1. Introduction
The development of a meat source that is minimally-dependent on livestock could contribute to satisfying the global demand for animal protein [
1]. One of the primary thrusts of the rapidly expanding field of cellular agriculture serves to produce large quantities of livestock skeletal muscle, the primary component of meat, using principles of tissue engineering and bioprocess engineering. Cultured meat companies are beginning to offer marketable products; however, an unfavorably high cost-to-yield ratio is preventing cultured meat from entering the competitive marketplace [
2]. Techno-economic analyses suggest that major improvements in bioproduction technology are needed to drive down the cost of a final meat product, produced at scale, before economic sustainability can be reached [
3].
Lab grown meat production is expected to encounter many of the same challenges that tissue engineering has encountered over the last few decades [
4]. These include recapitulating the complex native environment of three-dimensional tissues and providing such structured tissues with nutritional and oxygen support beyond their diffusion limits. For these reasons, bioprocess designs specifically concerned with yielding large quantities of cell mass may support a more rapid development of unstructured meat products. One such design, the suspension bioreactor, has been widely employed by the pharmaceutical industry to yield large quantities of cells [
5].
Suspension reactors use microcarriers to provide scaffolds for anchorage-dependent cell growth. Such systems are designed to generate fluid flow fields that maximize the number of cells cultured per unit volume while maintaining cell-to-cell homogeneity. A typical bioprocess for culturing mammalian cells in suspension may employ many suspension reactors of increasing size, referred to as seed reactors, within which cells are sequentially expanded to the limits offered by each unit before being inoculated into the larger systems [
6]. Stir-tank reactors are especially popular seed-reactors due to their volume capacity, and relatively simple inoculation and harvest procedures; however, their primary criticism, a high media volume to cell quantity ratio, has led researchers to explore the applicability of other bioreactor configurations when operating at scale, such as packed bed and hollow fiber reactors [
7].
Many of the target mammalian cell types for lab grown meat are anchorage-dependent and require a stiff substrate upon which they can adhere [
8]. Microcarriers are small beads of a wide variety of materials, shapes, sizes, and densities to support the growth of anchorage-dependent cells, such as primary bovine satellite cells, in suspension cultures. Microcarriers have been employed for unique biomedical applications, mainly the production of complex biomolecules such as recombinant proteins; however, there has yet to be a cell carrier designed specifically for lab grown meat [
9,
10,
11]. Carriers for use in cellular agriculture must be readily available, affordable, and support cell adhesion and viability during and post inoculation. It is also advantageous to design an edible cell carrier. While cells can be harvested from traditional, inedible carriers, this adds extra steps and associated costs. If carriers are to be integrated into a final unstructured cultured meat product, serving as a bulking agent, a fortifying agent, or a food stabilizer, then they must be both edible and acceptable by consumers.
Among plants, broccoli florets have many of the desired characteristics of a cell carrier for cellular agriculture; however, the interactions between native plant cells and seeded mammalian cells remain unknown. Decellularization, a technique used to isolate intact extracellular material from living tissues in their native morphology, has been investigated for applications in tissue engineering [
12,
13,
14]. This process involves the exposure of living tissues to a sequence of detergents to remove intracellular components, leaving behind intact extracellular matrix. Decellularization of plants has previously yielded scaffolds that supported adhesion and differentiation of primary bovine satellite cells [
15,
16]. Additionally, plant cell walls, the remaining material following plant decellularization process, are primarily composed of cellulose, pectin, and hemicellulose, all of which are considered as nutritionally beneficial in most diets [
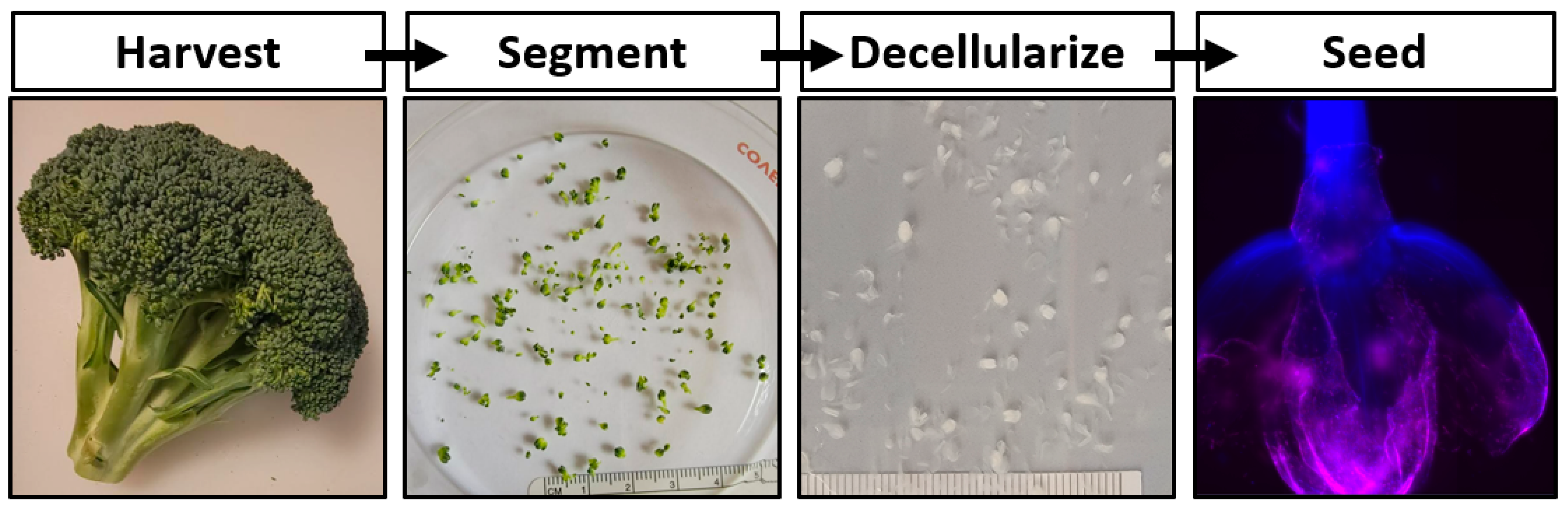
17]. In this work the development of a cultured meat bioprocess utilizing decellularized broccoli florets is proposed (
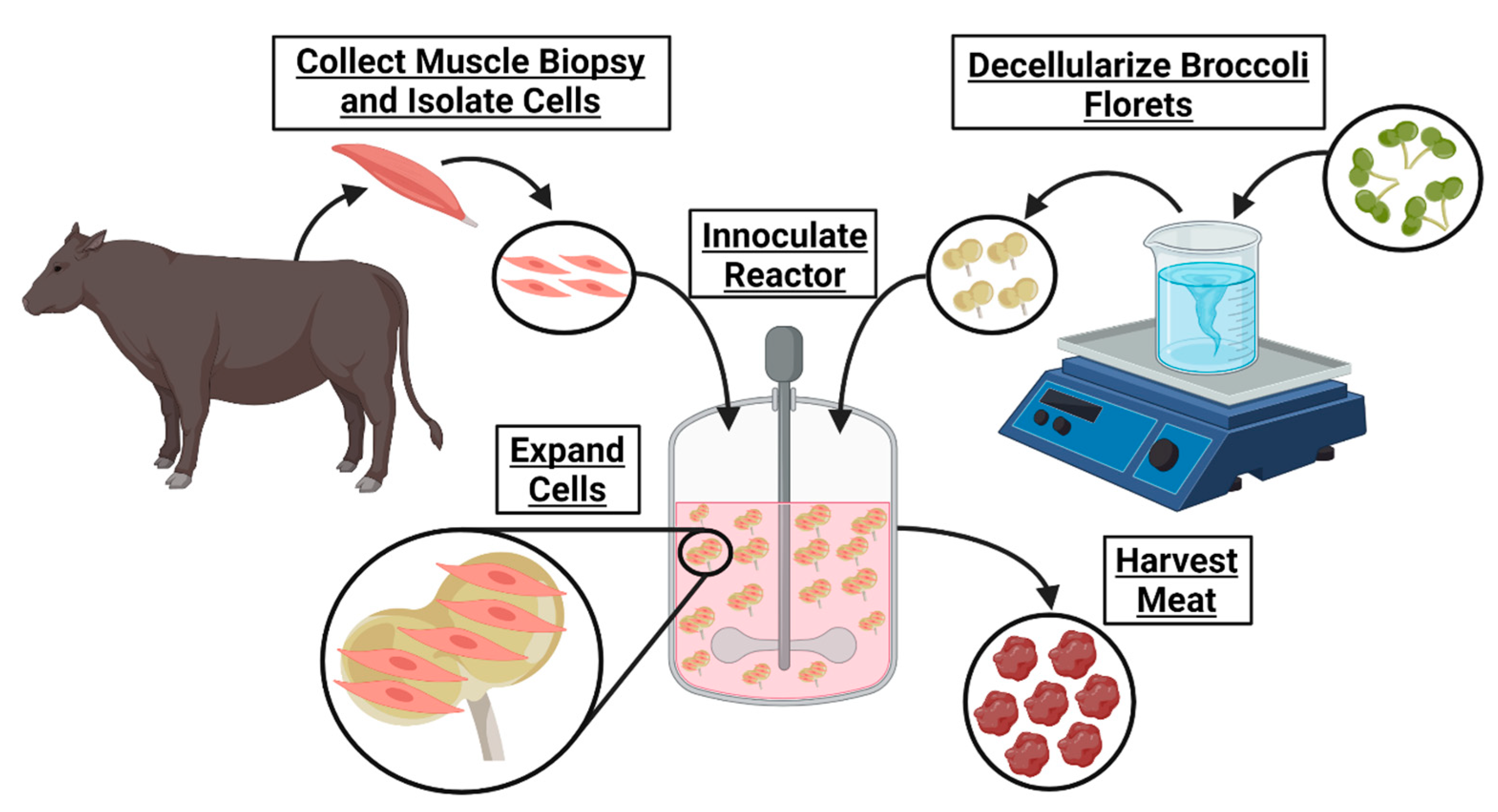
Figure 1).
Ultimately, cultured meat products cannot contribute to fighting climate change and will remain inaccessible until their cost of manufacturing is significantly decreased. The development of affordable cell carriers might reduce this cost. To determine if decellularized plants could serve as an edible cell carrier for cultured meat, broccoli florets were decellularized and seeded with bovine satellite cells. These carriers supported cell viability in suspension, suggesting a novel, edible scaffold to serve as a cell carrier for lab grown meat.
2. Materials and Methods
2.1. Satellite Cell Isolation and Culture
For all studies, primary bovine satellite cells, isolated from the rear calf muscles of adult cows <2 years were used (n = 3). Muscle samples were collected at a local slaughterhouse and transported on ice to a laboratory. Samples were briefly rinsed in 70% ethanol, before immersion into a rinse media (DMEM/F12 (Thermo Fisher Scientific, Waltham, MA, USA) and 1% Penicillin/Streptomycin (Thermo Fisher Scientific)) for 10 min. Samples were removed from the rinse media and recursive incisions were made using sterile scalpel blades. Muscle biopsies were collected from within the incision and minced using sterile iris scissors. The minced tissue was submerged in digestion medium (DMEM/F12, 1% Penicillin/Streptomycin (P/S), and 10% Collagenase (1800 units/mL) Type I derived from Clostridium Histolyticum (Worthington, Lakewood, NJ, USA)) and incubated at 37 °C for 90 min. Samples were agitated intermittently during incubation. The supernatant was then collected and sequentially passed through a 100 µm, 70 µm, and 40 µm cell strainer (VWR) before being centrifuged at 300× g for 5 min in a 50 mL conical tube. The resulting supernatant was removed and the pellet was resuspended in 10 mL growth medium (DMEM/F12, 10% heat-inactivated fetal bovine serum, Thermo Fisher Scientific, 1% P/S, 4 ng/mL recombinant human fibroblast growth factor–2 (FGF2), 2.5 ng/mL recombinant human hepatocyte growth factor (HGF), 10 ng/mL recombinant human epidermal growth factor (EGF), and 5 ng/mL recombinant human insulin-like growth factor–1 (IGF)). The resultant suspensions were seeded into T-75 tissue culture treated culture flasks and incubated at 37 °C with 5% CO2 until cell adhesion would occur.
Primary satellite cells were maintained in growth media on tissue-culture-treated flasks and media was replenished every 48 h. Cells were subcultured with 25% trypsin upon reaching 80% confluence to avoid contact inhibition and contact driven differentiation. To maintain cell purity, a preplating method for selective isolation of satellite cells from their potentially heterogenous population was employed during the first subculture. Within this procedure, trypsinized cells were incubated on tissue-culture-treated T-75 culture flasks for 30 min at 37 °C and 5% CO2 for 30 min. Cells remaining in suspension after this time period were removed from the flask and replated into new tissue-culture-treated T-75 culture flasks and were henceforth considered satellite cell cultures.
2.2. Scaffold Preparation
Fresh organic broccoli was purchased at a local marketplace. Florets were detached from the broccoli stalk using a scalpel blade, ensuring that each carrier consisted of a single bulb. Samples were rinsed thoroughly with distilled water. One gram of florets was placed in a 50 mL conical tube containing 45 mL of DI water, 10% SDS (Sigma-Aldrich, St. Louis, MO, USA), 3% Tween-20 (Sigma-Aldrich), and 10% bleach (The Clorox Co., Oakland, CA, USA) and gently agitated on a laboratory roller for 48 h. The solution was refreshed after 24 h. The decellularization solution was aspirated from the samples and the florets were then transferred to a 2 L beaker containing DI H2O for at least 1 h, while replacing the DI H2O every 15 min. The samples were stored in DI H2O at room temperature.
2.3. DNA Content
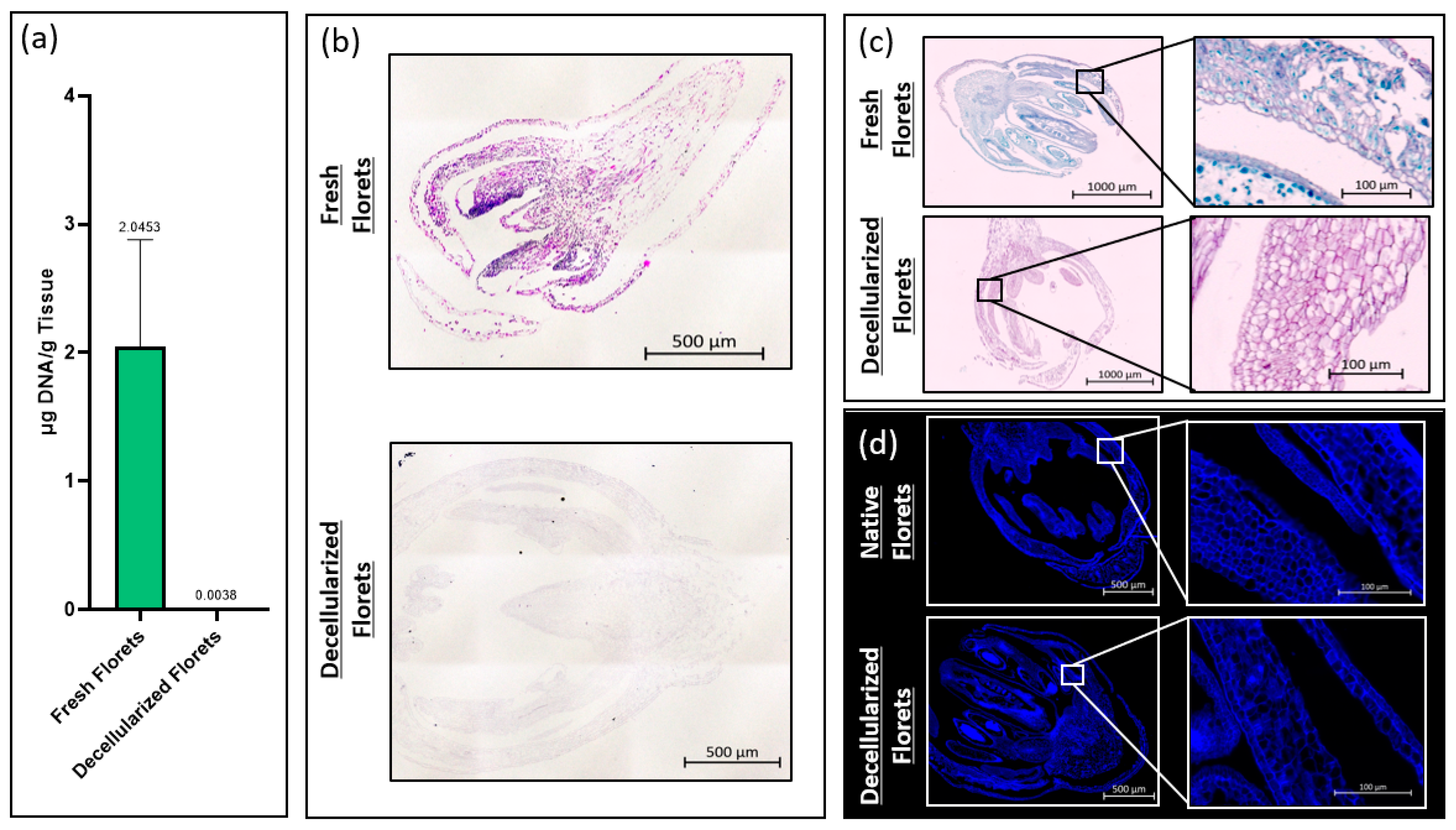
The total DNA content of fresh and decellularized scaffolds was measured using a CYQUANT® DNA assay kit (Thermo Fisher Scientific, Waltham, MA, USA). To ensure comprehensive DNA release, samples were snap-frozen using liquid nitrogen and homogenized using iris scissors. The total DNA content was calculated by comparing the intensity of sample fluorescence with that of a standard curve at an excitation wavelength of 480 nm and an emission wavelength of 520 nm per the manufacturer’s recommendation. Fluorescence intensity measurements were taken using a PerkinElmer Victor3 spectrophotometer (PerkinElmer, Waltham, MA, USA).
2.4. Histology
Fresh and decellularized samples were fixed in paraformaldehyde and embedded in paraffin wax using a Tissue-Tek VIP 6 AI tissue processor (Sakura Finetek USA, Torrance, CA, USA). During processing, samples were sequentially exposed to 70% ethanol and 80% ethanol for 30 min each, followed by two rinses of 95% ethanol and three rinses in 100% ethanol for 30 min each. Samples were then submerged in 3 xylene rinses for 20 min each, and then 4 paraffin rinses under vacuum for 30 min each. Paraffin blocks were sectioned at 6 um.
Samples were stained for hematoxylin and eosin by deparaffinizing in xylene, and then rehydrated by dipping the samples in 100% ethanol, 95% ethanol, and 70% ethanol, for two minutes each, then rinsed in running water for 5 min. Samples were placed in filtered Harris Hematoxylin for 10 min, differentiated in acid alcohol for 30 s, and dipped in ammonia water for 1 min. Samples were then counterstained with eosin for 1 min before being dehydrated via submersion in 95% ethanol, and twice in 100% ethanol for 1 min each. Samples were cleared in xylene, cover-slipped, and imaged using an Axioimager Z2 microscope (Zeiss, Oberkochen, Germany). Images were taken and stitched together after the background gradients were removed, creating a complete tiled image using ZEN 3.4 Blue Edition© imaging software (Carl Zeiss Microscopy).
Fresh and decellularized samples were stained using Sass’s Safranin and Fast Green protocol. Tissue sections were deparaffinized as described above and stained in Safranin-O (1% w/v) (Sigma-Aldrich). Samples were rinsed in running water until remaining dye was removed. The sections were then dehydrated in 70% ethanol and 90% ethanol for 1 min each before being dipped in Fast Green FCF (0.1% w/v in 95% ethanol) (Sigma-Aldrich) for 10 s. Samples were rinsed in 100% ethanol before clearing in xylene. Samples were cover slipped and imaged as described above.
Fresh and decellularized samples were stained using calcofluor white (Sigma-Aldrich), a fluorescent stain that marks cellulose. Tissue sections were deparaffinized as described above and submerged in calcofluor white stain (Sigma-Aldrich) for 30 s. Samples were rinsed in DI-H2O for 30 s before cover slipping and imaging as described above.
2.5. Scaffold Size Distribution
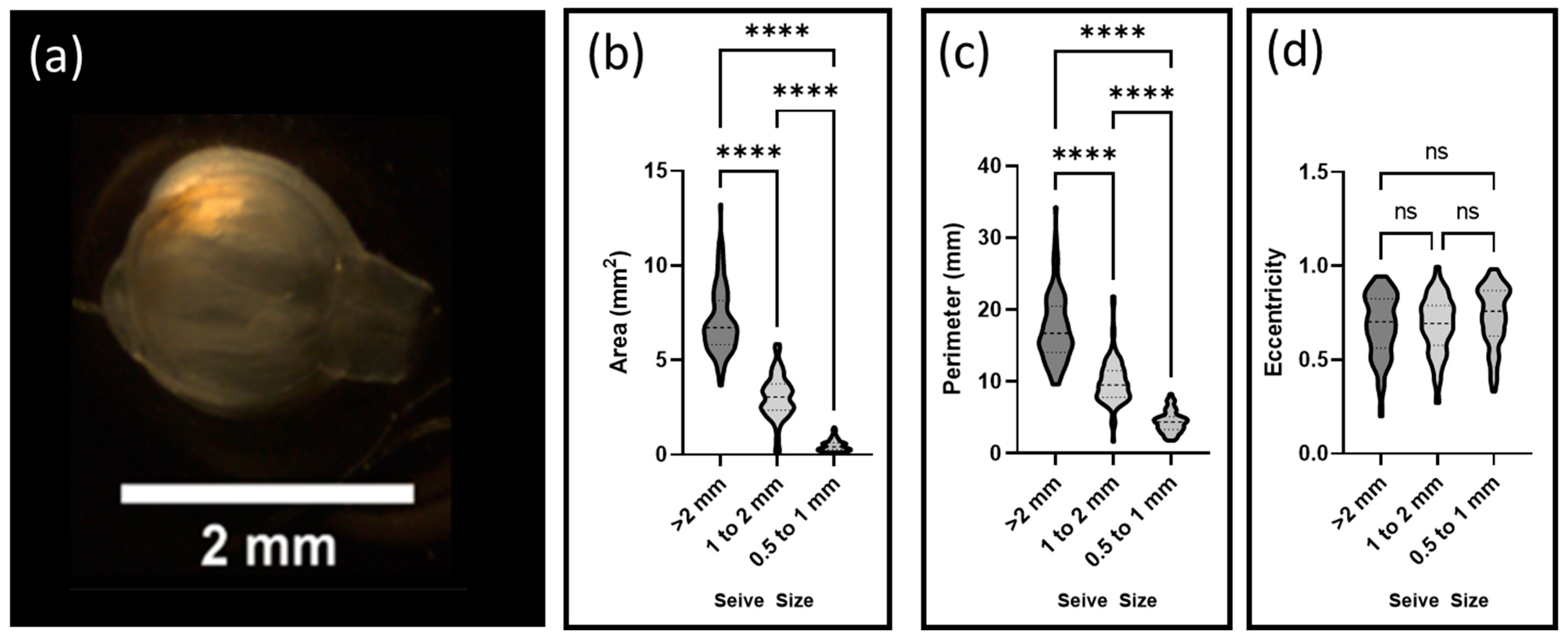
Decellularized floret scaffolds were sorted by size using sieves of decreasing diameter (3 mm, 2 mm, 1 mm, and 0.5 mm) (Tool USA, Long Beach, CA, USA). Scaffolds were placed on each sequential sieve and water was gently passed over the samples for two minutes. A total of 95 scaffolds were then collected from each sieve and imaged using a Stemi 2000-CS scope (Zeiss). Particle areas, perimeters, and eccentricities were calculated using FIJI-ImageJ 1.8.0_172 (National Institutes of Health (NIH), Rockville, MD, USA) and its associated segmentation software.
2.6. Scaffold Density
Density measurements of the decellularized scaffolds were made using a 10 mL pycnometer (Bomex, Bejing, China). Room temperature DI H2O was used as the medium for gravimetric displacement. The volume of the pycnometer was calculated by dividing the total mass of water that it could contain by the density of water. Next, the dried sample to be measured was placed inside the empty pycnometer and its mass was recorded. The pycnometer with the sample was filled with room temperature water. The total mass of the added water was recorded and divided by the density of water to calculate the total volume of added water. Next, the volume of the florets initially placed in the pycnometer was calculated by subtracting the volume of water added from the total volume of the pycnometer. Lastly, the density of the sample was calculated by dividing the mass of the sample by the volume of the sample.
2.7. Reactor Inoculation and Culture Conditions
Decellularized floret scaffolds were sterilized by submerging them in 70% ethanol for 30 min. Samples were washed three times, sequentially, in sterile 1X PBS, and then conditioned in growth media in 15 mL conical tubes for 1 h at 37 °C and 5% CO2. Subconfluent satellite cells were trypsinized and inoculated into the 15 mL conical tubes to a ratio of 106 cells/200 mg scaffold/1 mL growth media. The caps of the reactors were left untightened to ensure adequate gas exchange. The contents of conical tubes were intermittently suspended by rocking every 15 min for 2 h on the first day, and then every 24 h thereafter. An additional 9 mL of growth media was added to the reactor 24 h after the initial inoculation and growth media was replaced every 48 h.
2.8. Attachment Visualization
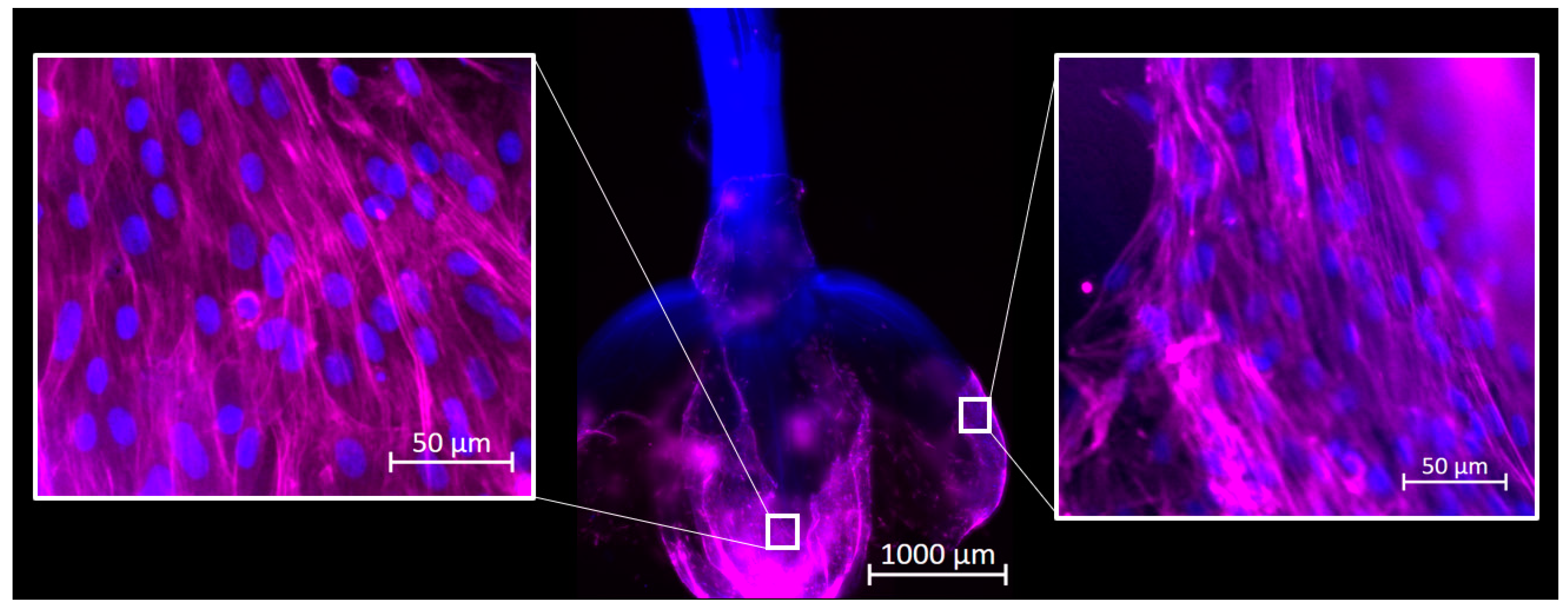
Reactors were inoculated as described above, and cultures were maintained for 72 h. Samples were taken from each reactor and fixed in 4% paraformaldehyde. Samples were stained for F-actin using Phalloidin 647 and for DNA with Hoechst 33342 (Thermo-Fisher Scientific) and imaged using an Axioimager Z2 microscope. Images were processed using ZEN 3.4 Blue Edition software (Carl Zeiss Microscopy, Jena, Germany).
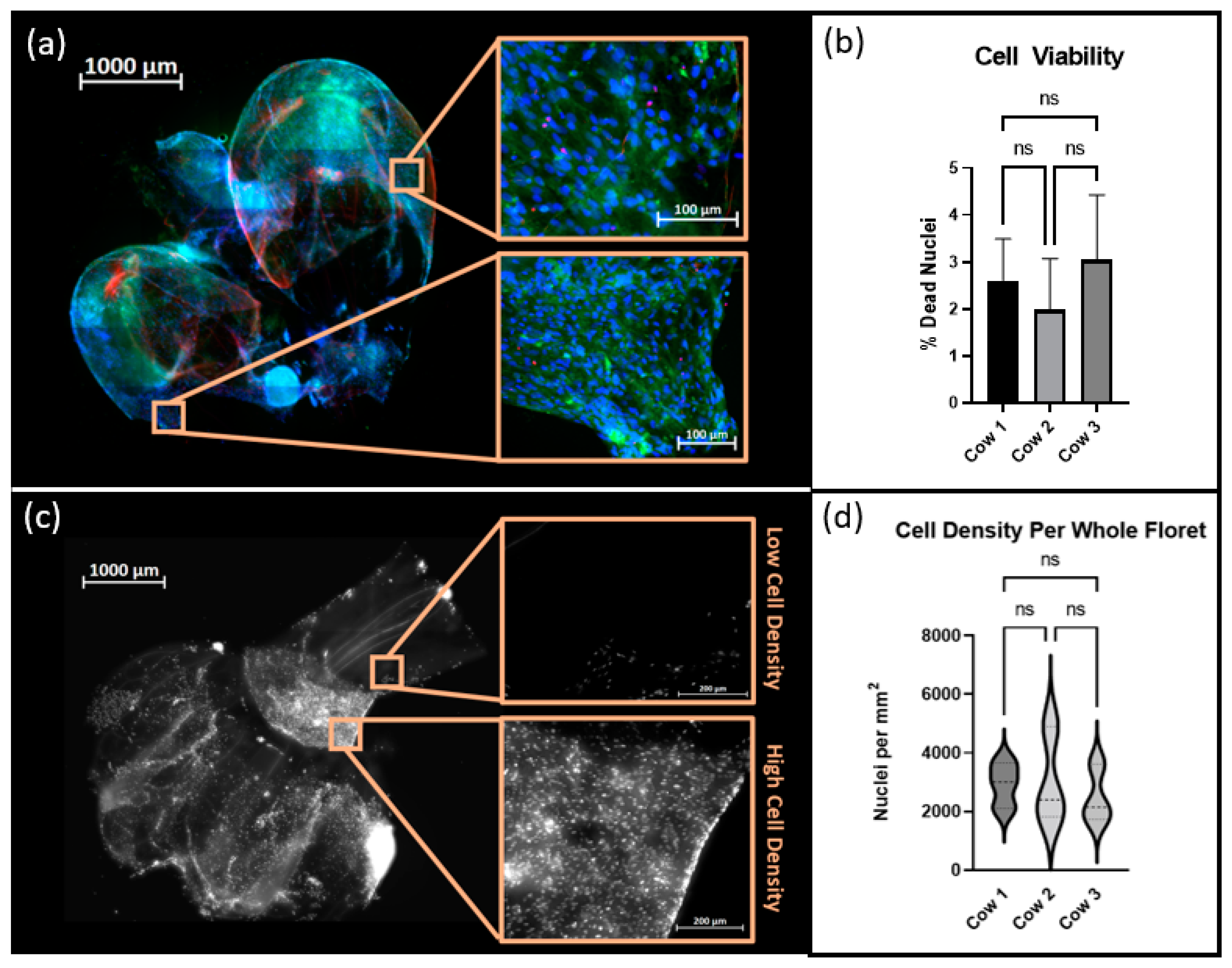
2.9. Cell Viability and Cell Density Analysis
Suspension cultures were maintained as described above for 120 h. Cells were stained using a LIVE/DEAD
® kit (Thermo Fisher Scientific) and Hoechst 33342 and subsequently fixed in 4% paraformaldehyde. Samples were imaged with an Axioimager Z2 microscope and maximum intensity images were created using the ZEN 3.4 Blue Edition software. FIJI-ImageJ 1.8.0_172, was used for image processing. The Trainable WEKA Segmentation Plug-in, which employs a supervised machine learning algorithm to classify image pixels, was used to distinguish regions of positive signal from autofluorescence inherent to the cellulose scaffold [
18]. The percentage of viable cells was calculated by quantifying the occurrence of Hoechst stained nuclei colocalized with positive signals of ethidium homodimer. Nuclei that did not display ethidium homodimer were considered to be viable cells. Additionally, each floret image was cropped into a minimum of 6 subregions. The total number of nuclei per subregion was counted and the cell density in nuclei/mm
2 was calculated per region using each crop’s respective metadata.
2.10. Statistics
All statistical analysis was performed using GraphPad Prism. Results were described as mean ± the standard deviations. All comparisons were made using a Brown–Forsythe and Welch one-way ANOVA with post-hoc t-testing. A p-value of 0.5 was the cutoff for significant differences. One asterisk denotes a p ≤ 0.05, two asterisks denote p ≤ 0.01, three asterisks denote p ≤ 0.001, and four asterisks denote p ≤ 0.0001.
4. Discussion
Meats and other animal products account for an estimated 43 percent of the global protein supply and are a robust nutritional resource; however, innovation to ensure the environmental sustainability of animal protein is necessary [
1,
19,
20]. With an increasing trend in meat consumption in developing countries, current livestock systems, which already demand approximately 25 percent of available land on Earth, may continue to contribute to freshwater depletion, biodiversity loss, and deforestation [
21,
22].
The realization of lab grown meat products is dependent on the development of more affordable, available, and relevant technologies for biomanufacturing at scale. One such technology, decellularized plant biomaterials, has been studied as cell scaffolds for a wide variety of biomedical applications due to their diversity of macro and microstructures [
15]. For example, decellularized spinach has been explored due to its robust vasculature and its ability to support a number of cell types. This same technology has been translated from biomedical applications to cultured meat applications [
16]. Decellularized spinach has recently been explored as a scaffold for bovine skeletal muscle in the context of lab grown meat. While this study demonstrated the ability to support satellite cell differentiation, and suggested that topological features of plant scaffolds may play a role in the alignment of muscle fibers, further integration of decellularized plant technology into potential bioprocesses is warranted.
Stir tank bioreactors and other suspension-style reactor designs, operating conditions, and inputs have yet to be designed with consideration of the economic demands of the cultured meat industry. Recent developments in serum-free culture, plant-derived proteins, and recombinant growth factors, coupled with advanced technologies in spent media recycling, might drive down the cost of operation of suspension bioreactors to an economically competitive level. Still, further investigation into their development is warranted [
23,
24,
25].
The simple decellularization process presented yielded plant-derived scaffolds with the capacity to support mammalian cells during culture (
Figure 6). Scaffold preparation was not dependent on any secondary bioprocesses, such as those dependent on bacterially derived materials; rather, the production of decellularized florets could be completed anywhere that broccoli can be grown, on a traditional farm, or an integrated hydroponics facility. Future studies exploring the capacity for locally abundant plant materials to serve as adequate microcarriers is encouraged to further expand the accessibility of cultured meat.
Histological analysis showed that decellularized florets are primarily composed of cellulose. This ultimately suggests that, if these scaffolds are not further modified during downstream processing, they may impart unto a final cultured meat product the nutritional benefit of insoluble fiber. Ultimately, an edible cell carrier will be considered in respect to the nutritional content of the total cultured meat serving. It would be advantageous if the scaffold used as a cell carrier during the suspension culture contributed positive nutritional attributes to the final product [
26]. Future studies examining the nutritional qualities of cultured meat grown on decellularized plant scaffolds should be encouraged.
While previous studies have demonstrated that decellularized plants can support the adhesion and viability of numerous cell types during extended culture, cell inoculation methods in those studies did not reflect the dynamic inoculation that cells are expected to experience upon introduction into a suspension-style bioreactor [
16]. Previously, cells were placed directly onto a well-supported, unmoving flat scaffold. Media was changed frequently; however, at no point were the scaffolds and cells agitated or suspended in the culture media, as they would be in a large suspension bioreactor. In this study, cells were inoculated directly into an environment already containing culture medium and unsupported scaffolds, and the environment was intermittently agitated to generate random flow fields that suspended the reactor contents. In this dynamic environment cells adhered to and remained viable on the decellularized plant-based scaffolds, and ultimately suggested that traditional cell inoculation strategies associated with microcarrier culture applied to primary bovine satellite cells and decellularized plant-based carriers. This method removed the time and user intensive process of cell seeding, which is important in reducing costs in the cellular agriculture industry.
Suspension style reactors generate fluid flow fields that suspend cell-populated microcarriers in the interest of maximizing the number of cells cultured per unit volume. It is important to minimize the fluidic stresses imparted upon cells by such flow fields, typically generated via stirring impeller, or rocking platform, as such stresses have been showed to impact mammalian cell expansion [
27]. Computational fluid dynamics was used to calculate the patterns of fluid flow within such reactors so that cell engineers could predict the stresses that their cell carriers might encounter during expansion [
28]. Data pertaining to the size, shape, and density of the microcarriers was paramount for predicting the minimum carrier suspension criteria, as well as the mechanics by which the carrier passed through the media during suspension [
29].
Microcarriers are currently fabricated with a density slightly greater than that of the media within which they are suspended. This ensures that microcarriers in a static environment will settle, allowing for initial cell attachment during inoculation, and can then be suspended with minimal energy input. The decellularized floret scaffolds were found to have a density of approximately 1.01 g/cm
3. This density measurement was comparable to microcarriers previously suggested for lab grown meat, and those historically used for suspension culture of anchorage-dependent cells for biomedical applications [
9]. These materials, comprised of edible polysaccharides and polypeptides varied in density between 1.03 g/cm
2 and 1.04 g/cm
2 [
30,
31,
32,
33] (
Figure 7). Cell culture media containing 10% FBS has been reported at 1.007 g/m
3 and decreased with lesser concentrations of FBS [
34]. Such similar density values implied that the suspension criteria for cells cultured on decellularized florets would be minimal when suspending the cell carriers.
These suspension criteria served as the conditional basis for computational fluid dynamics and generated the bioreactor’s velocity gradient, which would ultimately be used to identify the fluid flow profile of the bioreactor.
The size and shape of the carrier were key variables in predicting the stresses cells were expected to encounter [
29]. A larger carrier, must displace greater fluid volumes as they pass around it, and thus will encounter greater stress magnitudes. The ability to sort our decellularized floret carriers by size, as demonstrated, might be a means of minimizing stress-induced cell damage. It was interesting to note that the cross-sectional images generated via histological staining revealed relatively large spaces within the bulb of the floret scaffold. Porous microcarriers have been developed in the past as a means of protecting cells that have infiltrated the bead from shear stresses that they might encounter [
10]. It is recommended that the porosity of decellularized floret microcarriers, and of any future developed decellularize plant-derived microcarriers, is examined.
Additionally, the surface of a more eccentric particle is expected to encounter greater shear stresses than normal stresses as fluid volumes pass around it. The relatively high eccentricity of the decellularized florets, when compared to traditional, spherical microcarriers might be advantageous, as shear stresses have been shown to promote alignment of mammalian cells, a necessary characteristic of functional muscle tissue [
35].
The applicability of decellularized plant-derived cell carriers, in consideration of a scaled bioprocess, was studied. The decellularization process used in this paper reflected a suspension-style method previously reported; however, a few changes were made [
16]. First, Triton X-100, which is not generally regarded as safe in food products was replaced with Polysorbate-20 (Tween), an emulsifier that has been approved as safe by the Food and Drug Administration [
36]. Secondly, in the interest of reducing the water use and time to yield decellularized plants, SDS, Polysorbate-20, and bleach were combined into one decellularization solution. This decreased the processing time from 7 days to 2 days and water use by a factor of 3.5. Further optimization of this decellularization process is encouraged for both broccoli and other widely available plant materials. The use of plant waste as cell carriers, such as wheat middlings, a byproduct of wheat processing, or aquatic plant waste, which is typically displaced via dredging or beach combing, may further support sustainability efforts.
The development of edible, nutritionally beneficial, scaffolds as presented in this study might further simplify a scaled cultured meat bioprocess. Synthetic microcarriers are inedible and would require a potentially expensive harvest procedure to ensure no inedible materials were incorporated into the final product. This harvest procedure may require digestive enzymes, such as animal derived trypsin. The purification of trypsin, which is dependent on secondary bioprocesses, will come with an associated cost, estimated at approximately 98 USD/liter [
37]. Additionally, trypsin harvesting from small scale microcarrier suspension cultures is reported to vary between 70–95%, suggesting that this step may waste cell biomass [
38].
Such processes will drive up the cost of manufacturing for a final product. While there are commercial microcarriers derived from edible materials, they were not designed in consideration of their nutritional composition. For example, Cytodex microcarriers are composed of dextran which is believed to convert to glucose upon ingestion, and may ultimately be a health detriment to diabetic consumers [
39]. The cell carriers proposed in this paper, being composed primarily of the insoluble dietary fiber cellulose, have well documented positive effect on health [
17]. Additionally, research into biofortification has yielded procedures to improve the nutritional characteristics of fresh plants. If these fortifications can be maintained through the decellularization process, the nutritional profile of a final product can be further improved [
40].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






