Abstract
Post-mortem interval (PMI) estimation is a critical task in forensic science. In this study, we used maggots collected from pig carcasses and applied an integrated proteomics and metabolomics approach to determine potential candidate substances for the estimation of PMI. After methanol precipitation, the supernatant containing metabolites and the protein pellet were separated and subjected to metabolomic and proteomic analyses using liquid chromatography-tandem mass spectrometry (LC-MS/MS). MS/MS data were analyzed for identification and quantification using Proteome Discoverer and Compound Discoverer software. A total of 573 metabolites and more than 800 porcine proteins were identified in maggots. This is the first dataset of proteins and metabolites in maggots collected from porcine carcasses. In this study, guanosine monophosphate, xanthine, inosine, adenosine, and guanine were detected with a similar tendency to increase during early days of maggot development and then decreased gradually. We broadly profiled various biomolecules through analysis in the spot of incident. Especially, we confirmed that proteome and metabolome profiling could be performed directly and indirectly.
1. Introduction
Forensic science is used as an investigative technique to establish facts in civil or criminal legal cases. From the perspective of solving crimes, provision and rigorous selection of measurable information to determine the time of death are highly useful [1]. However, the case where a forensic investigation is necessary often includes unintentional death. In many cases, therefore, variables affecting results due to prolonged exposure to poor environments or if measured information is inaccurate or limited must be considered [2]. To overcome such a limit, more accurate and reliable data should be collected and analyzed based on technology of applied science such as chemistry and biology because a victim can die from various causes with body decomposition [1]. Post-mortem interval (PMI) is an index estimated through establishment of the relative time after death [3,4,5]. This parameter can have a large error due to various external factors that can influence the body condition and change chemical compositions of a human body [6,7]. Other methods to estimate the PMI such as biochemical approaches using thanatomicrobiome and epinecrotic community analysis [8], postmortem protein degradation [9], and application of endogenous nuclease activity and hydrolysis property related to DNA [10] and RNA trait [11,12] are continuously published. In addition, various studies such as decay–decomposition time measurement [13] for specific targets have been conducted. In one of forensic science fields, the estimation of PMI through forensic entomology is widely used [14]. Especially, developmental progression of insects has been applied over several decades [15]. However, the insect behavior in various environments is inconsistent. Furthermore, a novel approach is required to overcome more intrinsic difficulties, such as different growth rates from 2 to 10 times depending on the ambient temperature [16]. In this regard, the studies including variables such as changes in temperature at the place and whether or not insects can be easily accessible themselves have been reported [17,18,19]. However, physicochemical analysis studies of a breeding insect at the incident site have not been reported yet.
To estimate correct PMI, currently advanced techniques based on analytical chemistry are applied to obtain data from an entomological and forensic investigation [20]. Diverse methods such as supposition confirming the species after building the sample specimen standard [21], calculating inversely by checking with many factors such as environmental conditions (e.g., the site temperature) [22], and estimating PMI by comparing stages of development with collected storage samples [23,24] have been reported. A method using a constant temperature range related to growth and the base temperature effectively applied to the growth has the problem of over-underestimation of the factor since it is just a measured value [23]. Due to characteristics of maggots, which are a metamorphic species [25,26], the parameter to be reflected in PMI estimation poses many difficulties because frictional heat between crowding can induce an increase in body temperature [27]. Therefore, an additional high-level scientific approach is needed to provide a more reliable data set. The credibility of data is underscored in the biomedical analysis of a corpse for case resolution [1]. Since material identification using LC-MS/MS has high accuracy and specific information, it can provide a reliable quantitative and qualitative evaluation of the material [28]. Proteomics is the study of all proteins existing in samples [29,30]. It is a powerful tool for generating large datasets that represent characteristics such as patterns and interactions of expressed proteins [31,32]. Proteomics is a confirmatory and orthogonal strategy to perform genomic analyses in forensic sciences, particularly when samples lack nucleic acids, such as hair and bone samples [31]. Metabolomics also contributes to explaining cellular functions at a direct level in conjunction with proteomics, including overall chemical and biological substances that interact with each other at the same time [33]. It is an excellent tool for presenting whole systems of a biological individual. This approach can also be applied to forensic research besides health and environmental sciences. Broad profiling is an analysis of a scientifically significant change pattern and enables rapid selection of specific unknown marker candidates [3,34,35,36]. In this study, broad profiling was performed using an integrated workflow for simultaneous analysis of proteins and metabolites from maggots and porcine corpse. To the best of our knowledge, this is the first proteome and metabolome dataset on maggots collected from porcine corpses. We performed quantitative–qualitative analysis to suggest specific marker candidates from biomolecules in insects accumulated through ingestion.
2. Materials and Methods
2.1. Materials
Acetonitrile, methanol, and high-performance liquid chromatography (HPLC) grade water were obtained from J.T.Baker (Philipsburg, NJ, USA). Tris (2-carboxyethyl) phosphine (TCEP) was purchased from Thermo Fisher Scientific (Rockford, IL, USA). Iodoacetamide (IAA), ammonium bicarbonate (ABC), and urea were bought from Sigma (St. Louis, MO, USA). Formic acid was obtained from Fluka (Charlotte, NC, USA). Sequencing-grade modified trypsin was purchased from Promega (Madison, WI, USA). Microcon YM-30 filter was supplied by Milipore (Burlington, MA, USA). cOmplete™ Protease inhibitor cocktail (tablet type) was purchased from Roche Biochemicals (Basel, Switzerland).
2.2. Experimental Design
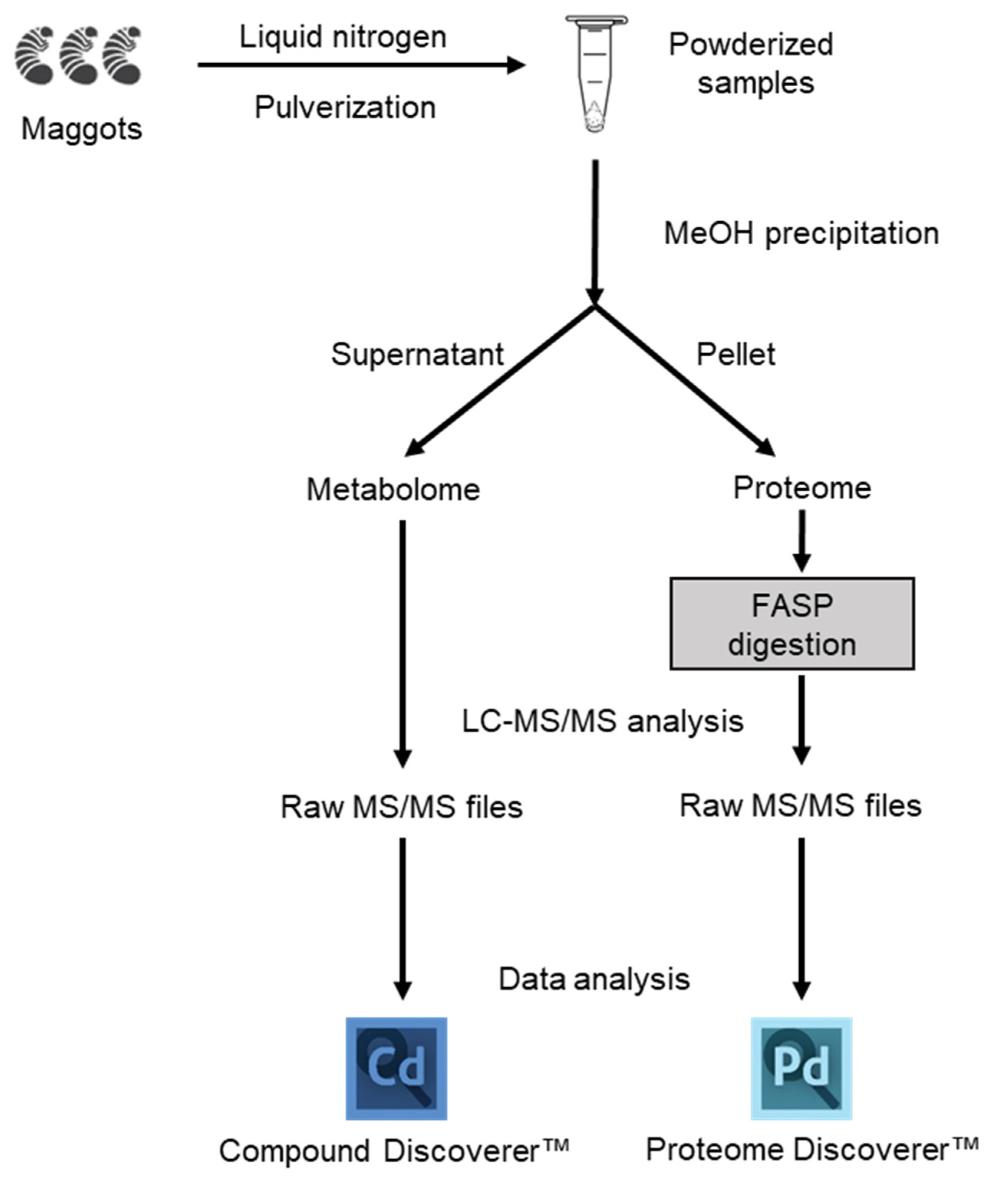
In this study, we used available workflows for universal proteomics and metabolomics analysis [37]. If brief, as shown in Figure 1, after precipitation with MeOH, the supernatant was collected for metabolomic analysis while the protein pellet was used in a simultaneous workflow for proteomic analysis. The protein digestion step was performed with The Filter Aided Sample Preparation (FASP) method, which allows the removal of detergent and isolation of peptides from undigested material [38].
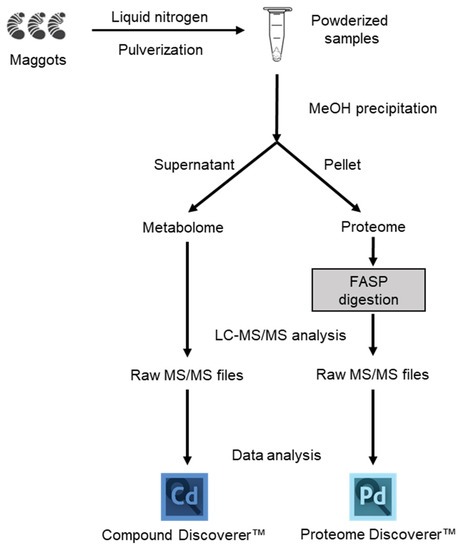
Figure 1.
Schematic overview of the workflow used for proteomic and metabolomic analysis of maggot sample. Both omics were carried out simultaneously from a single precipitation procedure.
2.3. Sample Preparation
2.3.1. Sample Collection
Maggots were sampled from a mountain at the Korean Police Investigation Academy, Korea. Three pigs (Sus scrofa L.) were obtained from Korean Police Investigation Academy. These pigs were sacrificed using chemical drugs and the euthanasia procedure following animal ethics guidelines. The animal experiment was approved by Institutional Animal Care and Use Committee of National Institute of Scientific Investigation (NISI-IACUC). Victims were left on a flatland located inside a mountainside forest to facilitate access to insects without artificial handling in order to proceed as similar as possible to natural conditions. The sampling area has a temperate monsoon climate. Its seasonal characteristics are not strong. The average monthly temperature was recorded at about 22 °C during the experiment.
The soil breeding insects including maggots was collected into a 50 mL corning tube with exudate from pig carcasses. To allow equivalency, samples were evenly collected immediately below hind limbs, abdomen, forelimbs, and head of pig carcasses and subsequently combined. Samples were collected at four different time points: day 3, day 5, day 9, and day 11. Due to low amounts of maggots, samples collected on the same day were pooled. These collected samples were freeze-dried and stored at −80 °C to maintain the similar water-holding capacity.
2.3.2. Sample Pretreatment
Maggot samples were washed with HPLC water to remove contaminants, such as soil. They were then cryo-pulverized with liquid nitrogen using a Covaris Tissue CryoPrep system (Covaris, Woburn, MA, USA). Powdered samples were collected and placed into glass tubes.
2.3.3. Extraction of Metabolites
A methanol precipitation method was used for the separation of proteins and metabolites [39,40]. In brief, pulverized samples were thawed on ice. Then 1.6 mL of cold 80% methanol was immediately added for protein precipitation. The resulting mixture was incubated at −20 °C for 16 h. After centrifugation at 2000× g for 10 min, the protein pellet and the supernatant containing metabolites were separated. The supernatant was transferred to a new 1.5 mL tube and completely dried using a HyperVAC-MAX VC2200 centrifugal vacuum concentrator (Hanil Scientific Inc., Korea). Dried metabolite contents were reconstituted in 100 μL of 0.1% formic acid in water and then subjected to LC-MS/MS analysis.
2.3.4. Protein Digestion
The protein pellet was suspended in 1 mL of lysis buffer (8 M urea–0.1 M Tris-HCl buffer, pH 8.5). After adding 40 μL of protease inhibitor cocktail (25X stock), the sample was sonicated for 20–40 min at 15 °C using a Covaris S2 Focused-Ultrasonicator (Covaris, Woburn, MA, USA). Protein concentration was quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The digestion step was performed using FASP on Microcon 30 K centrifugal filter device (Millipore, Billerica, MA, USA). Each 150 μg of protein sample was adjusted to a volume of 200 μL with lysis buffer. Proteins were reduced by incubating with 5 mM TCEP at 37 °C for 30 min and alkylated with 50 mM IAA at 25 °C for an hour in dark conditions. After washing with lysis buffer and 50 mM ABC sequentially, proteins were digested with trypsin (enzyme to protein ratio of 1:50; w/w) at 37 °C for 18 h. Resulting peptide mixtures were collected to new tubes and trypsin was inactivated by acidifying with 10 μL of formic acid. These peptide mixtures were desalted with a Sep-Pak tC18 96-well plate (Waters, Milford, MA, USA). Elutions were completely dried using a centrifugal vacuum concentrator.
2.4. LC-MS/MS Analyses of Metabolites and Proteins
2.4.1. Proteomic Analysis
LC-MS analysis for proteomics was performed on a nano-LC-MS/MS system consisting of a Q Exactive™ Hybrid Quadrupole-Orbitrap MS coupled with a Dionex Ultimate 3000 HPLC (Thermo Fisher Scientific, MA, USA). Peptide mixtures were analyzed in a data-dependent acquisition (DDA) approach and loaded onto an Acclaim™ PepMap™ 100 C18 nano-trap column (75 μm × 2 cm, 3 μm particles, 100 Å pore size) using a mobile phase at a flow rate of 2.5 μL/min for 10 min. After a trapping step, samples were subsequently separated using an Acclaim™ PepMap™ C18 100 Å RSLC nano-column (75 μm × 50 cm, 2 μm particles, 100 Å pores, Thermo Fisher Scientific) with 0.1% formic acid in water (solvent A) and 0.1% formic acid in 80% acetonitrile (solvent B) at a fixed flow rate of 300 nL/min. The gradient of mobile phase was as follows: 4% solvent B in 4 min, 4–40% solvent B in 116 min, 40–96% solvent B in 10 min, 96–4% solvent B in 0.1 min, and 4% solvent B for 19.9 min. The injection quantity was 1 μg, which was determined with a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific). The electrospray ionization source was set up as follows: positive mode, spray voltage of 2.2 kV, and capillary temperature of 320 °C Properties of DDA Full Ms / dd-MS2 were set up as follows: full-MS scans, 400 to 2000 m/z of scan range, 70,000 of resolution at 400 m/z, 3 × 106 of AGC target, and maximum injection time (IT) of 100ms. MS2 scans used the following parameters: 17,500 of resolution at 400 m/z, 1 × 105 of AGC target, maximum IT of 100 ms, ±2 m/z of isolation width, 20 s of a dynamic exclusion time, and normalized collision energy (NCE) for dd-MS2 of 27%.
2.4.2. Metabolomic Analysis
LC-MS analysis for metabolomics was performed using a Q Exactive™ Hybrid Quadrupole-Orbitrap MS (Thermo Fisher Scientific, MA, USA) coupled with a 1290 Infinity UHPLC (Agilent, CA, USA). Metabolite mixtures were loaded using a ZORBAX Eclipse Plus C18 Rapid Resolution High Definition (RRHD) column (2.1 × 150 mm, 1.8 μm particles) with solvent A at a flow rate of 0.2 mL/min. The separation of metabolites was as follows: 2.5% solvent B in 5 min, 2.5–12.5% solvent B in 29 min, 12.5–37.5% solvent B in 22 min, 80% solvent B in 14 min, and 2.5% solvent B for 20 min. The electrospray source was equipped with a Heated Electrospray ionization (HESI-II) Probe combined with the standard Thermo Scientific™ Ion Max source. Parameters were set as follows: positive mode, spray voltage; 4 kV, capillary temperature; and 320℃. Properties of full MS/dd-MS2 were set up as follows: full-MS scans, 150 to 2000 m/z of scan range, 70,000 of resolution at 400 m/z, 1 × 106 of AGC target, and maximum IT of 60 ms. For MS2 scans, the following parameters were used: 17,500 of resolution at 400 m/z, 2 × 105 of AGC target, maximum IT was 250 ms, ±2 m/z of isolation width, and NCE for dd-MS2 of 30%.
2.5. Data Analysis and Statistical Interpretation
2.5.1. Proteomics Data Analysis
Thermo MS/MS raw files of each triplicated analysis were searched using a Proteome Discoverer™ software (ver. 2.4). The Sus scrofa database was downloaded from Uniprot (released April, 2021), and included 1511 of the reviewed entries in database. An appropriate consensus workflow included a peptide-spectrum match (PSM) validation step and SEQUEST HT process for detection as a database search algorithm. Search parameters were set up as follows: 10 ppm of tolerances of precursor ion masses, 0.02 Da fragment ion mass, and maximum of two missed cleavages with trypsin enzyme. Dynamic modification for peptide sequence was as follows: static carbamidomethylation of cysteine (+57.012 Da), variable modifications of methionine oxidation (+15.995 Da), and carbamylation of protein in N-term (+43.0006 Da). After searching, results showing FDR below 1% were selected and filtered with a length of at least six peptides. The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [41] partner repository, with the dataset identifier PXD028013.
2.5.2. Metabolomics Data Analysis
Obtained UHPLC-Orbitrap-MS/MS RAW files were processed using Compound Discoverer™ 3.2.0.421 (Thermo Fisher Scientific, Waltham, MA, USA). Untargeted metabolomics workflow was used to perform retention time alignment and compound identification. Identification of compounds using mzCloud and ChemSpider. Chemical background was removed using a blank file. A Supplementary Method S1 of the entire workflow of Compound Discoverer is attached. In the obtained data set, identification was matched with levels 2 and 3 by filtering using annotation implemented with Metabolomics Standards Initiative (MSI) of the Metabolomics Society [42]. Level 2 matched the exact mass (10 ppm) with a fragmentation score over 80 of the mzCould database and Level 3 matched with the exact mass (5 ppm) in an MS 1 database (Chemspider). In all samples, 455 and 100 metabolites matched with levels 3 and 2, respectively. Principal component analysis (PCA) and heatmap was conducted using Metaboanalyst (http://www.metaboanalyst.ca/MetaboAnalyst, accessed on 26 August 2021, ver. 5.0) [43].
3. Results
3.1. Metabolomics
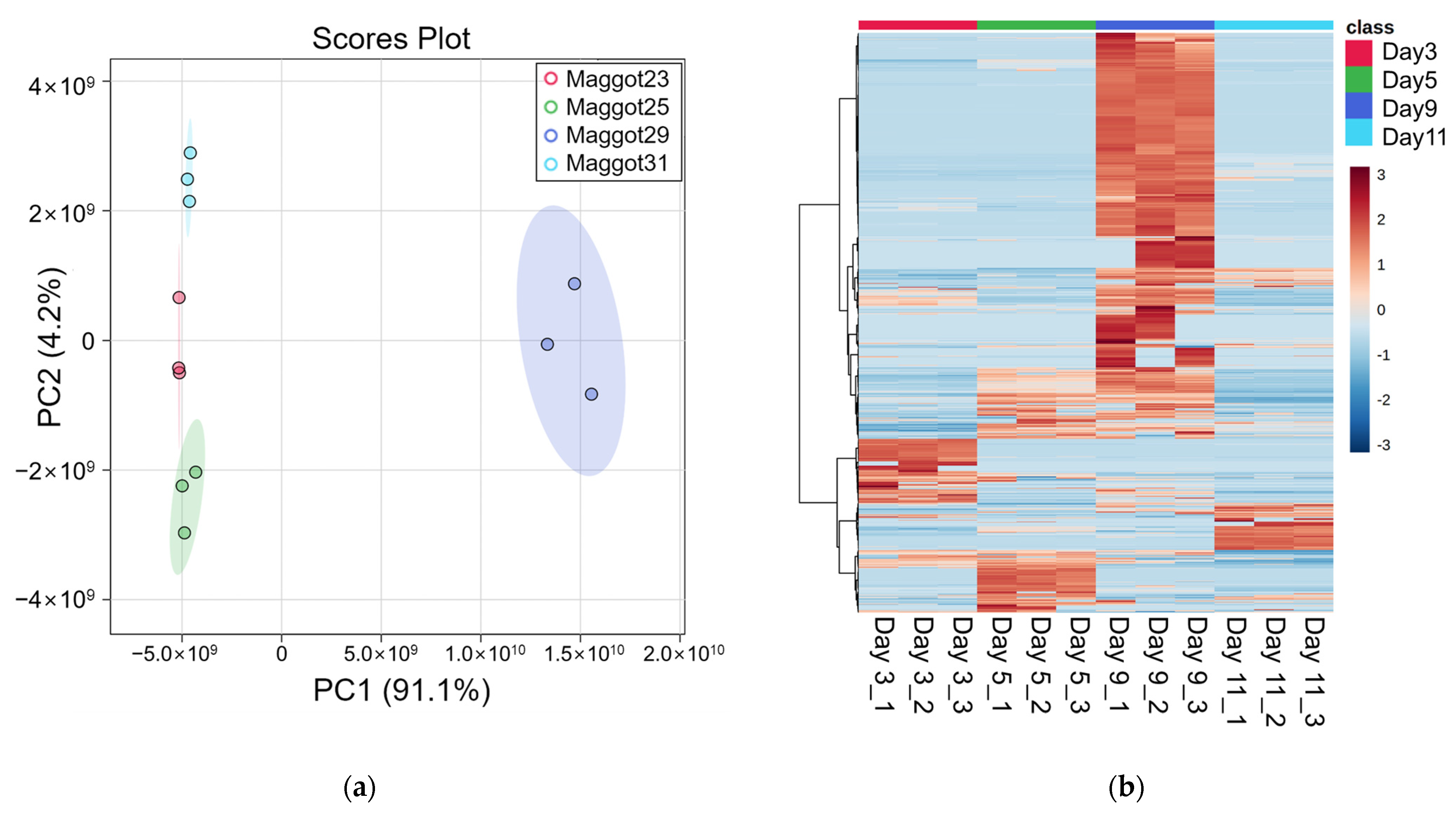
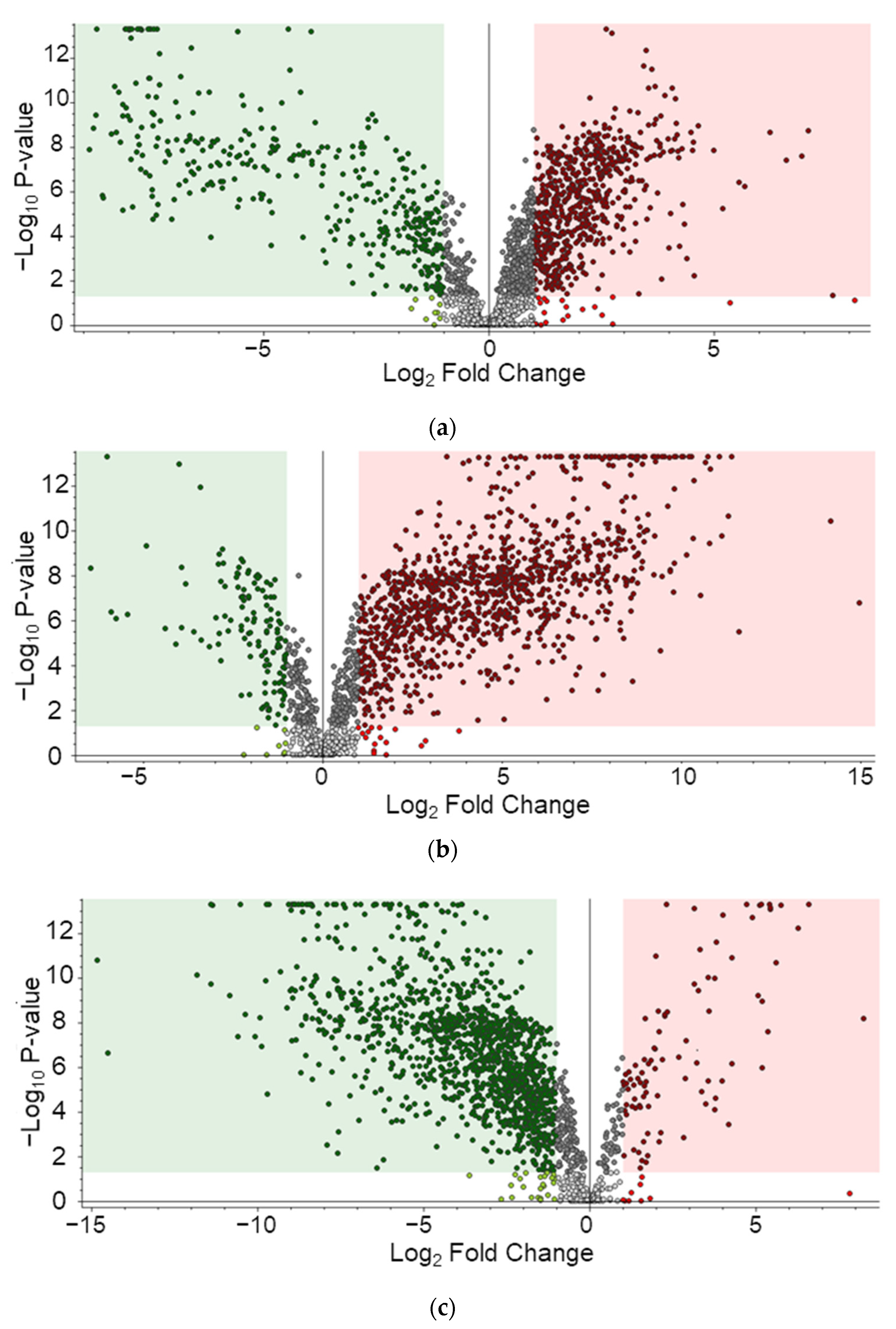
Metabolites identified in samples were composed of those derived from porcine carcasses ingested by maggots and metabolites of maggots. The identification of analytes was based on the MS2 database but limited to classification by species because the structure was matched based on the elemental composition levels of small molecules. To build a list of differential metabolites (DMs) for each group, pelletized proteins were denatured through a focused lysis procedure and the initial protein concentration was determined using the BCA assay. Concentrations of metabolites were estimated in proportion to concentrations of proteins. Metabolites were dried, resuspended, and used for analysis. Using LC-MS/MS analysis, 212, 222, 368, and 194 metabolites were identified on day 3, day 5, day 9, and day 11, respectively (Table S1). By filtering using annotation implemented with the Metabolomics Standards initiative (MSI) of the Metabolomics Society [42], 134, 153, 258, and 194 metabolites were identified with level 2 and 33, 37, 57, and 31 metabolites were identified with level 3 on day 3, day 5, day 9, and day 11, respectively. Figure S1 presents peak area of the top 20 metabolites. These are average values of triplication results. These metabolites identified at different sampling time points were visualized by PCA plot (Figure 2a), showing differences among the four time points. A heatmap was generated for 573 metabolites (Figure 2b). According to the time of sampling given in this study, Figure 3 shows down- and up-regulated metabolites indicated by green and red, respectively. First, in the comparison of day 5 vs. day 3, 309 metabolites were differentially expressed (210 up- and 99 down-regulated). Subsequently, 366 metabolites (315 up- and 51 down-regulated) and 457 (41 up- and 416 down-regulated) metabolites were differentially expressed in the comparison of day 9 vs. day 5 and the comparison of day 11 vs. day 9, respectively. DMs are listed in Table S2.
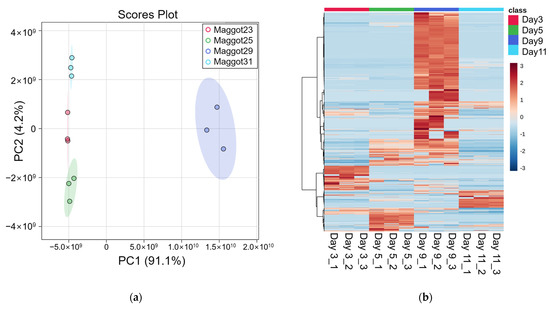
Figure 2.
Metabolomics analysis results. (a) PCA plot, (b) Heatmap.
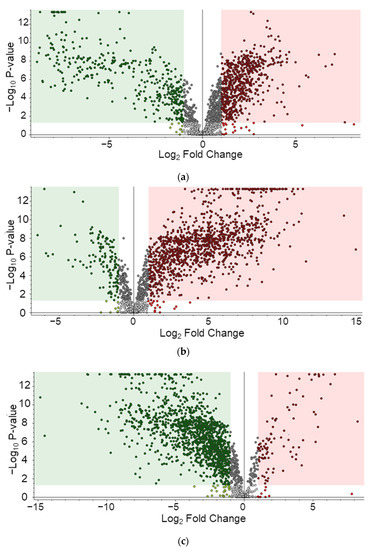
Figure 3.
Differentially expressed metabolites. (a) Day 5 vs. day 3, (b) Day 9 vs. day 5, (c) Day 11 vs. day 9.
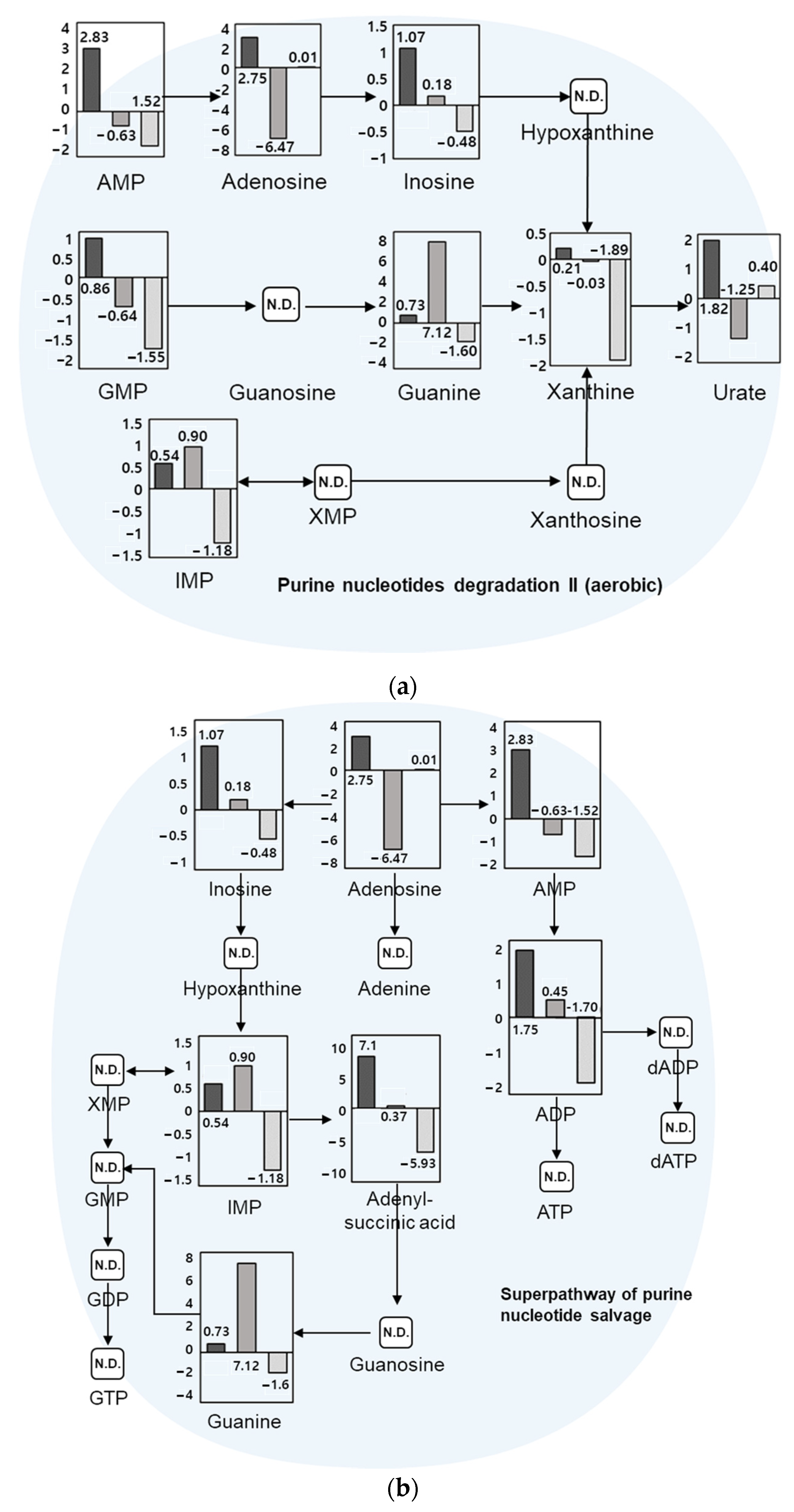
Among 267 pathways identified from the metabolite dataset, top 20 pathways are listed in Table 1. Quantitative metabolites were significantly matched and potentially correlated with nucleotide-related pathways such as purine nucleotides degradation II (aerobic) and purine nucleotide salvage in Compound Discoverer through the KEGG database. Quantitative values for metabolites in each pathway are shown in Figure 4. Changes in quantitative values of day 5 vs. day 3, day 9 vs. day 5, and day 11 vs. day 9 are shown on the y-axis as log2 fold change. In the purine nucleotides degradation II pathway, 16 referenced compounds among 27 compounds in the pathway were matched, with adenosine 5’-monophosphate (AMP), guanosine monophosphate (GMP), and xanthine commonly increased until day 5, which is the initial stage of growth. They showed a tendency to decrease until day 11, the time of being an adult insect. In the superpathway of purine nucleotide salvage pathway, 14 referenced compounds out of 54 compounds in the pathway were matched, of which purine nucleotide compounds such as inosine, adenosine, and guanine matched with a similar tendency.

Table 1.
List of top 20 pathways from metabolite datasets.
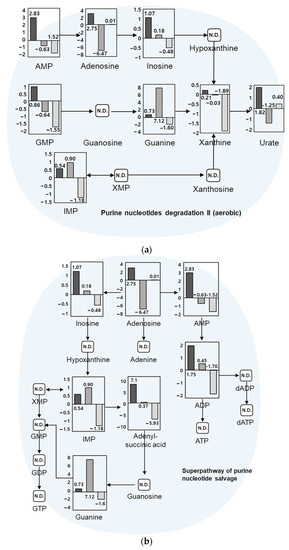
Figure 4.
Metabolic pathways through the KEGG database, including (a) purine nucleotides degradation II (aerobic) and (b) superpathway of purine nucleotide salvage. Log2 fold-change quantitative values were for day 5 vs. day 3, day 9 vs. day 5, and day 11 vs. day 9, from left to right, respectively. AMP: adenosine monophosphate; ADP: adenosine diphosphate; ATP: adenosine triphosphate; dADP: deoxyadenosine diphosphate; dATP: deoxyadenosine triphosphate; GMP: guanosine monophosphate; GDP: guanosine diphosphate; GTP: guanosine triphosphate; IMP: inosine monophosphate; XMP: xanthosine monophosphate.
3.2. Proteomics
The workflow not only allowed metabolomic analysis, but also allowed proteomic analysis in an integrated manner. In sample preparation, metabolites were extracted and the remaining pellet was pretreated. Proteins were digested into peptides through the FASP method. Subsequently, a C18-based additional desalting step was performed for peptides. As a result, 827, 1005, 947, and 738 pig-derived proteins consumed by maggots were identified on day 3, day 5, day 9, and day 11, respectively. Corresponding proteins are listed in Table S3. Top-scoring proteins were mainly cytoskeletal proteins in abundance, such as ACTB (actin, cytoplasmic 1), ACTA1 (actin, 9alpha skeletal muscle), TUBB1 (tubulin beta chain), and TUBA1A/1B (tubulin alpha-1A/-1B). In addition, ribosomal proteins (40S/60S ribosomal proteins), which exist at the intersection of the microtrabecular that fixes the form of cell, are also identified with high-scoring. Additionally, all the proteomics raw files were analyzed with the merged insect data base, which includes Scathophaga stercoraria, Sarcophagidae, and Musca domestica. However, it is difficult to find a significant correlation with PMI due to the small number of identified proteins. A list of the corresponding proteins is attached to Table S4.
4. Discussion
Although analytical techniques in forensics science field have been steadily developed, obtaining reliable and intact information from neglected corpses remains difficult. Although it is possible to identify a body’s DNA, information at protein and metabolite levels have been steadily needed. Metabolites can be interpreted as an indicator by directly relating the phenotype of an object to time. Existing research results have proven the effectiveness of metabolomics in the estimation of PMI [44,45]. In addition, one study has been conducted to evaluate the overall molecular pattern of metabolites related to PMI [46]. In this study, metabolites composed of a mixture of those derived from porcine carcasses and maggot metabolites could be identified.
In previous studies, PMI was estimated with an estimation equation using a number of nucleic acids such as hypoxanthine at ambient temperature [47]. In addition, increases and decreases of free fatty acid and glycerophospholipid in human muscles have been detected during 24-days PMI with lipids infected by invasive microbes after death [48]. In other study, distinct patterns between volatile fatty acids in soil-extracted fluid were detected during chemical decomposition of cadavers [49]. Based on these studies, metabolites identified in this study could be selected as marker candidates for PMI determination by omics technology. However, it is difficult to be sure that selected metabolites are precisely derived from maggots even though species are different. Sometimes small molecules have similar or same elemental composition [50]. For this reason, a combined omics approach of proteomics and metabolomics was briefly used in this study to obtain a more definite and reliable data set. This approach simultaneously provides the proof that peptides of a victim, which are accumulated in maggots, could be identified based on the victim’s RNA FASTA sequence databases. As a result of this experiment, cytoskeletal proteins, and ribosomal related proteins known to interact closely with the cytoskeleton were mainly identified. They showed a high abundance in the in vivo proteome. This result suggests that it is possible to apply this approach to understand post-mortem proteolysis because actin is one of muscle-building substance that can cause rigor mortis, one of main cadaveric reactions [51]. As similar result, there are also studies showing that the presence of cytoskeletal proteins related to post-mortem proteolysis of structural transmembrane proteins [52]. These studies indicate that proteomic approaches could be usefully applied for PMI estimation, overcoming the limitation of irregular degeneration of corpses by intrinsic chemicals of maggots. Quantitative proteomics were not tried in this study, which could lead to large errors sometimes with conventional sample preparation. Moreover, the protein can be lost according to various causes of death and the surrounding environment. Hence, more precise experiments, such as labeling, should be performed to quantify candidates if necessary. PMI can be estimated by interpreting cadaveric reactions through entomology methods. However, the appraisal in terms of legality by these methods has limitations. Errors may be accompanied by variables that do not leave traces. Thus, the developed evaluation process can be newly applied to deal with various cases. Selecting targets based on omics research allows molecular-level research efficiency to be significantly enhanced. It also overcomes the weakness of entomology-based PMI estimation method caused by a short maggot life cycle. Moreover, this metabolomics experiment could help us build a quantitative database. It provides the listed of identified metabolites derived from a corpse or maggots. In addition, it suggests that species or type of a corpse could be identified through proteome detection of protein accumulated in maggots. In this way, past traces of an organism are not limited to the organism only through the omics technique. Various environmental factors can be extensively investigated. They will contribute to conceiving direct and indirect clues. The compatibility of mass spectrometry technology with high selectivity is expected to be more active as an ideal tool required in the field of forensic science.
5. Conclusions
In this study, we broadly profiled various biomolecules through analysis in the spot of the incident. Especially, we confirmed that proteome and metabolome profiling by direct and indirect omics approaches for objects and environments is also available for simultaneous proteomic and metabolomics analyses of maggots collected from porcine corpses using LC-MS/MS in an integrated workflow. A total of 573 metabolites and more than 800 pig-derived proteins were identified. From the metabolite dataset, nucleotide-related pathways such as purine nucleotides degradation II (aerobic) and purine nucleotide salvage were identified. We can deduce that these metabolites might be potential biomarker candidates for PMI based on its quantitative pattern by succession. Some metabolites could be potential biomarkers for PMI estimation, including guanosine monophosphate, xanthine, inosine, adenosine, and guanine. Based on this analysis, further studies that specify candidates capable of PMI estimation among profiled biomolecules are needed to obtain forensic evidence applicable to investigations.
Supplementary Materials
The following materials are available online at https://www.mdpi.com/article/10.3390/app11177885/s1, Method S1, Data analysis workflow of Compound Discoverer; Table S1, List of metabolites of whole triplicated and merged sample identified from Compound Discoverer™; Table S2, List of identified DMs list; Table S3, List of proteins of whole triplicated and merged sample identified with Proteome Discoverer™; Table S4, List of protein with the merged insect data base, which includes Scathophaga stercoraria, Sarcophagidae, and Musca domestica. Figure S1, Bar graphs of the peak areas of top 20 significant metabolites. The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028013. Reviewers may access it using username reviewer_pxd028013@ebi.ac.uk and password RoCSU59v).
Author Contributions
Conceptualization, J.-M.P., H.-J.L. and H.L.; methodology, H.-J.L. and H.L.; software, J.-H.M. and M.J.; validation, V.-A.D. and J.-M.P.; formal analysis, V.-A.D. and J.-M.P.; investigation, J.-H.M., M.J., S.C. and Y.-S.E.; resources, J.-M.P., Y.-S.E. and T.-H.S.; data curation, J.-H.M., M.J., V.-A.D. and J.-M.P.; writing—original draft preparation, J.-H.M. and V.-A.D.; writing—review and editing, H.-J.L. and H.L.; visualization, J.-H.M. and V.-A.D.; supervision, J.-M.P.; project administration, H.-J.L. and H.L.; funding acquisition, H.-J.L. and H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2017M3D9A1073784) and a Gachon University grant (Grant No. GCU-2018-0698).
Institutional Review Board Statement
The pig sample collection was conducted according to the guidelines of the Declaration of Helsinki. It was approved by NISI-IACUC (IACUC# KPIA-20-01).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, data collection, data analyses, or data interpretation, in the writing of the manuscript, or in the decision to publish the results.
References
- Houck, M.M.; Siegel, J.A. Fundamentals of Forensic Science; Academic Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Aubernon, C.; Hedouin, V.; Charabidze, D. The maggot, the ethologist and the forensic entomologist: Sociality and thermoregulation in necrophagous larvae. J. Adv. Res. 2019, 16, 67–73. [Google Scholar] [CrossRef]
- Choi, K.-M.; Zissler, A.; Kim, E.; Ehrenfellner, B.; Cho, E.; Lee, S.; Steinbacher, P.; Yun, K.N.; Shin, J.H.; Kim, J.Y.; et al. Postmortem proteomics to discover biomarkers for forensic PMI estimation. Int. J. Legal Med. 2019, 133, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, B. Flies as forensic indicators. J. Med. Entomol. 1991, 28, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Erzinçlioğlu, Z. Blowflies; The Richmond Publishing Co. Ltd.: Berkshire, UK, 1996. [Google Scholar]
- Gilbert, J.A.; Neufeld, J.D. Life in a world without microbes. PLoS Biol. 2014, 12, e1002020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, T.E.; Higley, L.G.; Baxendale, F.P. Maggot development during morgue storage and its effect on estimating the post-mortem interval. J. Forensic Sci. 2007, 52, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, H.R.; Das, S. Thanatomicrobiome and epinecrotic community signatures for estimation of post-mortem time interval in human cadaver. Appl. Microbiol. Biotechnol. 2020, 104, 9497–9512. [Google Scholar] [CrossRef] [PubMed]
- Zissler, A.; Stoiber, W.; Steinbacher, P.; Geissenberger, J.; Monticelli, F.C.; Pittner, S. Postmortem protein degradation as a tool to estimate the PMI: A systematic review. Diagnostics 2020, 10, 1014. [Google Scholar] [CrossRef]
- Tozzo, P.; Scrivano, S.; Sanavio, M.; Caenazzo, L. The role of DNA degradation in the estimation of post-mortem interval: A systematic review of the current literature. Int. J. Mol. Sci. 2020, 21, 3540. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.I.; Suárez-Peñaranda, J.M.; Otero, X.L.; Rodríguez-Calvo, M.S.; Costas, E.; Miguéns, X.; Concheiro, L. A new perspective in the estimation of Postmortem Interval (PMI) based on vitreous [K+]. J. Forensic Sci. 2001, 46, 209–214. [Google Scholar] [CrossRef]
- Scrivano, S.; Sanavio, M.; Tozzo, P.; Caenazzo, L. Analysis of RNA in the estimation of post-mortem interval: A review of current evidence. Int. J. Legal Med. 2019, 133, 1629–1640. [Google Scholar] [CrossRef]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Cadaver decomposition in terrestrial ecosystems. Naturwissenschaften 2006, 94, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Turchetto, M.; Vanin, S. Forensic entomology and climatic change. Forensic Sci. Int. 2004, 146, S207–S209. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Kunich, J.C. Entomology and the Law: Flies as Forensic Indicators; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Grassberger, M.; Reiter, C. Effect of temperature on Lucilia Sericata (Diptera: Calliphoridae) development with special reference to the Isomegalen- and Isomorphen-diagram. Forensic Sci. Int. 2001, 120, 32–36. [Google Scholar] [CrossRef]
- Jason, H.B.; Brundage, A. Forensic Entomology; CRC Press: Boca Raton, FL, USA, 2020; pp. 67–111. [Google Scholar]
- Anderson, G.S. The use of insects to determine time of decapitation: A case-study from British Columbia. J. Forensic Sci. 1997, 42, 947–950. [Google Scholar] [CrossRef]
- Lord, W.D.; Goff, M.L.; Adkins, T.R.; Haskell, N.H. The black soldier fly Hermetia Illucens (Diptera: Stratiomyidae) as a potential measure of human postmortem interval: Observations and case histories. J. Forensic Sci. 1994, 39, 215–222. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.N.; Logan, J.G. Exploiting insect olfaction in forensic entomology. In Current Concepts in Forensic Entomology; Amendt, J., Goff, M.L., Campobasso, C.P., Grassberger, M., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 205–221. [Google Scholar]
- Catts, E.P. Problems in estimating the postmortem interval in death investigations. J. Agric. Entomol. 1992, 9, 245–255. [Google Scholar]
- Jeong, S.J.; Park, S.H.; Park, J.E.; Park, S.H.; Moon, T.; Shin, S.E.; Lee, J.W. Extended model for estimation of ambient temperature for Postmortem Interval (PMI) in Korea. Forensic Sci. Int. 2020, 309, 110196. [Google Scholar] [CrossRef]
- Byrd, J.H.; Castner, J.L. Forensic Entomology: The Utility of Arthropods in Legal Investigations, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Shin, S.E.; Park, J.H.; Jeong, S.J.; Park, S.H. The growth model of forensically important Lucilia Sericata (Meigen) (Diptera: Calliphoridae) in South Korea. Insects 2021, 12, 323. [Google Scholar] [CrossRef]
- Bm, D.; De, M. Identification of eight homeobox-containing transcripts expressed during larval development and at metamorphosis in the gastropod Mollusc Haliotis Rufescens. Mol. Mar. Biol. Biotechnol. 1993, 2, 1–9. [Google Scholar]
- Giusti, A.F.; Hinman, V.F.; Degnan, S.M.; Degnan, B.M.; Morse, D.E. Expression of a Scr/Hox5 gene in the larval central nervous system of the Gastropod Haliotis, a non-segmented Spiralian Lophotrochozoan. Evol. Dev. 2000, 2, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Heaton, V.; Moffatt, C.; Simmons, T. Quantifying the temperature of maggot masses and its relationship to decomposition. J. Forensic Sci. 2014, 59, 676–682. [Google Scholar] [CrossRef]
- Xie, F.; Liu, T.; Qian, W.J.; Petyuk, V.A.; Smith, R.D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. Is proteomics the new genomics? Cell 2007, 130, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, V.-A.; Park, J.-M.; Lee, H. Review of three-dimensional liquid chromatography platforms for bottom-up proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [Green Version]
- Duong, V.-A.; Park, J.-M.; Lim, H.-J.; Lee, H. Proteomics in forensic analysis: Applications for human samples. Appl. Sci. 2021, 11, 3393. [Google Scholar] [CrossRef]
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197. [Google Scholar] [CrossRef]
- Shao, X.; Ji, F.; Wang, Y.; Zhu, L.; Zhang, Z.; Du, X.; Chung, A.C.K.; Hong, Y.; Zhao, Q.; Cai, Z. Integrative chemical proteomics-metabolomics approach reveals Acaca/Acacb as direct molecular targets of PFOA. Anal. Chem. 2018, 90, 11092–11098. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Peinado, L.S.; Luque de Castro, M.D. An overview on forensic analysis devoted to analytical chemists. Talanta 2017, 167, 181–192. [Google Scholar] [CrossRef]
- Kang, Y.-R.; Park, Y.S.; Park, Y.C.; Yoon, S.M.; JongAhn, H.; Kim, G.; Kwon, S.W. UPLC/Q-TOF MS based metabolomics approach to post-mortem-interval discrimination: Mass spectrometry based metabolomics approach. J. Pharm. Investig. 2012, 42, 41–46. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef]
- Chen, B.-S.; Wu, C.-C. Systems biology as an integrated platform for bioinformatics, systems synthetic biology, and systems metabolic engineering. Cells 2013, 2, 635–688. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Ma, Y.; Ling, T.; Su, X.; Jiang, B.; Nian, B.; Chen, L.; Liu, M.; Zhang, Z.; Wang, D.; Mu, Y.; et al. Integrated proteomics and metabolomics analysis of tea leaves fermented by Aspergillus Niger, Aspergillus Tamarii and Aspergillus Fumigatus. Food Chem. 2021, 334, 127560. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, D.; Su, X.; Duan, S.; Wan, J.; Yuan, W.; Liu, B.; Ma, Y.; Pan, Y. An integrated metagenomics/metaproteomics investigation of the microbial communities and enzymes in solid-state fermentation of Pu-Erh tea. Sci. Rep. 2015, 5, 10117. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Domingo-Almenara, X.; Guijas, C.; Palermo, A.; Rinschen, M.M.; Isbell, J.; Benton, H.P.; Siuzdak, G. Enhanced in-source fragmentation annotation enables novel data independent acquisition and autonomous METLIN molecular identification. Anal. Chem. 2020, 92, 6051–6059. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Vass, A.A.; Barshick, S.-A.; Sega, G.; Caton, J.; Skeen, J.T.; Love, J.C.; Synstelien, J.A. Decomposition chemistry of human remains: A new methodology for determining the postmortem interval. J. Forensic Sci. 2002, 47, 542–553. [Google Scholar] [CrossRef]
- Kaszynski, R.H.; Nishiumi, S.; Azuma, T.; Yoshida, M.; Kondo, T.; Takahashi, M.; Asano, M.; Ueno, Y. Postmortem interval estimation: A novel approach utilizing gas chromatography/mass spectrometry-based biochemical profiling. Anal. Bioanal. Chem. 2016, 408, 3103–3112. [Google Scholar] [CrossRef]
- Sato, T.; Zaitsu, K.; Tsuboi, K.; Nomura, M.; Kusano, M.; Shima, N.; Abe, S.; Ishii, A.; Tsuchihashi, H.; Suzuki, K. A preliminary study on postmortem interval estimation of suffocated rats by GC-MS/MS-based plasma metabolic profiling. Anal. Bioanal. Chem. 2015, 407, 3659–3665. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Xing, Y.; Xiong, X.; Gan, L.; Hu, L.; Zhao, F.; Tong, Y.; Deng, S. An electrochemical biosensor for hypoxanthine detection in vitreous humor: A potential tool for estimating the post-mortem interval in forensic cases. Microchem. J. 2020, 155, 104760. [Google Scholar] [CrossRef]
- Wood, P.L. Lipidomics analysis of postmortem interval: Preliminary evaluation of human skeletal muscle. J. Postgenomics Drug Biomark. Dev. 2012, 3, 127. [Google Scholar] [CrossRef] [Green Version]
- Vass, A.A.; Bass, W.M.; Wolt, J.D.; Foss, J.E.; Ammons, J.T. Time since death determinations of human cadavers using soil solution. J. Forensic Sci. 1992, 37, 1236–1253. [Google Scholar] [CrossRef]
- Pace, N.R. The universal nature of biochemistry. Proc. Natl. Acad. Sci. USA 2001, 98, 805–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, K.; Paul, R. Assessment of Post Mortem Interval, (PMI) from forensic entomotoxicological studies of larvae and flies. Entomol. Ornithol. Herpetol. Curr. Res. 2013, 2, 2161–2983. [Google Scholar]
- Leal-Gutiérrez, J.D.; Rezende, F.M.; Elzo, M.A.; Johnson, D.; Peñagaricano, F.; Mateescu, R.G. Structural equation modeling and whole-genome scans uncover chromosome regions and enriched pathways for carcass and meat quality in beef. Front. Genet. 2018, 9, 532. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).