
Phase Transfer Catalysts and Role of Reaction Environment in Nucleophilc Radiofluorinations in Automated Synthesizers


Abstract
:1. Introduction
2. [18F]Fluoride Activation for Aliphatic Nucleophilic SN2 Substitution
2.1. Azeotropic Drying Free Methods for Kryptofix-Mediated Radiofluorinations
2.2. Tetraalkylammonium Salts (Bu4N+, Et4N+) as the PTCs
2.3. PTC Free Cartridge-Based Radiofluorination of Aliphatic Substrates
3. [18F]Fluoride Activation for Conventional Aromatic Nucleophilic Substitution
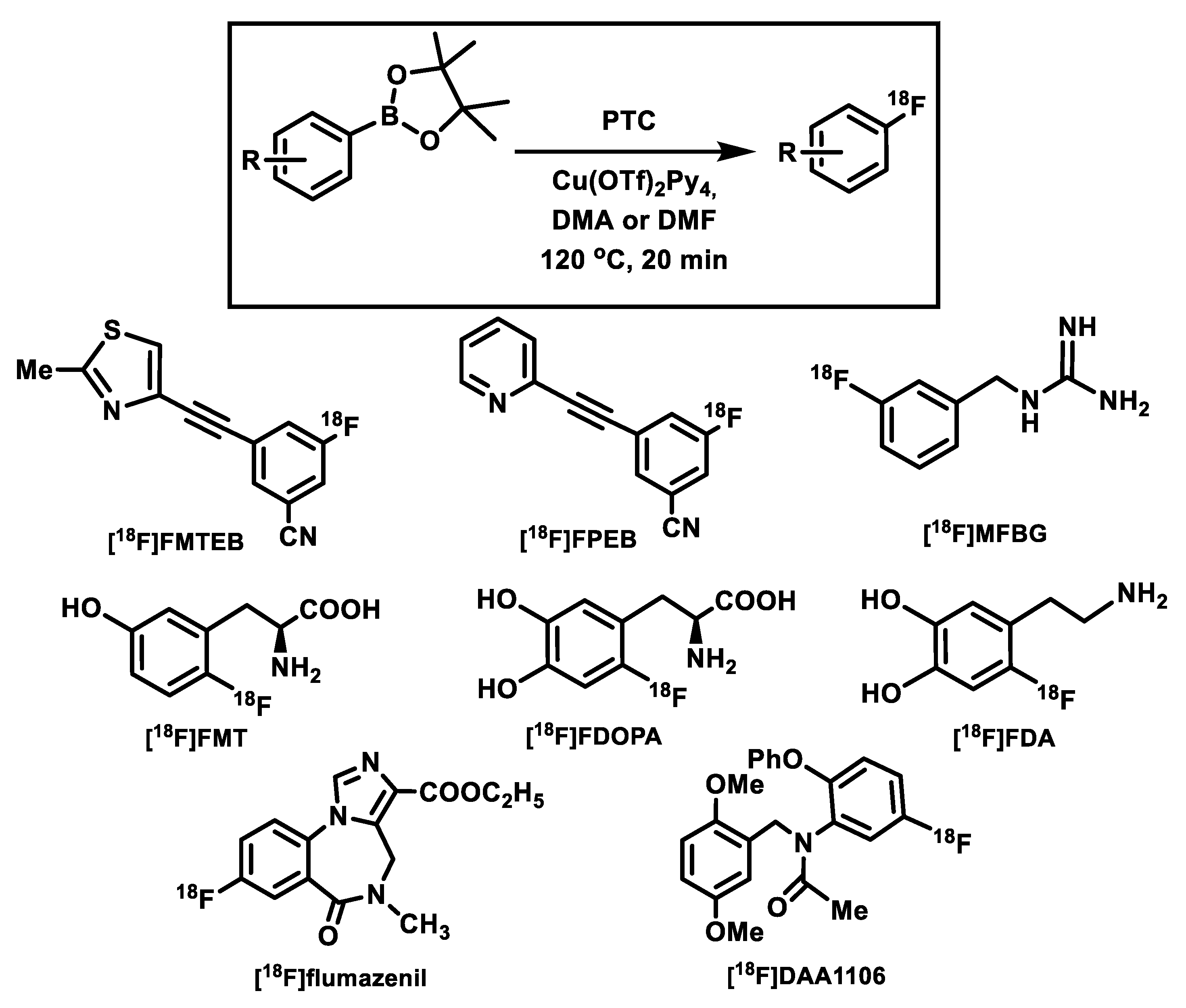
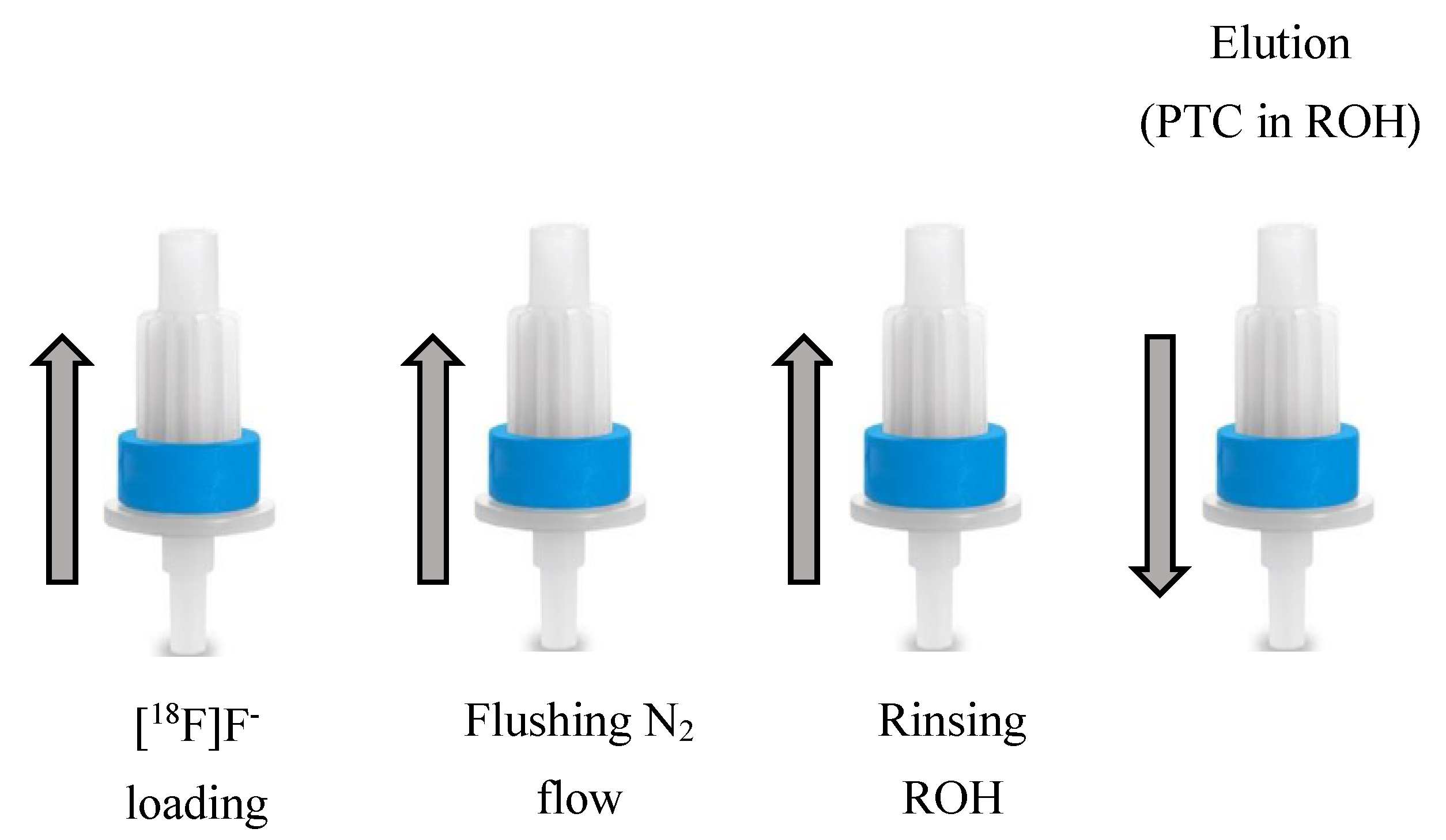
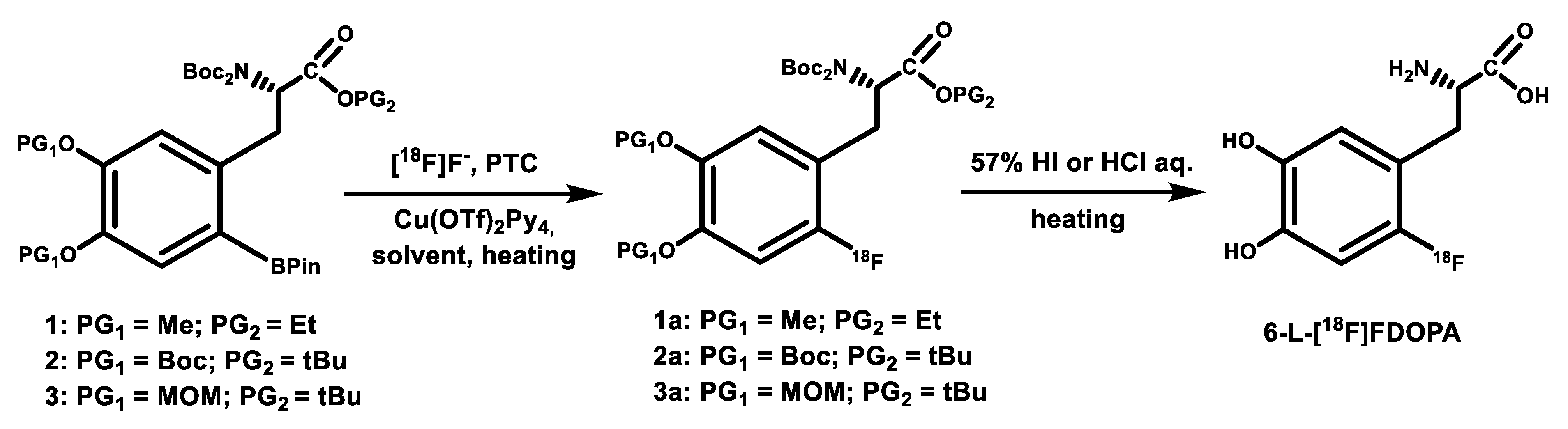
4. Copper-Mediated Late-Stage Radiofluorination of Non-Activated Arenes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eberl, S.; Eriksson, T.; Svedberg, O.; Norling, J.; Henderson, D.; Lam, P.; Fulham, M.J. High Beam Current Operation of a PETtraceTM Cyclotron for 18F− Production. Appl. Radiat. Isot. 2012, 70, 922–930. [Google Scholar] [CrossRef]
- Cai, L.; Lu, S.-Y.; Pike, V.W. Chemistry with [18F]Fluoride Ion. Eur. J. Org. Chem. 2008, 17, 2853–2873. [Google Scholar] [CrossRef]
- Coenen, H.H.; Ermert, J. 18F-labelling innovations and their potential for clinical application. Clin. Trans. Imaging 2018, 6, 16. [Google Scholar] [CrossRef]
- Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S.H. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed. 2019, 58, 2580–2605. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2014, 26, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schlyer, D.J.; Bastos, M.A.V.; Alexoff, D.; Wolf, A.P. Separation of [18F]fluoride from [18O]water using anion exchange resin. Appl. Radiat. Isot. 1990, 41, 531–533. [Google Scholar] [CrossRef]
- Sachinidis, J.I.; Poniger, S.; Tochon-Danguy, H.J. Automation for Optimised Production of Fluorine-18-Labelled Radiopharmaceuticals. Curr. Radiopharm. 2010, 3, 248–253. [Google Scholar] [CrossRef]
- Hjelstuen, O.K.; Svadberg, A.; Olberg, D.E.; Rosser, M. Standardization of fluorine-18 manufacturing processes: New scientific challenges for PET. Eur. J. Pharm. Biopharm. 2011, 78, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasikova, R. PET Radiochemistry automation: State of the art and future trends in 18F-nucleophilic fluorination. Curr. Org. Chem. 2013, 17, 2097–2107. [Google Scholar] [CrossRef]
- Coenen, H.H.; Schuller, M.; Stocklin, G.; Klatte, B.; Knochel, A. Preparation of N.C.A. [17-18F]-fluoroheptadecanoic acid in high yields via aminopolyether supported, nucleophilic fluorination. J. Labelled. Compds. Radiopharm. 1986, 23, 455–466. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.; Stocklin, G. Efficient stereospecifc synthesis of no-carrier-added 2-[F-18]-fuoro-2-deoxy-d-glucose using aminopolyether supported nucleophilic-substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar] [PubMed]
- Coenen, H.H.; Elsinga, P.H.; Iwata, R.; Kilbourn, M.R.; Pillai, M.R.; Rajan, M.G.; Wagner, H.N.; Zagnun, J.J. Fluorine-18 radiopharmaceuticals beyond [18F] FDG for use in oncology and neurosciences. Nucl. Med. Biol. 2010, 37, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Brodack, J.W.; Kilbourn, M.R.; Welch, M.J.; Katzenellenbogen, J.A. NCA 16 alpha-[18F]fluoroestradiol-17 beta: The effect of reaction vessel on fluorine-18 resolubilization, product yield, and effective specific activity. Int. J. Rad. Appl. Instrum. A 1986, 37, 217–221. [Google Scholar] [CrossRef]
- Zhang, M.-R.; Suzuki, K. [18F]Fluoroalkyl agents: Synthesis, reactivity and application for development of PET ligands in molecular imaging. Curr. Top. Med. Chem. 2007, 7, 1817–1828. [Google Scholar] [CrossRef]
- Van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.G. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomzina, N.A.; Zaitsev, V.V.; Krasikova, R.N. Optimization of nucleophilic fluorination step in the synthesis of various compounds labelled with fluorine-18 for their use as PET radiotracers. J. Label. Compds. Radiopharm. 2001, 44 (Suppl. S1), S895–S897. [Google Scholar] [CrossRef]
- Gomzina, N.A.; Vassiliev, D.A.; Krasikova, R.N. Optimization of robotic preparation of 2-[18F]fluoro-2-deoxy-D-glucose based on alkali hydrolysis. Radiochemistry 2002, 44, 366–372. [Google Scholar] [CrossRef]
- Fedorova, O.S.; Vaitekhovich, F.P.; Krasikova, R.N. Automated synthesis of [18F] fluoromethylcholine for positron emission tomography imaging. Pharm. Chem. J. 2018, 52, 730–734. [Google Scholar] [CrossRef]
- Fedorova, O.; Nikolaeva, V.; Krasikova, R. Automated SPE-based synthesis of 16α-[18F]fluoroestradiol without HPLC purification step. Appl. Radiat. Isot. 2018, 141, 57–63. [Google Scholar] [CrossRef]
- Li, G.Y.; Vaulina, D.D.; Li, J.J.; Fedorova, O.S.; Wang, H.E.; Liu, R.S.; Krasikova, R.N.; Chen, C.L. Synthesis and biological evaluation of 2-(3,4-dimethoxyphenyl)-6-(2-[18F]fluoroethoxy)benzothiazole ([18F]FEDBT) for PET imaging of breast cancer. Bioorg. Med. Chem. Lett. 2017, 27, 3460–3463. [Google Scholar] [CrossRef]
- Ryzhikov, N.N.; Seneca, N.; Krasikova, R.N.; Gomzina, N.A.; Shchukin, E.; Fedorova, O.S.; Vassiliev, D.A.; Gulyás, B.; Hall, H.; Savic, I.; et al. Preparation of high specific radioactivity [18F]flumazenil and its evaluation in cynomolgus monkey by positron emission tomography. Nucl. Med. Biol. 2005, 32, 109–116. [Google Scholar] [CrossRef]
- Stepanov, V.; Krasikova, R.; Raus, L.; Loog, O.; Hiltunen, J.; Halldin, C. An Efficient One-Step Radiosynthesis of [18F]FEPE2I, a PET Radioligand for Imaging of Dopamine Transporters. J. Label. Compd. Radiopharm. 2012, 55, 206–210. [Google Scholar] [CrossRef]
- Arakawa, R.; Takano, A.; Stenkrona, P.; Stepanov, V.; Nag, S.; Jahan, M.; Grybäck, P.; Bolin, M.; Chen, L.; Zhang, L.; et al. PET imaging of beta-secretase 1 in the human brain: Radiation dosimetry, quantification, and test-retest examination of [18F]PF-06684511. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2429–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnema, S.J.; Stepanov, V.; Nakao, R.; Sromek, A.W.; Zhang, T.; Neumeyer, J.L.; George, S.R.; Seeman, P.; Stabin, M.G.; Jonsson, C.; et al. (18)F-MCL-524, an (18)F-Labeled Dopamine D2 and D3 Receptor Agonist Sensitive to Dopamine: A Preliminary PET Study. J. Nucl. Med. 2014, 55, 1164–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogni, A.; Laera, L.; Cucchi, C.; Iwata, R.; Seregni, E.; Pascali, C. An improved automated one-pot synthesis of O’-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET) based on a purification by cartridges. Nucl. Med. Biol. 2019, 72–73, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, C.; Bogni, A.; Casanova, C.; Seregini, E.; Pascali, C. An improved one-pot preparation of [18F]FMISO based on solid phase extraction purification: Pitfalls on the analytical method reported in the Ph.Eur.’s monograph. J. Label. Compd. Radiopharm. 2021, 1–7. [Google Scholar] [CrossRef]
- Wessmann, S.H.; Henriksen, G.; Wester, H.J. Cryptate mediated nucleophilic 18F-fluorination without azeotropic drying. Nuklearmedizin 2012, 51, 1–8. [Google Scholar] [CrossRef]
- Wurzer, A.; Di Carlo, D.; Schmidt, A.; Beck, R.; Eiber, M.; Schwaiger, M.; Wester, H.-J. Radiohybrid Ligands: A Novel Tracer Concept Exemplified by 18F- or 68Ga-Labeled rhPSMA Inhibitors. J. Nucl. Med. 2020, 61, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Kostikov, A.P.; Chin, J.; Orchowski, K.; Niedermoser, S.; Kovacevic, M.M.; Aliaga, A.; Jurkschat, K.; Wängler, B.; Wängler, C.; Wester, H.J.; et al. Oxalic acid supported Si-18F-radiofluorination: One-step radiosynthesis of N-succinimidyl 3-(di-tert-butyl[18F]fluorosilyl)benzoate ([18F]SiFB) for protein labeling. Bioconjug Chem. 2012, 3, 106–114. [Google Scholar] [CrossRef]
- Iwata, R.; Pascali, C.; Terasaki, K.; Ishikawa, Y.; Furumoto, S.; Yanai, K. Minimization of the amount of Kryptofx 222—KHCO3 for applications to microscale 18F-radiolabeling. Appl. Radiat. Isot. 2017, 125, 113–118. [Google Scholar] [CrossRef]
- Song, R.; Tago, T.; Tatsuta, M.; Shiraishi, N.; Iwai, K.; Hirano, K.; Toyohara, D.; Tanaka, H. N-Alkyl 3-aminobut-2-enenitrile as a Non-radioactive Side Product in Nucleophilic 18F-Fluorination. ChemistrySelect 2021, 6, 2826–2831. [Google Scholar] [CrossRef]
- Kwon, Y.D.; Son, J.; Yun, M.J.; Chun, J.H. Azeotropic drying-free aliphatic radiofuorination to produce PET radiotracers in a mixed organic solvent system. Tetrahedron Lett. 2018, 59, 2848–2852. [Google Scholar] [CrossRef]
- Aerts, J.; Voccia, S.; Lemaire, C.; Giacomelli, F.; Goblet, D.; Thonon, D.; Plenevaux, A.; Warnock, G.; Luxen, A. Fast production of highly concentrated reactive [18F] fluoride for aliphatic and aromatic nucleophilic radiolabelling. Tetrahedron Lett. 2010, 51, 64–66. [Google Scholar] [CrossRef]
- Culbert, P.A.; Adam, M.J.; Hurtado, E.T.; Huser, J.M.A.; Jivan, S.; Lu, J.; Ruth, T.J.; Zeisler, S.K. Automated synthesis of [18F]FDG using tetrabutylammonium bicarbonate. Appl. Radiat. Isot. 1995, 46, 887–891. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H. Efficient routine production of the 18F-labelled amino acid O’-(2-[18F]fluoroethyl)-L-tyrosine. Appl. Radiat. Isot. 2002, 57, 853–856. [Google Scholar] [CrossRef]
- Krasikova, R.N.; Kuznetsova, O.F.; Fedorova, O.S.; Maleev, V.I.; Savel’eva, T.F.; Belokon, Y.N. No carrier added synthesis of O-(2′-[18F]fluoroethyl)-L-tyrosine via a novel type of chiral enantiomerically pure precursor, NiII complex of a (S)-tyrosine Schiff base. Bioorg. Med. Chem. 2008, 16, 4994–5003. [Google Scholar] [CrossRef]
- Jiang, X.; Li, Y.; Wang, X.; Shen, T.; Li, X.; Yao, Y.; Zhang, G.; Kou, Y.; Shen, J.; Luo, Z.; et al. Quick Automatic Synthesis of Solvent-Free 16α-[18F]Fluoroestradiol: Comparison of Kryptofix 222 and Tetrabutylammonium Bicarbonate. Front. Oncol. 2020, 10, 577979. [Google Scholar] [CrossRef]
- Krasikova, R.N.; Kuznetsova, O.F.; Fedorova, O.S.; Belokon, Y.N.; Maleev, V.I.; Mu, L.; Ametamey, S.; Schubiger, P.A.; Friebe, M.; Berndt, M.; et al. 4-[18F]fluoro glutamic acid (BAY 85-8050)—A new amino acid radiotracer for PET imaging of tumors: Synthesis and in vitro characterization. J. Med. Chem. 2011, 54, 406–410. [Google Scholar] [CrossRef]
- Brichard, L.; Aigbirhio, F.I. An Efficient Method for Enhancing the Reactivity and Flexibility of [18F]Fluoride Towards Nucleophilic Substitution Using Tetraethylammonium Bicarbonate. Eur. J. Org. Chem. 2014, 28, 6145–6149. [Google Scholar] [CrossRef]
- Cybulska, K.A.; Bloemers, V.; Perk, L.R.; Laverman, P. Optimised GMP-compliant production of [18F]DPA-714 on the Trasis AllinOne module. EJNMMI Radiopharm. Chem. 2021, 6, 20. [Google Scholar] [CrossRef]
- Bratteby, K.; Shalgunov, V.; Herth, M. Aliphatic 18F-Radiofluorination: Recent Advances in the Labeling of Base-Sensitive Substrates. ChemMedChem. 2021, 16, 2612–2622. [Google Scholar] [CrossRef]
- Orlovskaya, V.; Antuganov, D.; Fedorova, O.; Timofeev, V.; Krasikova, R. Tetrabutyl ammonium tosylate as inert phase-transfer catalyst: The key to high efficiency SN2 radiofluorinations. Appl. Radiat. Isot. 2020, 163, 109195. [Google Scholar] [CrossRef]
- Bratteby, K.; Shalgunov, V.; Battisti, U.M.; Petersen, I.N.; Broek, S.L.; Ohlsson, T.; Gillings, N.; Erlandsson, M.; Herth, M. Insights into Elution of Anion Exchange Cartridges: Opening the Path towards Aliphatic 18F-Radiolabeling of Base-Sensitive Tracers. ACS Pharmacol. Transl. Sci. 2021, 4, 1556–1566. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Akurathi, V.; Sromek, A.W.; Chen, Y.; Neumeyer, J.L.; Packard, A.B. A non-anhydrous, minimally basic protocol for the simplification of nucleophilic 18F-fluorination chemistry. Sci. Rep. 2020, 10, 6818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.W.; Lee, B.S.; Lee, S.J.; Oh, S.J.; Chi, D.Y. Fast and easy drying method for the preparation of activated [18F]fluoride using polymer cartridge. Bull. Korean Chem. Soc. 2011, 32, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-mediated aromatic radiofluorination revisited: Efficient production of PET tracers on a preparative scale. Chem. Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Orlovskaya, V.; Fedorova, O.; Nadporojskii, M.; Krasikova, R. A fully automated azeotropic drying free synthesis of O-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET) using tetrabutylammonium tosylate. Appl. Radiat. Isot. 2019, 152, 135–139. [Google Scholar] [CrossRef]
- Fedorova, O.S.; Orlovskaya, V.V.; Nadporojskii, M.; Krasikova, R.N. Automated synthesis of the 16α-[18F]fluoroestradiol ([18F]FES): Minimization of precursor amount and resulting benefits. Radiochim. Acta 2020, 108, 979–988. [Google Scholar] [CrossRef]
- Bratteby, K.; Denholt, C.L.; Lehel, S.; Petersen, I.N.; Madsen, J.; Erlandsson, M.; Ohlsson, T.; Herth, M.; Gillings, N. Fully Automated GMP-Compliant Synthesis of [18F]FE-PE2I. Pharmaceuticals 2021, 14, 601. [Google Scholar] [CrossRef]
- Inkster, J.A.H.; Sromek, A.W.; Akurathi, V.; Neumeyer, J.L.; Packard, A.B. The Non-Anhydrous, Minimally Basic Synthesis of the Dopamine D2 Agonist [18F]MCL-524. Chemistry 2021, 3, 1047–1056. [Google Scholar] [CrossRef]
- Toorongian, S.A.; Mulholland, K.; Jewett, D.M.; Bachelor, M.A.; Kilbourn, M.R. Routine production of 2-deoxy-2-[18F]fluoro-D-glucose by direct nucleophilic exchange on a quaternary 4-aminopyridinium resin. Int. J. Radiat. Appl. Instrum. B 1990, 17, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Kuge, Y.; Tsukamoto, E.; Katoh, C.; Seki, K.; Ohkura, K.; Ohmiya, Y.; Nishijima, K.; Tanaka, A.; Sasaki, M.; Tamaki, N. Synthesis of 18F-FDG with FDG MicroLab system: Basic studies for clinical application. Jpn. J. Nucl. Med. 1999, 36, 873–878. [Google Scholar] [PubMed]
- Mathiessen, B.; Zhuravlev, F. Automated Solid-Phase Radiofluorination Using Polymer-Supported Phosphazenes. Molecules 2013, 18, 10531–10547. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, C.F.; Aerts, J.; Voccia, S.; Libert, L.C.; Mercier, F.; Goblet, D.; Plenevaux, A.R.; Luxen, A.J. Fast Production of Highly Reactive No-Carrier-Added [18F]Fluoride for the Labeling of Radiopharmaceuticals. Angew. Chem. Int. Ed. 2010, 49, 3161–3164. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Liang, S.H. Aliphatic [18F]Fluorination Chemistry for Positron Emission Tomography. In Fluorination, Synthetic Organofluorine Chemistry 1; Hu, J., Umemoto, T., Eds.; Springer Nature Singapore Pte Ltd.: Singapore, 2018. [Google Scholar] [CrossRef]
- Liang, S.H.; Vasdev, N. Aliphatic 18F Bond Formation via Transition Metal Based [18F]Fluorination. Angew. Chem. Int. Ed. Engl. 2014, 53, 11416–11418. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.; Lee, S.J.; Jackson, I.M.; Ichiishi, N.; Brooks, A.F.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]-γ-Fluoro-α,β-unsaturated Esters and Ketones via Vinylogous 18F-Fluorination of α-Diazoacetates with [18F]AgF. Synthesis 2019, 51, 4401–4407. [Google Scholar] [CrossRef]
- Lemaire, C.; Cantineau, R.; Guillaume, M.; Plenevaux, A.; Christiaens, L. Fluorine-18-altanserin: A radioligand for the study of serotonin receptors with PET: Radiolabeling and in vivo biologic behavior in rats. J. Nucl. Med. 1991, 32, 2266–2272. [Google Scholar] [PubMed]
- Hasler, F.; Kuznetsova, O.F.; Krasikova, R.N.; Cservenyak, T.; Quednow, B.B.; Vollenweider, F.X.; Ametamey, S.M.; Westera, G. GMP-compliant radiosynthesis of [18F]altanserin and human plasma metabolite studies. Appl. Radiat. Isot. 2009, 67, 598–601. [Google Scholar] [CrossRef] [Green Version]
- Le, B.D.; Lemaire, C.; Ginovart, N.; Plenevaux, A.; Aerts, J.; Brihaye, C.; Hassoun, W.; Leviel, V.; Mekhsian, P.; Weissmann, D.; et al. High-yield radiosynthesis and preliminary in vivo evaluation of p-[18F]MPPF, a fuoro analog of WAY-100635. Nucl. Med. Biol. 1998, 25, 343–350. [Google Scholar] [CrossRef]
- Wong, D.F.; Waterhouse, R.; Kuwabara, H.; Kim, J.; Brašić, J.R.; Chamroonrat, W.; Stabins, M.; Holt, D.P.; Dannals, R.F.; Hamill, T.G.; et al. 18F-FPEB, a PET Radiopharmaceutical for quantifying metabotropic glutamate 5 receptors: A first-in-human study of radiochemical safety, biokinetics, and radiation dosimetry. J. Nucl. Med. 2013, 54, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Dollé, F. Fluorine-18-labelled fuoropyridines: Advances in radiopharmaceutical design. Curr. Pharm. Des. 2005, 11, 3221–3235. [Google Scholar] [CrossRef] [PubMed]
- Horti, A.; Koren, A.O.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; London, E.D.; Dannals, R.F. Synthesis of a radiotracer for studying nicotinic acetylcholine receptors: 2-[18F]Fluoro-3-(2(S)- azetidinylmethoxy)pyridine (2-[18F]F-A-85380). J. Label. Compds. Radiopharm. 1998, 41, 309–318. [Google Scholar] [CrossRef]
- Cselényi, Z.; Jönhagen, M.E.; Forsberg, A.; Halldin, C.; Julin, P.; Schou, M.; Johnström, P.; Varnäs, K.; Svensson, S.; Farde, L. Clinical validation of 18F-AZD4694, an amyloid-β-specific PET radioligand. J. Nucl. Med. 2012, 53, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, J.; Martin, R.; Remde, Y.; Schäfer, M.; Hienzsch, A.; Hübner, S.; Zerges, A.M.; Marx, H.; Hesse, R.; Weber, K.; et al. Procedures for the GMP-Compliant Production and Quality Control of [18F]PSMA-1007: A Next Generation Radiofluorinated Tracer for the Detection of Prostate Cancer. Pharmaceuticals 2017, 10, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olberg, D.E.; Arukwe, J.M.; Hjelstuen, O.K.; Solbakken, M.; Kindberg, G.M.; Cuthbertson, A. One Step Radiosynthesis of 6-[18F]Fluoronicotinic Acid 2,3,5,6-Tetrafluorophenyl Ester ([18F]F-Py-TFP): A New Prosthetic Group for Efficient Labeling of Biomolecules with Fluorine-18. J. Med. Chem. 2010, 53, 1732–1740. [Google Scholar] [CrossRef]
- Basuli, F.; Zhang, X.; Jagoda, E.M.; Choyke, P.L.; Swenson, R.E. Facile room temperature synthesis of fluorine-18 labeled fluoronicotinic acid-2,3,5,6-tetrafluorophenyl ester without azeotropic drying of fluorine-18. Nucl. Med. Biol. 2016, 43, 770–772. [Google Scholar] [CrossRef] [Green Version]
- Richarz, R.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Neumaier, B.; Zlatopolskiy, B.D. Neither azeotropic drying, nor base nor other additives: A minimalist approach to 18F-labeling. Org. Biomol. Chem. 2014, 12, 8094–8099. [Google Scholar] [CrossRef]
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P. Late-Stage [18F]fluorination: New Solutions to Old Problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Cole, E.L.; Stewart, M.N.; Littich, R.; Hoareau, R.; Scott, P. Radiosyntheses Using Fluorine-18: The Art and Science of Late Stage Fluorination. Curr. Top. Med. Chem. 2014, 14, 875–900. [Google Scholar] [CrossRef] [Green Version]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Clemente, G.S.; Elsinga, P.H.; Dömling, A. MCR Scaffolds Get Hotter With 18F-Labeling. Molecules 2019, 24, 1327. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.S.; Kaur, T.; Preshlock, S.; Tanzey, S.S.; Winton, W.P.; Sharninghausen, L.S.; Wiesner, N.; Brooks, A.F.; Sanford, M.S.; Scott, P. Copper-Mediated Late-Stage Radiofluorination: Five Years of Impact on Preclinical and Clinical PET Imaging. Clin. Transl. Imaging 2020, 8, 167–206. [Google Scholar] [CrossRef]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Cu-Catalyzed Fluorination of Diaryliodonium Salts With KF. Org. Lett. 2013, 15, 5134–5137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [Green Version]
- Pike, V.W.; Aigbirhio, F.I. Reactions of Cyclotron-Produced [18F]fluoride With Diaryliodonium salts—A novel Single-Step Route to No-Carrier-Added [18F]fluoroarenes. J. Chem. Soc. Chem. Commun. 1995, 21, 2215. [Google Scholar] [CrossRef]
- Pike, V.W. Hypervalent Aryliodine Compounds as Precursors for Radiofluorination. J. Label. Compd. Radiopharm. 2018, 61, 196–227. [Google Scholar] [CrossRef] [PubMed]
- Way, J.D.; Wuest, F. Automated radiosynthesis of no-carrier-added 4-[18F]fluoroiodobenzene: A versatile building block in 18F radiochemistry. J. Label. Compds. Radiopharm. 2014, 57, 104–109. [Google Scholar] [CrossRef]
- Vāvere, A.L.; Neumann, K.D.; Butch, E.R.; Hu, B.; DiMagno, S.G.; Snyder, S.E. Improved, one-pot synthesis of 6-[18 F]fluorodopamine and quality control testing for use in patients with neuroblastoma. J. Label. Comp. Radiopharm. 2018, 61, 1069–1080. [Google Scholar] [CrossRef]
- Kuik, W.-J.; Kema, I.P.; Brouwers, A.H.; Zijlma, R.; Neumann, K.D.; Dierckx, R.A.J.O.; DiMagno, S.G.; Elsinga, P.H. In Vivo Biodistribution of No-Carrier-Added 6-18F-Fluoro-3,4-Dihydroxy-L-Phenylalanine (18F-DOPA), Produced by a New Nucleophilic Substitution Approach, Compared With Carrier-Added 18F-L-DOPA, Prepared by Conventional Electrophilic Substitution. J. Nucl. Med. 2014, 56, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisonial, A.; Serre, A.; Ouadi, A.; Schmitt, S.; Canitrot, D.; Léal, F.; Miot-Noirault, E.; Brasse, D.; Marchand, P.; Chezal, J.M. Base/Cryptand/Metal-Free Automated Nucleophilic Radiofluorination of [18F]FDOPA from Iodonium Salts: Importance of Hydrogen Carbonate Counterion. Eur. J. Org. Chem. 2018, 2018, 7058–7065. [Google Scholar] [CrossRef]
- Krasikova, R.N. Nucleophilic Synthesis of 6-L-[18F]FDOPA. Is Copper-Mediated Radiofluorination the Answer? Molecules 2020, 25, 4365. [Google Scholar] [CrossRef] [PubMed]
- Orlovskaya, V.V.; Modemann, D.J.; Kuznetsova, O.F.; Fedorova, O.S.; Urusova, E.A.; Kolks, N.; Neumaier, B.; Krasikova, R.N.; Zlatopolskiy, B.D. Alcohol-Supported Cu-Mediated 18F-Fluorination of Iodonium Salts under “Minimalist” Conditions. Molecules 2019, 24, 3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Schimler, S.D.; Hanley, P.S.; Sanford, M.S. Cu(OTf)2-Mediated Fluorination of Aryltrifluoroborates with Potassium Fluoride. J. Am. Chem. Soc. 2013, 135, 16292–16295. [Google Scholar] [CrossRef] [PubMed]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Tredwell, M.; Huiban, M.; Hienzsch, A.; Gruber, S.; Wilson, T.C.; Taylor, N.J.; Cailly, T.; et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Orlovskaya, V.; Fedorova, O.; Kuznetsova, O.; Krasikova, R. Cu-Mediated Radiofluorination of Aryl Pinacolboronate Esters: Alcohols as Solvents with Application to 6-L-[18F]FDOPA Synthesis. Eur. J. Org. Chem. 2020, 45, 7079–7086. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Fedorova, O.; Orlovskaya, V.; Krasikova, R. Copper-mediated radiofluorination of aryl pinacolboronate esters: A straightforward protocol using pyridinium sulfonates. Eur. J. Org. Chem. 2019, 2019, 918–922. [Google Scholar] [CrossRef]
- Zhang, X.; Basuli, F.; Swenson, R.E. An azeotropic drying-free approach for copper-mediated radiofluorination without addition of base. J. Label. Comp. Radiopharm. 2019, 62, 139–145. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Ichiishi, N.; Makaravage, K.J.; Sanford, M.S.; Scott, P.J. Development of Customized [18F]Fluoride Elution Techniques for the Enhancement of Copper-Mediated Late-Stage Radiofuorination. Sci. Rep. 2017, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlatopolskiy, B.D.; Zischler, J.; Schäfer, D.; Urusova, E.A.; Guliyev, M.; Bannykh, O.; Endepols, H.; Neumaier, B. Discovery of 7-[18F]Fluorotryptophan as a Novel Positron Emission Tomography (PET) Probe for the Visualization of Tryptophan Metabolism in Vivo. Med. Chem. 2018, 61, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Neumaier, F.; Rüngeler, T.; Drewes, B.; Kolks, N.; Neumaier, B. Preparation of a First 18F-Labeled Agonist for M1 Muscarinic Acetylcholine Receptors. Molecules 2020, 25, 2880. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Endepols, H.; Krasikova, R.N.; Fedorova, O.S.; Neumaier, B. 11C- and 18F-labelled tryptophans as PET-tracers for imaging of altered tryptophan metabolism in age-associated disorders. Russ. Chem. Rev. 2020, 89, 879–896. [Google Scholar] [CrossRef]
- Craig, A.; Kolks, N.; Urusova, E.A.; Zischler, J.; Brugger, M.; Endepols, H.; Neumaier, B.; Zlatopolskiy, B.D. Preparation of labeled aromatic amino acids via late-stage 18F-fluorination of chiral nickel and copper complexes. Chem. Commun. 2020, 56, 9505–9508. [Google Scholar] [CrossRef] [PubMed]
- Orlovskaya, V.V.; Craig, A.S.; Fedorova, O.S.; Kuznetsova, O.F.; Neumaier, B.; Krasikova, R.N.; Zlatopolskiy, B.D. Production of 6-L-[18F]Fluoro-m-tyrosine in an Automated Synthesis Module for 11C-Labeling. Molecules 2021, 26, 5550. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Mossine, A.V.; Mahringer, A.; Aliaga, A.; Bailey, J.J.; Shao, X.; Stauff, J.; Arteaga, J.; Sherman, P.; Grand’Maison, M.; et al. Identification of [18F]TRACK, a Fluorine-18-Labeled Tropomyosin Receptor Kinase (Trk) Inhibitor for PET Imaging. J. Med. Chem. 2018, 61, 1737–1743. [Google Scholar] [CrossRef]
- Yuan, G.; Shoup, T.M.; Moon, S.H.; Brownell, A.L. A concise method for fully automated radiosyntheses of [18F]JNJ-46356479 and [18F]FITM via Cu-mediated 18F-fluorination of organoboranes. RSC Adv. 2020, 10, 25223–25227. [Google Scholar] [CrossRef]
- Mossine, A.V.; Tanzey, S.S.; Brooks, A.F.; Makaravage, K.J.; Ichiishi, N.; Miller, J.M.; Henderson, B.D.; Skaddan, M.B.; Sanford, M.S.; Scott, P.J.H. One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor. Org. Biomol. Chem. 2019, 17, 8701–8705. [Google Scholar] [CrossRef]
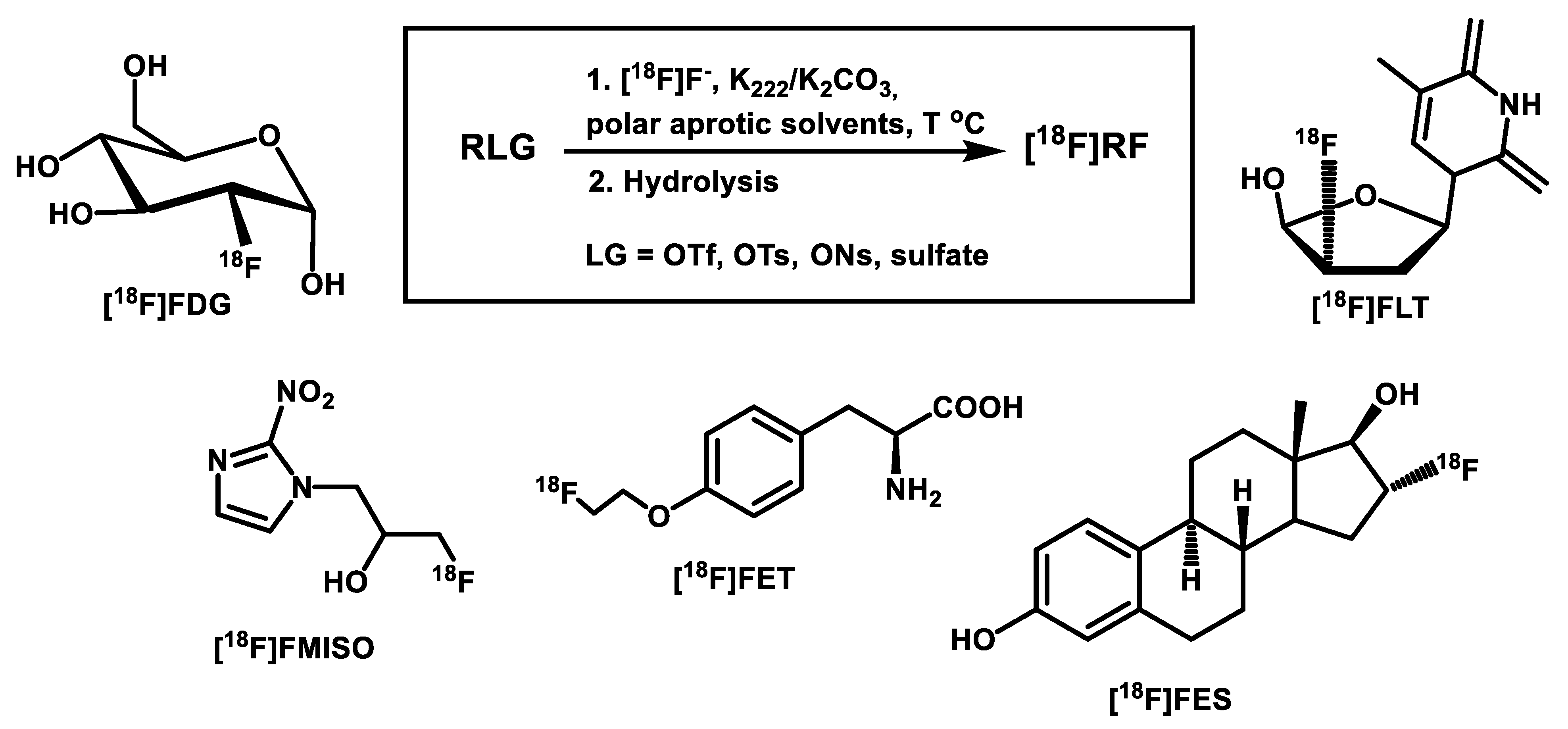
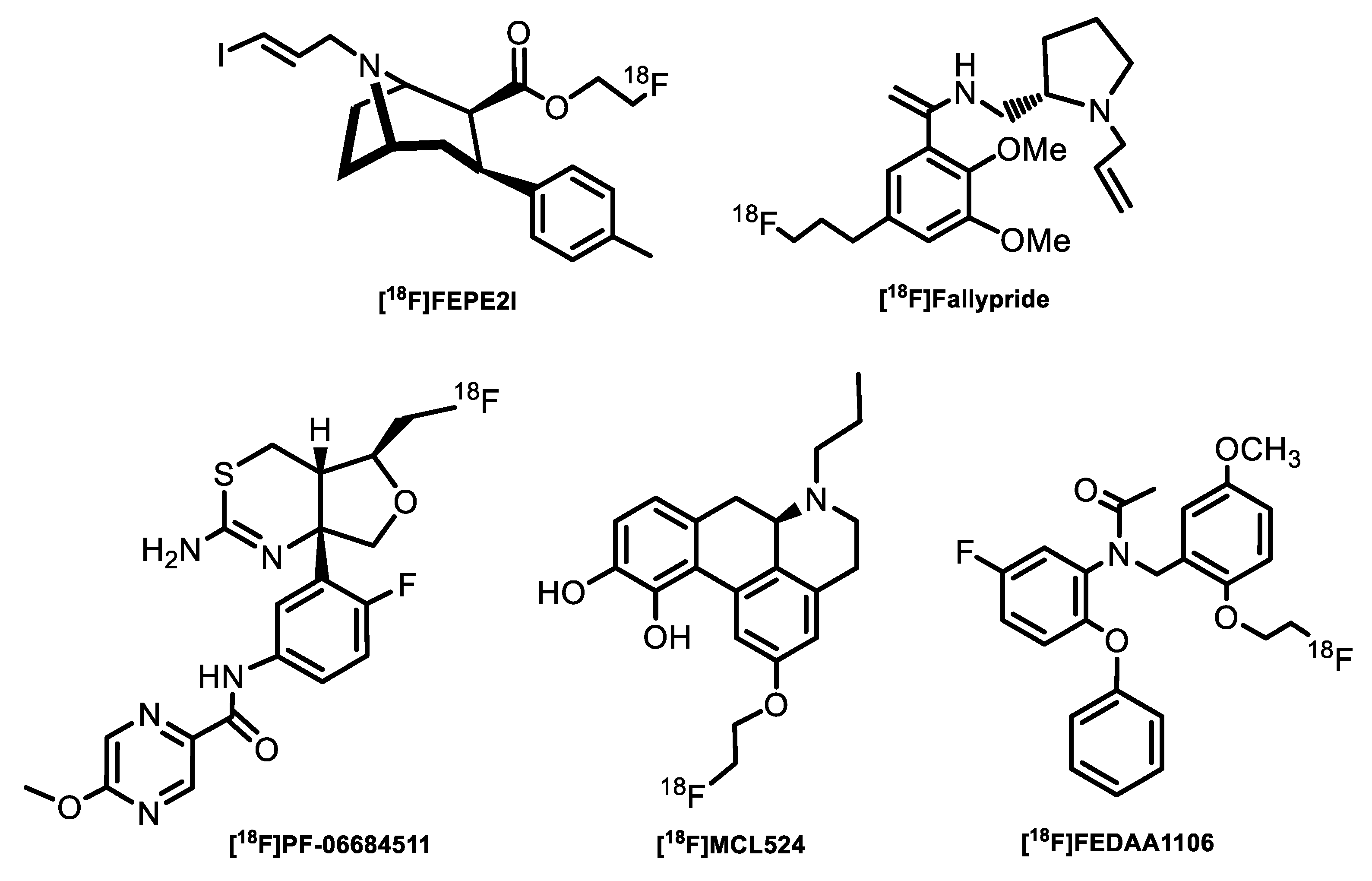
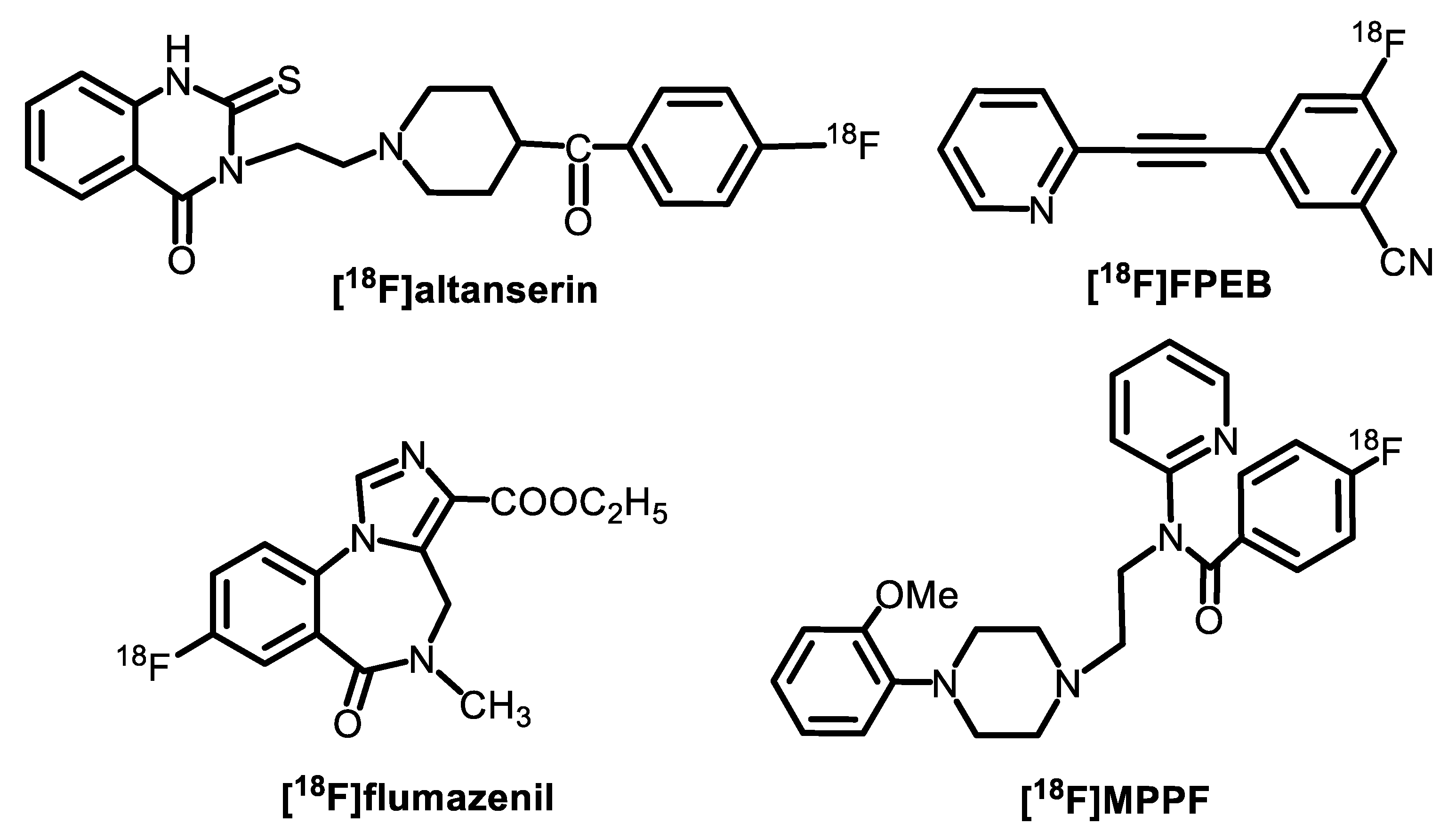
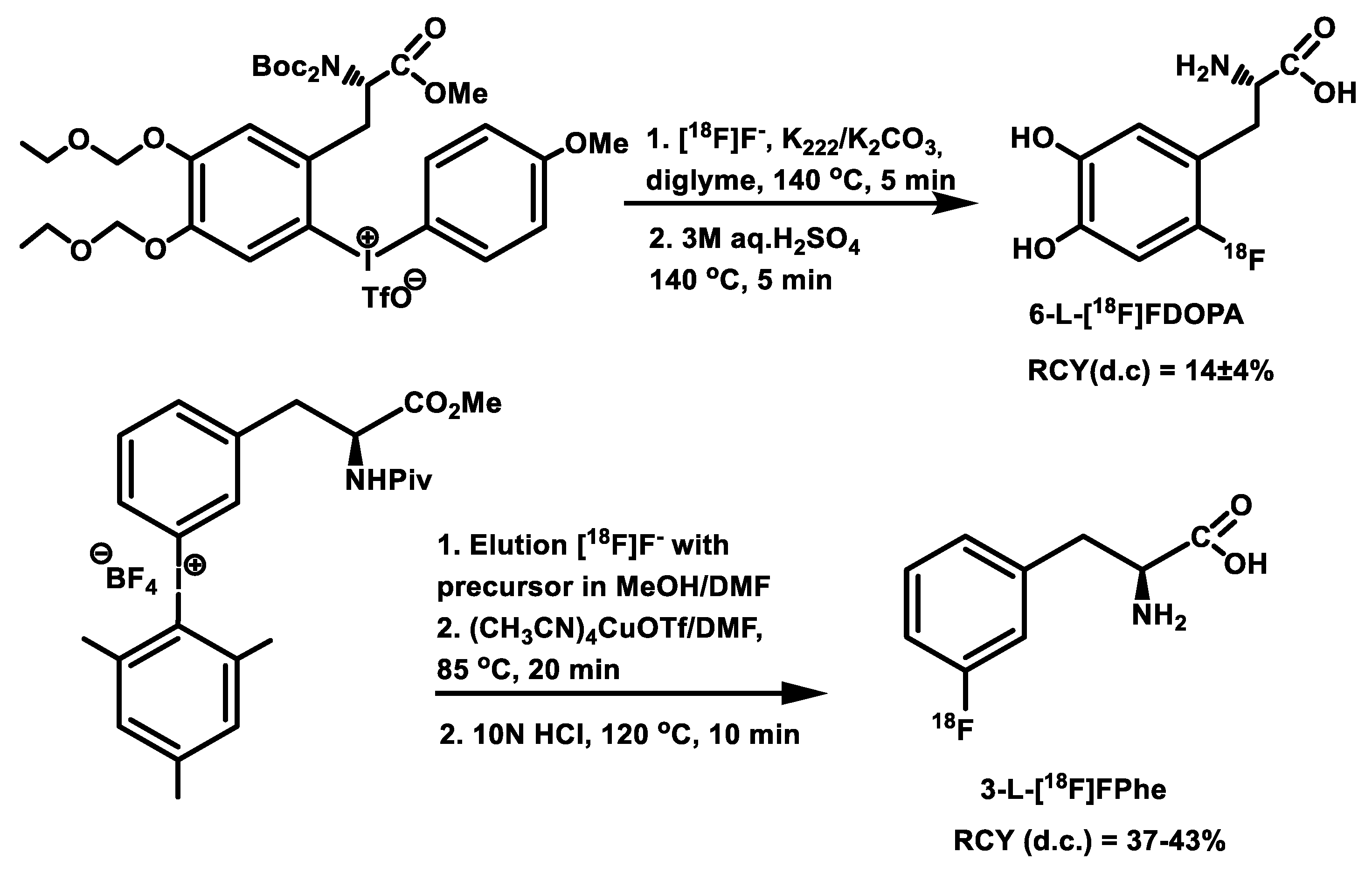

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radiotracer or Synthon | Precursor Amount, mg/µmol | RCC, % by TLC (n = 3) | RCY (%)/ Synthesis Time, Min |
|---|---|---|---|
| [18F]FETs | 0.2/0.5 | 96 ± 2 | - |
| [18F]FDG | 1.2/1.2 | 91 ± 1 | 60/25 [42] |
| [18F]FMISO | 0.2/0.5 | 74 ± 1 | - |
| [18F]FET | 0.4/0.6 * | 83 ± 1 | - |
| 2/0.3 ** | 93 ± 4 | 40/35 [47] | |
| [18F]FLT | 1/1.1 | 65 ± 4 | - |
| [18F]FES | 0.2/0.5 | 94 ± 1 | 33/32 [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasikova, R.N.; Orlovskaya, V.V. Phase Transfer Catalysts and Role of Reaction Environment in Nucleophilc Radiofluorinations in Automated Synthesizers. Appl. Sci. 2022, 12, 321. https://doi.org/10.3390/app12010321
Krasikova RN, Orlovskaya VV. Phase Transfer Catalysts and Role of Reaction Environment in Nucleophilc Radiofluorinations in Automated Synthesizers. Applied Sciences. 2022; 12(1):321. https://doi.org/10.3390/app12010321
Chicago/Turabian StyleKrasikova, Raisa N., and Viktoriya V. Orlovskaya. 2022. "Phase Transfer Catalysts and Role of Reaction Environment in Nucleophilc Radiofluorinations in Automated Synthesizers" Applied Sciences 12, no. 1: 321. https://doi.org/10.3390/app12010321
APA StyleKrasikova, R. N., & Orlovskaya, V. V. (2022). Phase Transfer Catalysts and Role of Reaction Environment in Nucleophilc Radiofluorinations in Automated Synthesizers. Applied Sciences, 12(1), 321. https://doi.org/10.3390/app12010321