Mucin-Grafted Polyethylene Glycol Microparticles Enable Oral Insulin Delivery for Improving Diabetic Treatment

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Empty MPs
2.3. Coating of the MPs
2.4. Loading of Insulin in MPs
2.5. Characterization of Insulin-Loaded MPs
2.5.1. Thermal Analysis
2.5.2. Recovery Values of Insulin-Loaded MPs
2.5.3. Encapsulation Efficiency (EE) and Drug Loading (DL) of Insulin-Loaded MPs
2.5.4. Morphology and Particle Size Evaluations
2.6. In Vitro Release Study
2.7. Antidiabetic Study
2.7.1. Induction of Diabetes
2.7.2. Antidiabetic Efficacy of Insulin-Loaded MPs
- -
- Group 1: received orally 50 IU /kg of batch A5;
- -
- Group 2: received orally 50 IU/kg of pure insulin solution;
- -
- Group 3: received subcutaneously 5 IU/kg of insulin;
- -
- Group 4: received orally 5 mL/kg of water;
- -
- Group 5: received 50 mg/kg of batch A0 (blank).
2.8. Statistical Analysis
3. Results and Discussion
3.1. Thermal Analysis
3.2. Morphology and Particle Size of Insulin-Loaded MPs
3.3. Encapsulation Efficiency, Drug Loading, and Recovery Yield of MPs
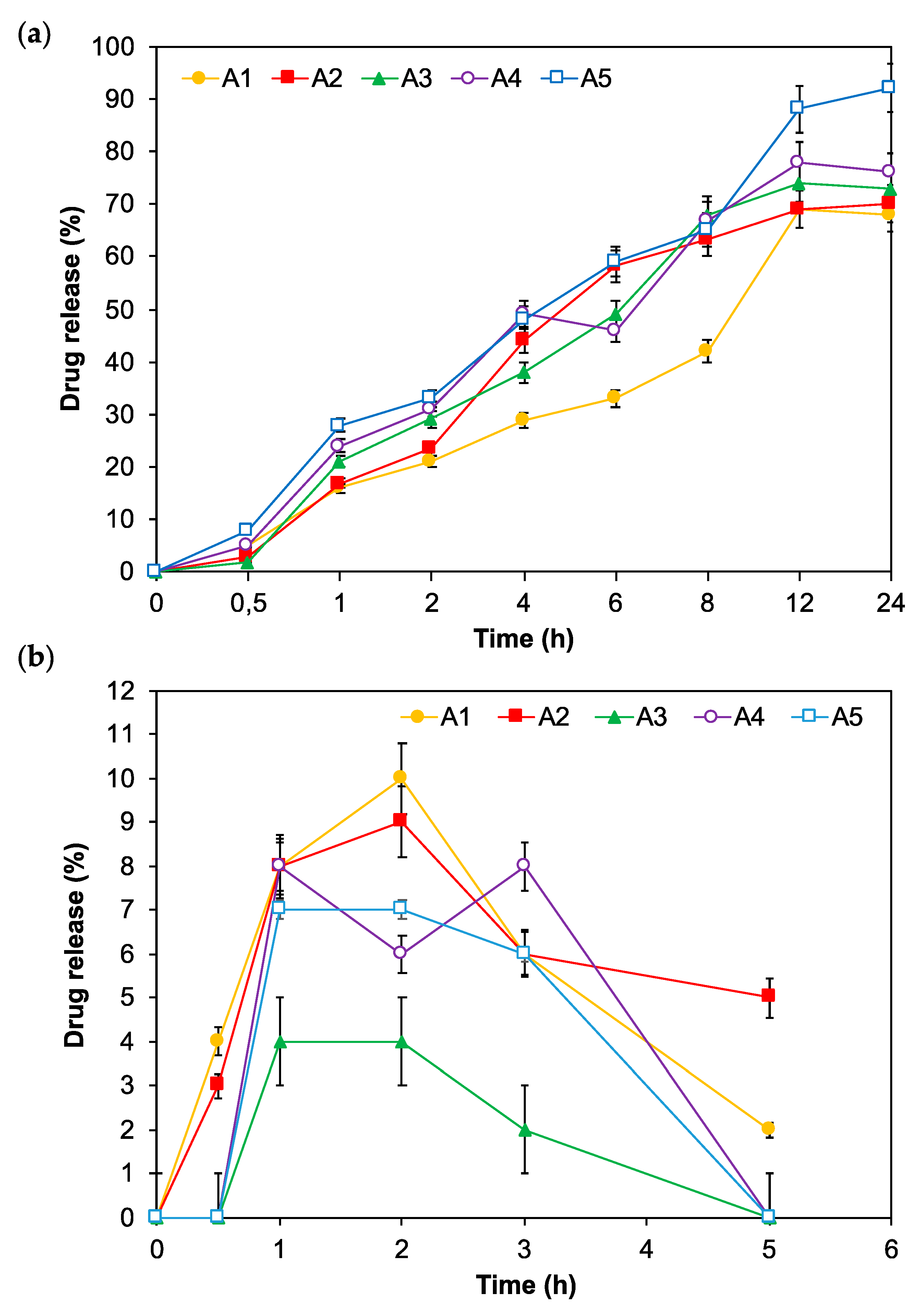
3.4. In Vitro Release of Insulin-Loaded MPs
3.5. In Vivo Antidiabetic and Toxicological Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sah, S.P.; Singh, B.; Choudhary, S.; Kumar, A. Animal models of insulin resistance: A review. Pharmacol. Rep. 2016, 68, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Fonte, P.; Araújo, F.; Silva, C.; Pereira, C.; Reis, S.; Santos, H.A. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnol. Adv. 2014, 33, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Rolla, A.R.; Rakel, R.E. Practical approaches to insulin therapy for type 2 diabetes mellitus with premixed insulin analogues. Clin. Ther. 2005, 27, 1113–1125. [Google Scholar] [CrossRef]
- Sastry, S.V.; Nyshadham, J.V.; Fix, J.A. Recent technological advances in oral drug delivery—A review. Pharmaceut. Sci. Tech. Today 2000, 3, 138–143. [Google Scholar] [CrossRef]
- Yaturu, S. Insulin therapies: Current and future trends at dawn. World J. Diabetes 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Builders, P.F.; Kunle, O.O.; Adikwu, M.U. Preparation and characterization of mucinated agarose: A mucin-agarose physical crosslink. Int. J. Pharm. 2008, 356, 174–180. [Google Scholar] [CrossRef]
- Elsayed, A.; Remawi, M.A.; Qinna, N.; Farouk, A.; Badwan, A. Formulation and characterization of an oily-based system for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2009, 73, 269–279. [Google Scholar] [CrossRef]
- Huang, Y.J.; Wang, C.H. Pulmonary delivery of insulin by liposomal carriers. J. Control Release 2006, 113, 9–14. [Google Scholar] [CrossRef]
- Pothal, R.K.; Sahoo, S.K.; Chatterjee, S.; Sahoo, D.; Barik, B.B. Preparation and evaluation of mucoadhesive microcapsules of theophylline. Ind. Pharm. 2004, 3, 74–79. [Google Scholar]
- Momoh, M.A.; Adedokun, M.O.; Adikwu, M.U.; Kenechukwu, F.C.; Ibezim, E.C.; Ugwoke, E.E. Design, characterization and evaluation of PEGylated-mucin for oral delivery of metformin hydrochloride. Afr. J. Pharm. Pharmacol. 2013, 7, 347–355. [Google Scholar]
- Mortazavi, S.A.; Carpenter, B.G.; Smart, J.D. Comparative study on the role played by mucus glycoprotein in the rheological behaviors of the mucoadhesive/mucosal interaction. Int. J. Pharm. 1992, 94, 195–201. [Google Scholar] [CrossRef]
- Adikwu, M.U.; Aneke, K.O.; Builders, P.F. Biophysical properties of mucin and its use as a mucoadhesive agent in drug delivery: Current development and future concepts. Nigerian J. Pharm. Res. 2005, 4, 60–69. [Google Scholar]
- Builders, P.F.; Kunle, O.O.; Okpaku, L.C.; Builders, M.I.; Attama, A.A.; Adikwu, M.U. Preparation and evaluation of mucinated sodium alginate microparticles for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2008, 70, 777–783. [Google Scholar] [CrossRef]
- Momoh, M.A.; Kenechukwu, F.C.; Ernest, O.C.; Oluseun, A.; Abdulmumin, B.; Youngson, D.C.; Kenneth, O.C.; Anthony, A.A. Surface-modified mucoadhesive microparticles as a controlled release system for oral delivery of insulin. Heliyon 2019, 5, e02366. [Google Scholar]
- Li, L.; Yang, L.; Li, M.; Zhang, L. A cell-penetrating peptide mediated chitosan nanocarriers for improving intestinal insulin delivery. Carbohydr. Polym. 2017, 174, 182–189. [Google Scholar] [CrossRef]
- Momoh, M.A.; Emmanuel, O.C.; Onyeto, A.C.; Darlington, Y.; Kenechukwu, F.C.; Ofokansi, K.C.; Attama, A.A. Preparation of snail cyst and PEG-4000 composite carriers via PEGylation for oral delivery of insulin: An in vitro and in vivo evaluation. Trop. J. Pharm. Res. 2019, 18, 919–926. [Google Scholar]
- Builders, P.F.; Ibekwe, N.; Okpaku, L.C.; Attama, A.A.; Kunle, O.O. Preparation and characterization of mucinated cellulose microparticles for therapeutic and drug delivery purposes. Eur. J. Pharm. Biopharm. 2009, 72, 34–41. [Google Scholar] [CrossRef]
- Chawla, A.; Sharma, P.; Pawar, P. Eudragit S-100 coated sodium alginate microspheres of naproxen sodium: Formulation, optimization and in vitro evaluation. Acta Pharm. 2012, 62, 529–545. [Google Scholar] [CrossRef]
- Ramasamy, T.; Ruttala, H.B.; Shanmugam, S.; Umadevi, S.K. Eudragit-coated aceclofenac-loaded pectin microspheres in chronopharmacological treatment of rheumatoid arthritis. Drug Deliv. 2013, 20, 65–77. [Google Scholar] [CrossRef]
- Bin, X.; Guohua, J.; Weijiang, Y.; Depeng, L.; Yongkun, L.; Xiangdong, K.; Juming, Y. Preparation of poly(lactic-co-glycolic acid) and chitosan composite nanocarriers via electrostatic self assembly for oral delivery of insulin. Mater. Sci. Eng. C 2017, 78, 420–428. [Google Scholar]
- European Community, Council Directive on the Ethics of Experiments Involving Laboratory Animals (86/609/EEC). 2003. Available online: http://data.europa.eu/eli/dir/1986/609/oj (accessed on 9 April 2020).
- Kenechukwu, F.C.; Umeyor, C.E.; Momoh, M.A.; Ogbonna, J.D.N.; Chime, S.A.; Nnamani, P.O.; Attama, A.A. Evaluation of gentamicin-entrapped solid lipid microparticles formulated with a biodegradable homolipid from Capra hircus trop. J. Pharm. Res. 2014, 13, 1999-1205. [Google Scholar]
- Attama, A.A.; Okafor, C.E.; Builders, P.F.; Okorie, O. Formulation and in vitro evaluation of a PEGylated microscopic lipospheres delivery system for ceftriaxone sodium. Drug Deliv. 2009, 16, 448–457. [Google Scholar] [CrossRef]
- Potta, S.G.; Minemi, S.; Nukala, R.K.; Peinado, C.; Lamprou, D.A.; Urquhart, U. Development of solid lipid nanoparticles for enhanced solubility of poorly soluble drugs. J. Biomed. Nanotech. 2010, 6, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Momoh, M.A.; Ossai, E.C.; Omeje, C.E.; Omenigbo, O.P.; Kenechukwu, F.C.; Ofokansi, K.C.; Attama, A.A.; Olobayo, K.O. A new lipid-based oral delivery system of erythromycin for prolong sustain release activity. Mater. Sci. Eng. C 2019, 97, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Des Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.J. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, F.; Monica, L.; Claudio, C.; Massimo, M.; Maurizio, D.; Davide, M. Investigation of size, surface charge, PEGylation degree and concentration on the cellular uptake of polymer nanoparticles. Colloid Surf. B 2014, 123, 639–647. [Google Scholar]
- Kenneth, O.; Gerhard, W.; Gert, F.; Conrad, C. Matrix-loaded biodegradable gelatin nanoparticles as new approach to improve drug loading and delivery. Eur. J. Pharm. Biopharm. 2010, 76, 1–9. [Google Scholar]
- Abdallah, M.; Yuichi, T.; Hirofumi, T. Design and evaluation of novel pH-sensitive chitosan nanoparticles for oral insulin delivery. Eur. J. Pharm. Sci. 2011, 42, 445–451. [Google Scholar]
- Sarmento, B.; Veiga, F.; Ferreira, D. Development and characterization of new insulin containing polysaccharide nanoparticles. Colloids Surf. B 2006, 53, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Ziv, E.; Bendayan, M. Intestinal absorption of peptides through the enterocytes. Microsc. Res. Tech. 2000, 49, 346–352. [Google Scholar] [CrossRef]
- Salatin, S.; Maleki, D.S.; Yari, K.A. Effect of the surface modification, size, and shape on cellular uptake of nanoparticles. Cell Biol. Int. 2015, 39, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lim, T.M.; Lim, L.Y. Pharmacological activity of peroral chitosan-insulin nanoparticles in diabetic rats. Int. J. Pharm. 2005, 293, 271–280. [Google Scholar]
- Simon-Giavarotti, K.A.; Giavarotti, L.; Gomes, L.F.; Lima, A.F.; Veridiano, A.M.; Garcia, E.A.; Mora, O.A.; Fernández, V.; Viodela, L.A.; Junqueira, V.B. Enhancement of lindane-induced liver oxidative stress and hepatotoxicityby thyroid hormone is reduced by gadolinium chloride. Free Radic. Res. 2000, 36, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Kasarala, G.; Tillmann, H.L. Standard liver tests. Clin. Liver Dis. 2016, 8, 13–18. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Batch | Mucin (g) | PEG-4000 (g) | Insulin (mL) |
|---|---|---|---|
| A0 | 1 | 1 | 0.0 |
| A1 | 1 | 1 | 15.0 |
| A2 | 1 | 2 | 15.0 |
| A3 | 1 | 3 | 15.0 |
| A4 | 1 | 4 | 15.0 |
| A5 | 1 | 5 | 15.0 |
| Batch 2 | EE (%) | DL (%) | PS (µm) | RV (%) |
|---|---|---|---|---|
| A0 | -- | -- | 276 ± 0.03 | 66 ± 0.14 |
| A1 | 82 ± 0.12 | 18 ± 0.01 | 311 ± 0.22 | 75 ± 0.13 |
| A2 | 81 ±0.11 | 26 ± 0.24 | 321 ± 0.13 | 80 ± 0.14 |
| A3 | 83 ±0.23 | 28 ± 0.04 | 328 ± 0.11 | 83 ± 0.17 |
| A4 | 84 ±0.16 | 35 ± 0.17 | 346 ± 0.05 | 86 ± 0.13 |
| A5 | 92 ± 0.12 | 39 ± 0.12 | 398 ± 0.21 | 89 ± 0.12 |
| Batch 2 | SGPT (IU/L) | SGOT (IU/L) | ALP (IU/L) |
|---|---|---|---|
| A1 | 38.0 ± 0.16 | 66 ± 0.01 | 113 ± 0.11 |
| A2 | 39.5 ± 0.21 | 65 ± 0.33 | 118 ± 0.23 |
| A3 | 36.5 ± 0.15 | 67 ± 0.13 | 120 ± 0.21 |
| A4 | 37.0 ± 0.11 | 66 ± 0.04 | 115 ± 0.22 |
| A5 | 38.5 ± 0.13 | 68 ± 0.34 | 116 ± 0.52 |
| Control (water) | 36.0 ± 0.30 | 64 ± 0.12 | 115 ± 0.11 |
| Reference value | 10-40 | 50-150 | 30-130 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
A. Mumuni, M.; E. Calister, U.; Aminu, N.; C. Franklin, K.; Musiliu Oluseun, A.; Usman, M.; Abdulmumuni, B.; Y. James, O.; C. Ofokansi, K.; A. Anthony, A.; et al. Mucin-Grafted Polyethylene Glycol Microparticles Enable Oral Insulin Delivery for Improving Diabetic Treatment. Appl. Sci. 2020, 10, 2649. https://doi.org/10.3390/app10082649
A. Mumuni M, E. Calister U, Aminu N, C. Franklin K, Musiliu Oluseun A, Usman M, Abdulmumuni B, Y. James O, C. Ofokansi K, A. Anthony A, et al. Mucin-Grafted Polyethylene Glycol Microparticles Enable Oral Insulin Delivery for Improving Diabetic Treatment. Applied Sciences. 2020; 10(8):2649. https://doi.org/10.3390/app10082649
Chicago/Turabian StyleA. Mumuni, Momoh, Ugwu E. Calister, Nafiu Aminu, Kenechukwu C. Franklin, Adedokun Musiliu Oluseun, Mohammed Usman, Barikisu Abdulmumuni, Oyeniyi Y. James, Kenneth C. Ofokansi, Attama A. Anthony, and et al. 2020. "Mucin-Grafted Polyethylene Glycol Microparticles Enable Oral Insulin Delivery for Improving Diabetic Treatment" Applied Sciences 10, no. 8: 2649. https://doi.org/10.3390/app10082649
APA StyleA. Mumuni, M., E. Calister, U., Aminu, N., C. Franklin, K., Musiliu Oluseun, A., Usman, M., Abdulmumuni, B., Y. James, O., C. Ofokansi, K., A. Anthony, A., C. Ibezim, E., & Díaz Díaz, D. (2020). Mucin-Grafted Polyethylene Glycol Microparticles Enable Oral Insulin Delivery for Improving Diabetic Treatment. Applied Sciences, 10(8), 2649. https://doi.org/10.3390/app10082649