3.2. Inline Monitoring and Polymerisation Rate Determination
Similar to the widespread application of NIR spectroscopy for in situ monitoring of chemical reactions, particularly polymerisation reactions [
23,
24], Raman spectroscopy can be employed. Previous research by Klement et al. has shown that the integral of the C=C stretching vibration band can be correlated linearly to the acrylic acid (AA) concentration in aqueous solutions from 20 g/L up to 400 g/L [
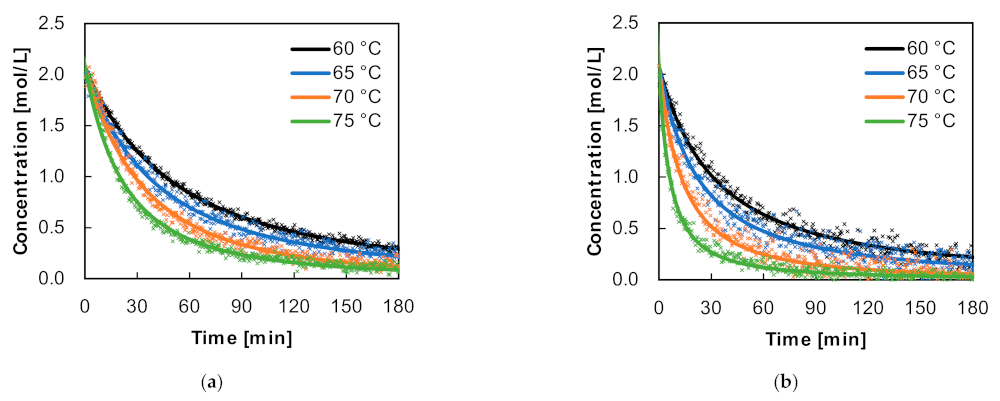
25]. Because the Raman spectrum of isopropyl alcohol does not overlap with this band, the AA concentration could be measured throughout the whole polymerisation reaction. AA concentration profiles of the reactions carried out with 0.5 mol% and 1.0 mol% AIBN regarding the AA content are shown in
Figure 4.
Here, the shown data points were acquired by multiplying the normalised concentration acquired from the band integration with the initial AA concentration. Raman measurements can have some fluctuations due to temperature changes, variations in viscosity, ascending bubbles, and the stirred reaction mass. However, depending on the desired spectra resolution, Raman laser intensity and the optical path length of the immersion probe, a single spectrum can be acquired in a fraction of a second. Over the course of the reaction, if spectra are acquired every 32 s, the data are sufficient to fit the concentration profiles using ideal kinetics. Under the initially classically expected assumption of a monomer reaction order of 1, however, curve fitting did not lead to a coverage of all measurement values during the reactions. Therefore, only if the reaction is to be examined up to lower conversions of about 30%, is it tenable to express the reaction with an order of 1 (see
Figure 5a). The fit error
σ2 is calculated as the sum of squares described as follows (Equation (1)):
where
P denotes the fitted value (in this case the normalised concentration),
N the amount of data points and
K the number of parameters to adjust the fit. In order to determine to what extent the reaction with a fit according to a reaction order
n = 1 is valid, the data points were only fitted up to a certain conversion section (in 10% steps); this is plotted in
Figure 5a. The error resulting from this fit is called the local error of the local fit. From these so-called local fits, the error that results when all measuring points of a reaction, i.e., the entire conversion range, are considered is then determined. This is called the global error of the local fit. In addition, these errors were compared with the fit used for the later described final evaluation according to reaction orders of
n > 1 (Equation (2)). For this purpose the fit applied to the entire reaction was used, but the error was determined in the respective conversion interval.
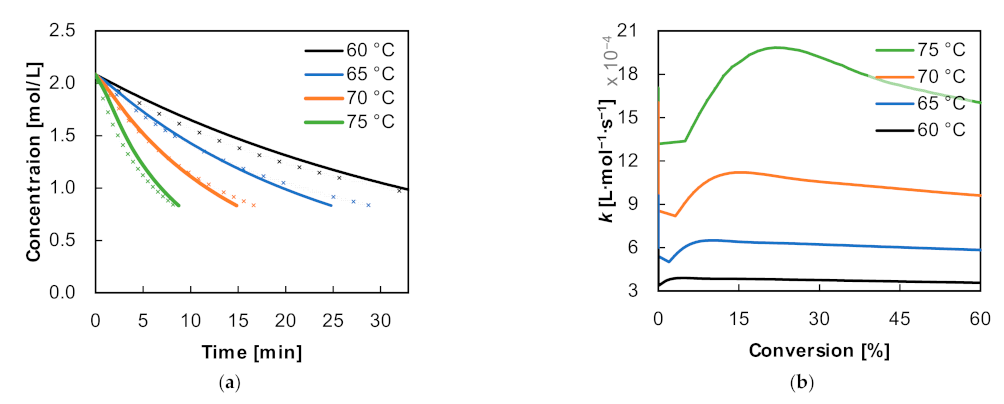
It can be concluded from the graph in
Figure 5a that the local fit errors for
n = 1, for the fit up to a 20% conversion to the fit up to a 70% conversion, are similar in magnitude. However, if the error of these fits is determined for the whole reaction range (>95% conversion) (local fit, global error), a constant yet significantly higher error can be achieved when fitting at least up to a conversion of 30%. Higher errors are to be expected for fits up to higher conversions due to the increasing relevance of the Raman measurement fluctuations. The error for the fit up to a conversion of 10% is not displayed on the graph because of its high value of 0.54. If the error of the global fit for the reaction order
n > 1 is considered in the respective conversion ranges, a constantly decreasing error results, which on the one hand is always lower than the global error for the fits according to reaction order 1 and from a fit up to 80% conversion is also lower than the corresponding local error for
n = 1.
As an example, for fitting the concentration profiles using the first order, the fit up to a conversion of 30% is shown. Up to that point an approximately constant local and global error can be obtained. This is shown in
Figure 5b. It shows that although the beginning of the reaction can be well represented, the monomer content is overestimated towards the middle of the reaction and underestimated in the end. This error increases with increasing temperature. Explanations as to why a reaction order of 1 cannot be assumed are discussed in more detail in the following.
The raw data was fitted using the integrated differential equation for reactions of any reaction order
n with [
AA]
0 and [
AA] denoting the initial and current acrylic acid concentration, respectively, and
kn being the reaction rate constant:
Here, the average deviation from the calculated concentration to the fitted ideal kinetics according to Equation (2) is 4%. Since the data can be well represented by ideal kinetics, it can be concluded that the Trommsdorff–Norrish effect plays a subordinate role in the performed reactions. This suggests that the expected chain-regulation function of the solvent could provide for low molar masses and therefore uniform mixing. Based on the accuracy of fit of the data in
Figure 4, the conversion of the reactions can be calculated by dividing each concentration data point by the maximum measured concentration, shown in
Figure 6.
The conversion chart shows that for 0.5 mol% of initiator used, a conversion of 80% was reached after 151, 111, 76 and 59 min, respectively, for each temperature. Doubling the AIBN concentration to 1.0 mol% led to noticeably steeper curves, shortening the reaction time up to an 80% conversion 1.5- to 3-fold and allowing for a conversion close to 100% after the displayed 180 min for 70 °C and 75 °C.
In free radical homopolymerisations, the polymerisation rate
Rp is defined as the decay of the monomer concentration over time. In complex forms of polymerisations where the steady-state radical concentration also depends on the monomer concentration [
M], the polymerisation rate is proportional to the initiator and monomer concentration with their respective reaction orders
α and
β [
10,
13,
26] and can be generally defined as:
In this study, AIBN was used as an initiator. Because it is widely used and well known for free radical polymerisations, it is safe to assume classical kinetic considerations. The square root proportionality of the propagation rate to the initiator concentration (
α = 0.5) is implied [
27]. Since the polymerisations shown in this work were also carried out at three different initiator concentrations, this assumption can also be confirmed by the straight line in
Figure 7. Ideally, the straight line in the graph would intersect the origin, which is not the case here. This is presumably due to the fact that no lower initiator concentrations than 0.5 mol% were used. Another possible reason would be that radicals are being scavenged by, e.g., MEHQ (4-methoxyphenol), which was included in the acrylic acid as a stabiliser. However, this effect should be negligible in the absence of dissolved molecular oxygen [
28] and was also confirmed again for our experimental set-up (
Figure A2).
From this, the rate constant
k is defined as a product of the chain propagation rate constant
kp with the classical description for the concentration of growing chains depending on the square root of the initiator concentration [
27]. Here,
f is the initiator efficiency,
kd and
kt stand for the initiator decomposition and polymer chain termination rate constant, respectively:
It is worth noting, however, that kinetics in the case of AA polymerisations are much more complex. Because growing AA polymer chains undergo backbiting and transfer reactions, it is important to differentiate between secondary and tertiary radicals and their different rate constants [
3]. Additionally, it has been found that the polymerisation rate of AA is highly dependent on the degree of neutralisation [
2,
4,
10,
11,
14]. This is explained by the increase of ionised species with an increasing pH, resulting in different types of propagation rate constants between the differently ionised species [
2,
14]. In our study, the acrylic acid solution was not neutralised and only a single initial monomer concentration was used. Nonetheless, the pH value was measured throughout the reaction. It should be noted that the pH value obtained here cannot be directly compared with the meaning of the pH value in aqueous solutions [
29], but it serves as a reproducible reference value in the experimental procedure. In the beginning, the solution of AA in isopropanol (150 g/L) had a pH of 3.0. After 15 min (60 °C, 0.5 mol% AIBN), the pH had risen to 3.6 and a final value of 4.0 was reached at the end of the reaction.
While the classical assumption for the monomer reaction order
β in Equation (3) would suggest a value of 1, previous studies have suggested orders above 1 due to an interaction of the monomer with the initiator itself or its caged radicals [
10]. While Cutié et al., Manickam et al. and Kabanov et al. found the order to be 1.5 [
10,
11,
30,
31], others such as Ito et al. and Scott et al., respectively, suggested values of 2 and 1.16–1.25 [
2,
32]. In this work, we could confirm a reaction order of 1.73 ± 0.15 in isopropyl alcohol across three different initiator concentrations. The resulting data from the experiments for the reaction order
n and
kn are listed in
Table 1.
In comparison to data found in the literature [
10,
11,
31,
32,
33] for the acidic (non-neutralised) AA polymerisation in water across different pH values at mostly temperatures between 50–60 °C, the rate constants found for isopropyl alcohol in this work are noticeably (up to 10-fold) slower. In acidic water (pH < 4) with different initiators and different concentrations thereof, rate constants were ranging from 0.14 to 0.90 L·mol
−1·min
−1 at 50–60 °C [
31,
32,
33]. The observed rates in isopropanol seem more comparable to neutralised AA polymerisations in water, ranging from 0.003 to 0.045 L·mol
−1·min
−1, also at 50–60 °C [
31,
32,
33]. However, it must be kept in consideration that AA polymerisation is strongly pH-dependent, but that the pH scale also behaves differently in solvents other than water [
29].
Using the data from
Figure 4 and
Figure 6, the rate of polymerisation can additionally be shown as a function of conversion (
Figure 8).
This graph shows that the maximum polymerisation rate was also the initial polymerisation rate. Initial polymerisation rates of 0.1–0.04 mol⸱L
−1⸱min
−1 with 0.5 mol% of AIBN and 0.35–0.06 mol⸱L
−1⸱min
−1 with 1.0 mol% of AIBN were observed, displaying a strong temperature dependence. The rates decreased with an increasing conversion, corresponding to the depletion of AA monomers. The commonly known autoacceleration of the polymerisation rate after the start of polymerisation due to the gel-effect [
34] was not present here due to the short polymer chains. Rather, an almost linear decrease in the polymerisation rate, especially at 60 °C, could be observed.
From the initial polymerisation rates,
k values were calculated according to Equation (3) and their natural logarithm was plotted against the reciprocal reaction temperatures to solve the Arrhenius equation. Due to the fact that the classical Arrhenius plot could be distorted by the temperature drop caused by the initiator addition and the subsequent temperature stroke, the Arrhenius parameters were estimated within a basic simulation using Predici 11. For this purpose, the reactions carried out were fed into Predici with their recipes and the experimental temperature and concentration curves. Based on the values obtained from the Arrhenius plots, the initial values for the parameter estimation were determined. The algorithm tried to map the experimental concentration profiles with the simulated ones up to a conversion of 60% (to cover the temperature profiles deviating from the ideal temperatures in
Figure 3). This is shown in
Figure 9 together with the development of the
k values.
The parameter estimation resulted in activation energies
Ea of 57.6 ± 0.8 kJ/mol for 0.5 mol% of AIBN and 88.5 ± 1.5 kJ/mol for 1.0 mol% of AIBN. These values are lower than the activation energies determined by the classical linear regression (65 ± 11 kJ/mol for 0.5 mol% of AIBN and 102 ± 14 kJ/mol for 1.0 mol% of AIBN). The frequency factors were estimated to be 2.69 × 10
5 ± 3.80 × 10
4 L·mol
−1·s
−1 and 2.65 × 10
10 ± 6.61 × 10
9 L·mol
−1·s
−1, respectively. In the literature, reaction temperatures starting at 70 °C and above have showed a shift in the Arrhenius plot, resulting in different slopes [
2,
30]. The higher temperature most likely induced a higher solution viscosity and thus an autoaccelerating behaviour. In this work, this was circumvented by the chain transferring solvent. Nevertheless, the activation energies found in this work are in accordance with the literature data, which lie between 40.1 and 98.1 kJ/mol [
2,
10,
11,
13,
30].
3.3. Calorimetry: Heat of Reaction Curves, Reaction Enthalpy
The reaction calorimetry, which was used to complement the Raman spectroscopy, is another important analytical tool to evaluate the safety of chemical reactions and optimise industrial processes. Especially since the thermal runaway of acrylic acid polymerisations have caused many accidents in the chemical industry, it is important to know and understand thermal behaviour during controlled polymerisations. A detailed analysis of runaway scenarios of bulk acrylic acid has already been performed using accelerating rate calorimetry (ARC) by Fujita et al. [
15].
The reaction heat was recorded for the polymerisation of acrylic acid in isopropanol and is shown for different temperatures in
Figure 10. The time point
t = 0 does not correspond to the respective start of the polymerisation but is set so that the negative heat input of the injection of the initiator solution is shown. In addition, the time for each curve was shifted to align with the reaction start according to the calorimetric global conversion.
It was shown that the initiator injection with differently pre-warmed initiator suspensions led to a drop in the reaction heat by up to 100 W in the worst case. This was due to the difficulties in handling the initiator suspension and could be avoided by using an initiator that dissolves well in isopropyl alcohol, such as V-601 (Dimethyl 2,2′-azobis(2-methylpropionate)). Nevertheless, it can be observed that the maximum reaction heat increased with an increasing temperature. Additionally, the maximum heat was reached later with higher temperatures. A drastic increase occurred at temperatures above 65 °C as expected due to this being the 10-h half-life decomposition temperature of AIBN. The heat of the reaction curves did not reach 0, indicating that the reaction never came to a halt and thus the conversion could not reach 100%. As expected, due to the higher conversion and hence higher viscosity, the final heat was lower for higher temperatures. In the end all reactions were so slow that the heat recordings were stopped prematurely, and the end-point calibrations of the heat flow were allowed, leading to minor deviations. Diagrams for the heat of reaction were not shown in the literature for the polymerisation of AA in neither water nor isopropanol.
From the plotted reaction heat, the integral under the curves can be calculated. With respect to the amount of monomer used, this corresponds to the enthalpy generated during the reaction Δ
HR. Using the conversion determined by Raman spectroscopy, the values obtained were related to a reaction turnover of 100% and the average was calculated. Thus, for the polymerisation of acrylic acid in IPA at 0.5, 1.0 and 1.5 mol% of AIBN in the temperature range of 60–75 °C, a reaction enthalpy of 67.37 ± 4.83 kJ/mol was determined. This value is in accordance with the data for the aqueous polymerisation found in the literature. It should also be noted that, in general, the polymerisations with higher initiator concentrations also gave slightly higher results for the enthalpy of reaction. Staudinger and Schläpfer determined a heat of combustion of 62.76 kJ/mol in 1939 [
35]. In 1947, Evans and Tyrall [
36] then obtained a value of 77.36 kJ/mol by calorimetric determination in water. For comparative reasons, reactions in water were also carried out in this work using isopropanol only as a chain-transferring agent. These were carried out in the temperature range of 40–55 °C with 0.5 mol% VA-057 as the initiator. The recorded reaction enthalpy was 75.4 ± 3.6 kJ/mol, which aligns with the literature data as well but is marginally higher than the enthalpy found for the polymerisation in isopropanol.
Assuming that the polymerisation reaction is the main reaction occurring, the polymerisation rate
Rp can be defined as the quotient of the reaction heat
and the reaction enthalpy −Δ
H [
37]:
Furthermore, the calorimetric global conversion
Xcalor can be calculated by integrating the polymerisation rate over the time
t and dividing it by the initially fed amount of monomer
M0 [
37,
38]:
The calorimetric conversion was used here as an aid to determine the start of polymerisation in the plotted curves for reaction heat. However, since only Raman spectroscopic conversion determination was used for the evaluations within this article, the following figure compares the conversion curves that can be obtained from calorimetry with those from Raman spectroscopy.
Figure 11 shows an example of this for the polymerisations in isopropanol with 1.0 mol% AIBN. It is noticeable that the conversion curves coincide somewhat better at higher temperatures than at lower temperatures. The course at 75 °C is almost completely congruent. Since isothermal calorimetry in this case involved small differences between
TR and
TJ, and only Raman spectroscopy specifically measures the decrease in the monomer band in the reaction mass, deviations are conceivable here.
3.4. Effects on Molecular Weight
Since the polymerisation of AA was carried out in a chain-transferring solvent, it is to be expected that polymers with low molecular weights were produced. At the end of each of the performed reactions, a sample was taken from the reactor outlet and analysed via size exclusion chromatography (SEC). In addition, polymerisations in water with the addition of 2-propanol (IPA/AA = 0.16) as a chain-transferring agent (CTA) were carried out for comparison purposes. A reaction in pure water (without CTA) was also meant to serve as a further comparison, but under comparable conditions a highly viscous reaction mass was produced in the batch reactor. Because of this, neither the stirrer could ensure reliable mixing, nor could the Raman spectra provide trustworthy spectra, since the measurements could only show one spot in an unevenly mixed medium. Comparative literature data from AA polymerisation in water or in water with the addition of a CTA were used but these were mostly carried out in a continuous or semi-batch process with different initiators and concentrations. Moreover, in contrast to the literature, here the molecular weight was considered after close to complete conversion for the most part.
In the following
Figure 12, first the influence of the initiator concentration, as well as the solvent on the molecular weight is shown.
First, it is shown in
Figure 12a that the number-average molecular weight is relatively low at 1.5 × 10
4 g/mol. No effect of initiator concentration was observed here for different initiator concentrations of 0.5, 1.0 and 1.5 mol% of the AIBN. The right part of the figure shows the corresponding molar mass distribution. The reaction in pure isopropanol led to a distribution with an average PDI of 1.64 ± 0.05 (1.67 in
Figure 12b). This is compared to different solvents under similar reaction conditions. The polymerisation in water with the addition of isopropanol as the CTA (IPA/AA = 0.16) is not much broader with an average PDI of 2.06 ± 0.31 (1.77 in
Figure 12b), but the molecular weight is 25 times higher (
Mn = 3.8 × 10
5 g/mol) than the molecular weight of the polymers made in pure isopropanol. The molar mass distribution of the reaction in pure water could not be directly compared with the other results here, as it did not show a completely unimodal distribution. Additionally, the observations during the reaction and sample preparation indicated a very high gel fraction, which presumably was adhered to the guard column during the SEC measurement due to reduced solubility in the eluent. In the literature, values for
Mn range up to 2 × 10
6 g/mol during AA polymerisation in water [
10].
Figure 13 shows the effect of temperature and initial monomer concentration on molecular weight.
In both graphs an expected linear dependence can be observed. Additional graphs for the temperature dependence of the polymerisations with 0.5 mol% and 1.5 mol% of AIBN are shown in
Figure A1. Here, the initial monomer concentration had a stronger influence on the molecular weight.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}