D.L., Geeson, D.A., Hanrahan, J.R.J., Eds.; American Nuclear Society Incorporated: La Grange Park, IL, USA, 2018. [Google Scholar]
- Hernandez, S.C.; Freibert, F.J.; Uberuaga, B.P.; Wills, J.M. Role of electronic and magnetic interactions in defect formation and anomalous diffusion in δ-Pu. J. Nucl. Mater. 2020, 532, 152027. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % at. Ga | a0 (Å) | ||
|---|---|---|---|
| 0 | 4.524 | - | - |
| 0.926 | 4.520 | −0.010 | −0.944 |
| 1.852 | 4.515 | −0.019 | −1.807 |
| 2.778 | 4.501 | −0.030 | −2.867 |
| at. % Ga | Distance from Ga | (eV) |
|---|---|---|
| 0.926 | 1nn | 1.39 |
| 2nn | 1.48 | |
| 6nn | 1.25 | |
| 1.852 | 1nn | 1.49 |
| 1nn between 2 Ga | 1.58 | |
| 2nn | 1.50 | |
| 2nn between 2 Ga | 1.61 | |
| 7nn | 1.22 | |
| 2.778 | 1nn | 1.39 |
| 1nn between 2 Ga | 1.57 | |
| 2nn | 1.44 | |
| 2nn between 2 Ga | 1.53 | |
| 2nn between 3 Ga | 1.75 | |
| 4nn | 1.10 | |
| 7nn | 1.17 |
| Pu-Ga Distance (Å) | Pu-vacancy Distance (Å) | ||||||
|---|---|---|---|---|---|---|---|
| at. % Ga | Defect Distance to Ga | Pu-1 | Pu-2 | Pu-3 | Pu-1 | Pu-2 | Pu-3 |
| 0.926 | No Defect | 3.07 | 3.14 | 10.58 | - | - | - |
| 1.852 | No Defect | 3.08 | 3.18, 3.18 | 8.47 | - | - | - |
| 2.778 | No Defect | 3.06 | 3.16, 3.15 | 8.42 | - | - | - |
| 0.926 | 1nn | 3.07 | 3.88 | 10.60 | 2.57 | 3.10 | 10.59 |
| 1.852 | 1nn | 3.06 | 3.10, 3.17 | 8.48 | 2.53 | 3.16 | 10.59 |
| 2.778 | 1nn | 3.10 | 5.40 | 8.39 | 2.97 | 3.00 | 10.57 |
| 2.788 | 1nn bw 2Ga | 3.03 | 3.12, 3.19 | 8.39 | 3.03 | 2.99 | 10.57 |
| 2.788 | 2nn bw 3Ga | 3.13 | 5.49 | 8.39 | 3.12 | 3.19 | 10.54 |
| 2.788 | 4nn | 3.02 | 5.53 | 5.45 | 3.79 | 1.94 | 5.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez, S.C.; Freibert, F.J. Ab Initio Study of the Effect of Mono-Vacancies on the Metastability of Ga-Stabilized δ-Pu. Appl. Sci. 2020, 10, 7628. https://doi.org/10.3390/app10217628
Hernandez SC, Freibert FJ. Ab Initio Study of the Effect of Mono-Vacancies on the Metastability of Ga-Stabilized δ-Pu. Applied Sciences. 2020; 10(21):7628. https://doi.org/10.3390/app10217628
Chicago/Turabian StyleHernandez, Sarah C., and Franz J. Freibert. 2020. "Ab Initio Study of the Effect of Mono-Vacancies on the Metastability of Ga-Stabilized δ-Pu" Applied Sciences 10, no. 21: 7628. https://doi.org/10.3390/app10217628