Magnolol, A Novel Antagonist of Thrombin and PAR-1, Inhibits Thrombin-Induced Connective Tissue Growth Factor (CTGF) Expression in Vascular Smooth Muscle Cells and Ameliorate Pathogenesis of Restenosis in Rats

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Animals
2.3. Cell Culture
2.4. Cell Viability and Cell Toxicity Assay
2.5. Immunoblotting
2.6. Immunofluorescent Staining
2.7. Thrombin Activity Assay
2.8. Protein-Ligand Docking
2.9. Transfection and Luciferase Reporter Assays
2.10. Wound Healing Assay
2.11. Animal Model of Balloon Angioplasty
2.12. Statistical Analysis
3. Results
3.1. The Treatment of Magnolol Will Reduce the Thrombin-Mediated CTGF Expression in VSMCs
3.2. Magnolol Hindered the VSMCs from Thrombin-Mediated CTGF Expression via Thrombin Activity Interfering, PAR-1 Cleavage Inhibiting and PAR-1 Active Site Blocking
3.3. Magnolol Performed Different Effect on Thrombin Induced JNK-1 and JNK-2 in Vascular Smooth Muscle Cells
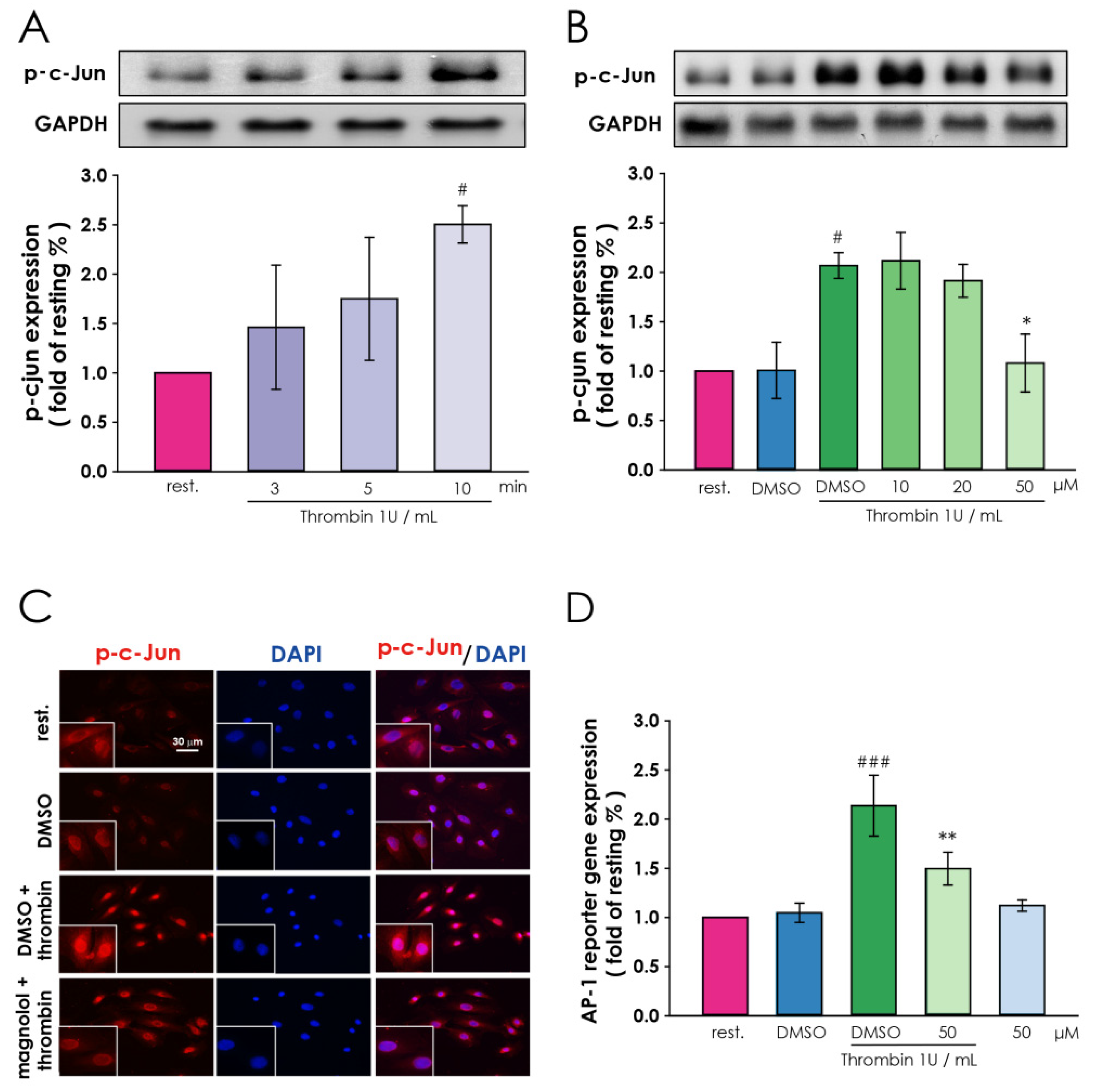
3.4. Magnolol Down-Regulate Thrombin-Induced Transcription Factor Activation in Vascular Smooth Muscle Cells
3.5. Treatment of Magnolol Moderates the Migration of VSMCs and Mitigates Restenosis
4. Discussions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stone, G.W.; Maehara, A.; Lansky, A.J.; de Bruyne, B.; Cristea, E.; Mintz, G.S.; Mehran, R.; McPherson, J.; Farhat, N.; Marso, S.P.; et al. A prospective natural-history study of coronary atherosclerosis. N. Engl. J. Med. 2011, 364, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Curcio, A.; Torella, D.; Indolfi, C. Mechanisms of Smooth Muscle Cell Proliferation and Endothelial Regeneration After Vascular Injury and Stenting: Approach to Therapy &ndash. Circ. J. 2011, 75, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; Hadri, L.; Lopez, J.J.; Hajjar, R.J.; Bobe, R. Benefit of SERCA2a gene transfer to vascular endothelial and smooth muscle cells: A new aspect in therapy of cardiovascular diseases. Curr. Vasc. Pharmacol. 2013, 11, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular Smooth Muscle Cell Proliferation in Restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef]
- Farb, A.; Kolodgie, F.D.; Hwang, J.-Y.; Burke, A.P.; Tefera, K.; Weber, D.K.; Wight, T.N.; Virmani, R. Extracellular matrix changes in stented human coronary arteries. Circulation 2004, 110, 940–947. [Google Scholar] [CrossRef]
- Gupta, S.; Clarkson, M.R.; Duggan, J.; Brady, H.R. Connective tissue growth factor: Potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000, 58, 1389–1399. [Google Scholar] [CrossRef]
- Oemar, B.S.; Werner, A.; Garnier, J.-M.; Do, D.-D.; Godoy, N.; Nauck, M.; Marz, W.; Rupp, J.; Pech, M.; Luscher, T.F. Human Connective Tissue Growth Factor Is Expressed in Advanced Atherosclerotic Lesions. Circulation 1997, 95, 831–839. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Wu, S.-B.; Kau, H.-C.; Wei, Y.-H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef]
- Yao, E.-H.; Fukuda, N.; Ueno, T.; Matsuda, H.; Nagase, H.; Matsumoto, Y.; Sugiyama, H.; Matsumoto, K. A pyrrole–imidazole polyamide targeting transforming growth factor-β1 inhibits restenosis and preserves endothelialization in the injured artery. Cardiovasc. Res. 2008, 81, 797–804. [Google Scholar] [CrossRef]
- Gallo, R.; Padurean, A.; Toschi, V.; Bichler, J.; Fallon, J.T.; Chesebro, J.H.; Fuster, V.; Badimon, J.J. Prolonged thrombin inhibition reduces restenosis after balloon angioplasty in porcine coronary arteries. Circulation 1998, 97, 581–588. [Google Scholar] [CrossRef][Green Version]
- Fager, G. Thrombin and proliferation of vascular smooth muscle cells. Circ. Res. 1995, 77, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Reisine, T. GPCRs Revisited: New Insights Lead to Novel Drugs. Pharmaceuticals 2011, 4, 244–272. [Google Scholar] [CrossRef]
- Soh, U.J.; Dores, M.R.; Chen, B.; Trejo, J. Signal transduction by protease-activated receptors. Br. J. Pharmacol. 2010, 160, 191–203. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; De Ceunynck, K. Targeting PAR1: Now What? Trends Pharmacol. Sci. 2017, 38, 701–716. [Google Scholar] [CrossRef]
- Macfarlane, S.R.; Seatter, M.J.; Kanke, T.; Hunter, G.D.; Plevin, R. Proteinase-Activated Receptors. Pharmacol. Rev. 2001, 53, 245–282. [Google Scholar]
- Hirano, K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 27–36. [Google Scholar] [CrossRef]
- Ranaware, A.M.; Banik, K.; Deshpande, V.; Padmavathi, G.; Roy, N.K.; Sethi, G.; Fan, L.; Kumar, A.P.; Kunnumakkara, A.B. Magnolol: A Neolignan from the Magnolia Family for the Prevention and Treatment of Cancer. Int. J. Mol. Sci. 2018, 19, 2362. [Google Scholar] [CrossRef]
- Marunnan, S.M.; Pulikkal, B.P.; Jabamalairaj, A.; Bandaru, S.; Yadav, M.; Nayarisseri, A.; Doss, V.A. Development of MLR and SVM Aided QSAR Models to Identify Common SAR of GABA Uptake Herbal Inhibitors used in the Treatment of Schizophrenia. Curr. Neuropharmacol. 2017, 15, 1085–1092. [Google Scholar] [CrossRef]
- Dreier, D.; Latkolik, S.; Rycek, L.; Schnurch, M.; Dymakova, A.; Atanasov, A.G.; Ladurner, A.; Heiss, E.H.; Stuppner, H.; Schuster, D.; et al. Linked magnolol dimer as a selective PPARgamma agonist—Structure-based rational design, synthesis, and bioactivity evaluation. Sci. Rep. 2017, 7, 13002. [Google Scholar] [CrossRef]
- Chen, J.-H.; Wu, C.-C.; Hsiao, G.; Yen, M.-H. Magnolol induces apoptosis in vascular smooth muscle. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.-C.; Chen, B.-C.; Hsu, M.-J.; Tsai, C.-T.; Hong, C.-Y.; Lin, C.-H. Thrombin induced connective tissue growth factor expression in rat vascular smooth muscle cells via the PAR-1/JNK/AP-1 pathway. Acta Pharmacol. Sin. 2012, 33, 49. [Google Scholar] [CrossRef] [PubMed]
- LeadIT, B. Available online: http://www.biosolveit.de/LeadIT (accessed on 12 January 2019).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Yoshida, T.; Azuma, H.; Aihara, K.; Fujimura, M.; Akaike, M.; Mitsui, T.; Matsumoto, T. Vascular smooth muscle cell proliferation is dependent upon upregulation of mitochondrial transcription factor A (mtTFA) expression in injured rat carotid artery. Atherosclerosis 2005, 178, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chandrasekharan, U.M.; Bandyopadhyay, S.; Morris, S.M., Jr.; DiCorleto, P.E.; Kashyap, V.S. Thrombin induces endothelial arginase through AP-1 activation. Am. J. Physiol. Cell Physiol. 2010, 298, C952–C960. [Google Scholar] [CrossRef]
- Yu, C.C.; Hsu, M.J.; Kuo, M.L.; Chen, R.F.; Chen, M.C.; Bai, K.J.; Yu, M.C.; Chen, B.C.; Lin, C.H. Thrombin-induced connective tissue growth factor expression in human lung fibroblasts requires the ASK1/JNK/AP-1 pathway. J. Immunol. 2009, 182, 7916–7927. [Google Scholar] [CrossRef]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Dolan, L.; Davis, R.J.; Ronai, Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene 1996, 13, 1531–1535. [Google Scholar]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Pan, H.; Boucher, M.; Kaunelis, D. PAR-1 Antagonists: An Emerging Antiplatelet Drug Class. In CADTH Issues in Emerging Health Technologies; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2016. [Google Scholar]
- Executive, T.C.; Steering, C. The Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA*CER) trial: Study design and rationale. Am. Heart J. 2009, 158, 327–334. [Google Scholar] [CrossRef]
- Chieng-Yane, P.; Bocquet, A.; Letienne, R.; Bourbon, T.; Sablayrolles, S.; Perez, M.; Hatem, S.N.; Lompre, A.M.; Le Grand, B.; David-Dufilho, M. Protease-activated receptor-1 antagonist F 16618 reduces arterial restenosis by down-regulation of tumor necrosis factor alpha and matrix metalloproteinase 7 expression, migration, and proliferation of vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 2011, 336, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Blanton, R.M.; Keaney, J.F., Jr.; Kant, S. JNK and cardiometabolic dysfunction. BioSci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Li, W.; Shao, Q.; Dwyer, T.; Huang, H.; Yang, T.; Liu, G. c-Jun N-terminal kinase 1 (JNK1) is required for coordination of netrin signaling in axon guidance. J. Biol. Chem. 2013, 288, 1883–1895. [Google Scholar] [CrossRef]
- Sluss, H.K.; Barrett, T.; Derijard, B.; Davis, R.J. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell. Biol. 1994, 14, 8376–8384. [Google Scholar] [CrossRef]
- Bugaud, F.; Nadal-Wollbold, F.; Levy-Toledano, S.; Rosa, J.P.; Bryckaert, M. Regulation of c-jun-NH2 terminal kinase and extracellular-signal regulated kinase in human platelets. Blood 1999, 94, 3800–3805. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, L.; Hochleitner, B.W.; Xu, Q. Activation of mitogen-activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2808–2816. [Google Scholar] [CrossRef]
- Schaefer, F.M.; Peng, J.; Hu, W.; Drvarov, O.; Nevzorova, Y.A.; Zhao, G.; Masaoudi, M.A.; Davis, R.J.; Trautwein, C.; Cubero, F.J. Bone marrow-derived c-jun N-terminal kinase-1 (JNK1) mediates liver regeneration. Biochim. Biophys. Acta 2015, 1852, 137–145. [Google Scholar] [CrossRef]
- Shibata, W.; Maeda, S.; Hikiba, Y.; Yanai, A.; Sakamoto, K.; Nakagawa, H.; Ogura, K.; Karin, M.; Omata, M. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008, 68, 5031–5039. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Natriuretic peptides suppress vascular endothelial cell growth factor signaling to angiogenesis. Endocrinology 2001, 142, 1578–1586. [Google Scholar] [CrossRef][Green Version]
- Babaev, V.R.; Yeung, M.; Erbay, E.; Ding, L.; Zhang, Y.; May, J.M.; Fazio, S.; Hotamisligil, G.S.; Linton, M.F. Jnk1 Deficiency in Hematopoietic Cells Suppresses Macrophage Apoptosis and Increases Atherosclerosis in Low-Density Lipoprotein Receptor Null Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, Z.; Liu, H.; Ren, Y.; Shao, D.; Zhang, W.; Lin, J.; Wolfram, J.; Wang, F.; Nie, S. Connective tissue growth factor stimulates the proliferation, migration and differentiation of lung fibroblasts during paraquat-induced pulmonary fibrosis. Mol. Med. Rep. 2015, 12, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pongkitwitoon, S.; Lu, H.; Lee, C.; Gelberman, R.; Thomopoulos, S. CTGF induces tenogenic differentiation and proliferation of adipose-derived stromal cells. J. Orthop. Res. 2019, 37, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Cabreira, M.d.G.; Wang, Z.; Ma, W.; Benito, J.; Ruvolo, P.P.; Davis, R.E.; Konopleva, M.; Andreeff, M. Connective Tissue Growth Factor (CTGF) Is Essential for Self Renewal and Proliferation of Mesenchymal Stromal Cells (MSCs) and Affects Leukemia-Stromal Interactions. Blood 2010, 116, 3845. [Google Scholar] [CrossRef]
- Game, B.A.; He, L.; Jarido, V.; Nareika, A.; Jaffa, A.A.; Lopes-Virella, M.F.; Huang, Y. Pioglitazone inhibits connective tissue growth factor expression in advanced atherosclerotic plaques in low-density lipoprotein receptor-deficient mice. Atherosclerosis 2007, 192, 85–91. [Google Scholar] [CrossRef]
- Vainio, L.E.; Szabo, Z.; Lin, R.; Ulvila, J.; Yrjola, R.; Alakoski, T.; Piuhola, J.; Koch, W.J.; Ruskoaho, H.; Fouse, S.D.; et al. Connective Tissue Growth Factor Inhibition Enhances Cardiac Repair and Limits Fibrosis After Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 83–94. [Google Scholar] [CrossRef]
- Chen, S.C.; Chang, Y.L.; Wang, D.L.; Cheng, J.J. Herbal remedy magnolol suppresses IL-6-induced STAT3 activation and gene expression in endothelial cells. Br. J. Pharmacol. 2006, 148, 226–232. [Google Scholar] [CrossRef]
- Van den Eshof, B.L.; Hoogendijk, A.J.; Simpson, P.J.; van Alphen, F.P.J.; Zanivan, S.; Mertens, K.; Meijer, A.B.; van den Biggelaar, M. Paradigm of Biased PAR1 (Protease-Activated Receptor-1) Activation and Inhibition in Endothelial Cells Dissected by Phosphoproteomics. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1891–1902. [Google Scholar] [CrossRef]
- Lv, L.; Zhang, J.; Zhang, L.; Xue, G.; Wang, P.; Meng, Q.; Liang, W. Essential role of Pin1 via STAT3 signalling and mitochondria-dependent pathways in restenosis in type 2 diabetes. J. Cell. Mol. Med. 2013, 17, 989–1005. [Google Scholar] [CrossRef]
- Maruyama, I.; Shigeta, K.; Miyahara, H.; Nakajima, T.; Shin, H.; Ide, S.; Kitajima, I. Thrombin activates NF-kappa B through thrombin receptor and results in proliferation of vascular smooth muscle cells: Role of thrombin in atherosclerosis and restenosis. Ann. N. Y. Acad. Sci. 1997, 811, 429–436. [Google Scholar] [CrossRef]
- Wu, C.H.; Chen, C.W.; Chen, H.C.; Chang, W.C.; Shu, M.J.; Hung, J.S. Elucidating the inhibitory mechanisms of magnolol on rat smooth muscle cell proliferation. J. Pharmacol. Sci. 2005, 99, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Whellan, D.J.; Tricoci, P.; Chen, E.; Huang, Z.; Leibowitz, D.; Vranckx, P.; Marhefka, G.D.; Held, C.; Nicolau, J.C.; Storey, R.F.; et al. Vorapaxar in acute coronary syndrome patients undergoing coronary artery bypass graft surgery: Subgroup analysis from the TRACER trial (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome). J. Am. Coll. Cardiol. 2014, 63, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Ansell, J.E. Direct thrombin inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Saver, J.L.; Hong, K.S.; Wu, H.C.; Ovbiagele, B. Risk of intracranial hemorrhage with protease-activated receptor-1 antagonists. Stroke 2012, 43, 3189–3195. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z.; Huang, X.; Shi, W.; Zhang, R.; Chen, M.; Huang, H.; Wu, L. Insights on the Multifunctional Activities of Magnolol. BioMed Res. Int. 2019, 2019, 1847130. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, W.-C.; Tsai, C.-T.; Hsu, K.-C.; Cheng, Y.-C.; Lin, T.E.; Chen, Y.-L.; Hong, C.-Y.; Lu, W.-J.; Shih, C.-M.; Yen, T.-L. Magnolol, A Novel Antagonist of Thrombin and PAR-1, Inhibits Thrombin-Induced Connective Tissue Growth Factor (CTGF) Expression in Vascular Smooth Muscle Cells and Ameliorate Pathogenesis of Restenosis in Rats. Appl. Sci. 2020, 10, 8729. https://doi.org/10.3390/app10238729
Ko W-C, Tsai C-T, Hsu K-C, Cheng Y-C, Lin TE, Chen Y-L, Hong C-Y, Lu W-J, Shih C-M, Yen T-L. Magnolol, A Novel Antagonist of Thrombin and PAR-1, Inhibits Thrombin-Induced Connective Tissue Growth Factor (CTGF) Expression in Vascular Smooth Muscle Cells and Ameliorate Pathogenesis of Restenosis in Rats. Applied Sciences. 2020; 10(23):8729. https://doi.org/10.3390/app10238729
Chicago/Turabian StyleKo, Wen-Chin, Chia-Ti Tsai, Kai-Cheng Hsu, Yu-Che Cheng, Tony Eight Lin, Yi-Ling Chen, Chuang-Ye Hong, Wan-Jung Lu, Chun-Ming Shih, and Ting-Lin Yen. 2020. "Magnolol, A Novel Antagonist of Thrombin and PAR-1, Inhibits Thrombin-Induced Connective Tissue Growth Factor (CTGF) Expression in Vascular Smooth Muscle Cells and Ameliorate Pathogenesis of Restenosis in Rats" Applied Sciences 10, no. 23: 8729. https://doi.org/10.3390/app10238729
APA StyleKo, W.-C., Tsai, C.-T., Hsu, K.-C., Cheng, Y.-C., Lin, T. E., Chen, Y.-L., Hong, C.-Y., Lu, W.-J., Shih, C.-M., & Yen, T.-L. (2020). Magnolol, A Novel Antagonist of Thrombin and PAR-1, Inhibits Thrombin-Induced Connective Tissue Growth Factor (CTGF) Expression in Vascular Smooth Muscle Cells and Ameliorate Pathogenesis of Restenosis in Rats. Applied Sciences, 10(23), 8729. https://doi.org/10.3390/app10238729