
A Probe-Based qPCR Method, Targeting 16S rRNA Gene, for the Quantification of Paenibacillus larvae Spores in Powdered Sugar Samples
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Inspection and Sample Collection
2.2. Microbiological Analysis
2.3. Molecular Analysis
2.3.1. DNA Extraction
2.3.2. TaqMan® Real-Time PCR Assay for the Detection of P. larvae DNA in Sugar Samples
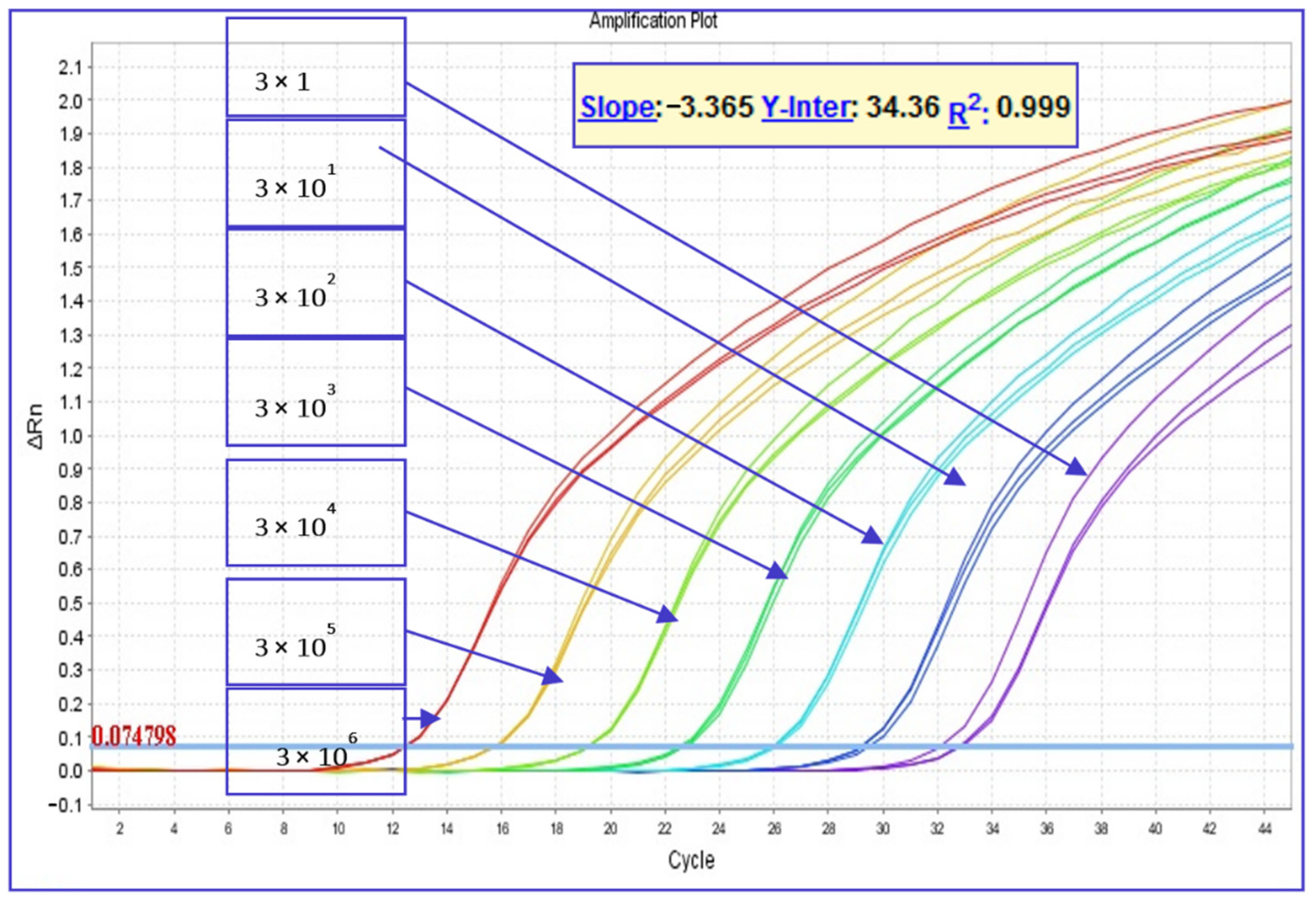
2.3.3. Standard Curve and Quantitative TaqMan® Real-Time PCR Assay for the Absolute Quantification of P. larvae Spores
2.3.4. ERIC Genotyping
2.4. Data Analysis
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Genersch, E.; Forsgren, E.; Pentikäinen, J.; Ashiralieva, A.; Rauch, S.; Kilwinski, J.; Fries, I. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int. J. Syst. Evol. Microbiol. 2006, 56, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Anonymous. American Foulbrood of Honey Bees (Infection of Honey Bees with Paenibacillus Larvae). In OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; 2018; pp. 719–735, Chapter 3.2.2; Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.02.02_AMERICAN_FOULBROOD.pdf (accessed on 27 September 2022).
- Ellis, J.D.; Munn, P.A. The worldwide health status of honey bees. Bee World 2005, 86, 88–101. [Google Scholar] [CrossRef]
- Hansen, H.; Brødsgaard, C.J. American foulbrood: A review of its biology, diagnosis and control. Bee World 1999, 80, 5–23. [Google Scholar] [CrossRef]
- Genersch, E. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 2010, 103, S10–S19. [Google Scholar] [CrossRef]
- Morrissey, B.J.; Helgason, T.; Poppinga, L.; Fünfhaus, A.; Genersch, E.; Budge, G.E. Biogeography of P aenibacillus larvae, the causative agent of American foulbrood, using a new multilocus sequence typing scheme. Environ. Microbiol. 2014, 17, 1414–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descamps, T.; De Smet, L.; Stragier, P.; De Vos, P.; de Graaf, D.C. Multiple Locus Variable number of tandem repeat Analysis: A molecular genotyping tool for Paenibacillus larvae. Microb. Biotechnol. 2016, 9, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Beims, H.; Bunk, B.; Erler, S.; Mohr, K.I.; Spröer, C.; Pradella, S.; Günther, G.; Rohde, M.; von der Ohe, W.; Steinert, M. Discovery of Paenibacillus larvae ERIC V: Phenotypic and genomic comparison to genotypes ERIC I-IV reveal different inventories of virulence factors which correlate with epidemiological prevalences of American Foulbrood. Int. J. Med. Microbiol. 2020, 310, 151394. [Google Scholar] [CrossRef] [PubMed]
- Loncaric, I.; Derakhshifar, I.; Oberlerchner, J.T.; Köglberger, H.; Moosbeckhofer, R. Genetic diversity among isolates of Paenibacillus larvae from Austria. J. Invertebr. Pathol. 2009, 100, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Pentikäinen, J.; Kalliainen, E.; Pelkonen, S. Molecular epidemiology of Paenibacillus larvae infection in Finland. Apidologie 2008, 40, 73–81. [Google Scholar] [CrossRef]
- Schäfer, M.O.; Genersch, E.; Fünfhaus, A.; Poppinga, L.; Formella, N.; Bettin, B.; Karger, A. Rapid identification of differentially virulent genotypes of Paenibacillus larvae, the causative organism of American foulbrood of honey bees, by whole cell MALDI-TOF mass spectrometry. Vet. Microbiol. 2014, 170, 291–297. [Google Scholar] [CrossRef]
- Bassi, S.; Formato, G.; Milito, M.; Trevisiol, K.; Salogni, C.; Carra, E. Phenotypic characterization and ERIC–PCR based genotyping of Paenibacillus larvae isolates recovered from American foulbrood outbreaks in honey bees from Italy. Vet. Q. 2014, 35, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Biová, G.; Bzdil, J.; Dostálková, S.; Petřivalský, M.; Brus, J.; Carra, E.; Danihlík, J. American foulbrood in the Czech Repub-lic: ERIC II genotype of Paenibacillus larvae is prevalent. Front. Vet. Sci. 2021, 8, 698976. [Google Scholar] [CrossRef] [PubMed]
- Hirai, Y.; Suzuki, T.; Inaba, N.; Minoguchi, N.; Takamatsu, D. Existence of Paenibacillus larvae genotypes ERIC I-ST2, ERIC I-ST15 and ERIC II-ST10 in the western region of Aichi prefecture, Japan. J. Vet. Med. Sci. 2016, 78, 1195–1199. [Google Scholar] [CrossRef] [Green Version]
- Križanová, H.; Halaša, N.; Roško, L.; Zubaj, J. Results of studies on the causative agent of American foulbrood. Vet. Med. 1988, 33, 633–640. [Google Scholar]
- Drobníková, V.; Richter, V.; Häusler, J.; Pytelová, I. Characterization of Bacillus larvae and related bacilli by chromatography of cell fatty acids. J. Apic. Res. 1994, 33, 69–74. [Google Scholar] [CrossRef]
- Rauch, S.; Ashiralieva, A.; Hedtke, K.; Genersch, E. Negative Correlation between Individual-Insect-Level Virulence and Colony-Level Virulence of Paenibacillus larvae, the Etiological Agent of American Foulbrood of Honeybees. Appl. Environ. Microbiol. 2009, 75, 3344–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genersch, E. Paenibacillus larvae and American Foulbrood—Long since known and still surprising. J. Consum. Prot. Food Saf. 2008, 3, 429–434. [Google Scholar] [CrossRef]
- Genersch, E. Amerikanische Faulbrut: Oft anders als im Lehrbuch. Dtsch. Bienen J. 2009, 8, 4–6. [Google Scholar]
- Anonymous. Report of the Meeting of the OIE ad hoc Group on Diseases of Honey Bees. 31 January–2 February, Paris, France. The World Organisation for Animal Health, OIE. 2012. Available online: https://www.oie.int/fileadmin/Home/eng/Internationa_Standard_Setting/docs/pdf/SCAD/A_SCAD_Feb2012.pdf (accessed on 27 September 2022).
- Hansen, H.; Rasmussen, B. The investigation of honey from bee colonies for Bacillus larvae. Dan. J. Plant. Soil. Sci. 1986, 90, 81–86. Available online: https://dcapub.au.dk/pub/planteavl_90_81.pdf (accessed on 27 September 2022).
- Hornitzky, M.A.Z.; Clark, S. Culture of Bacillus larvae from bulk honey samples for the detection of American foulbrood. J. Apic. Res. 1991, 30, 13–16. [Google Scholar] [CrossRef]
- Steinkraus, K.H.; Morse, R.A. American foulbrood incidence in some US and Canadian honeys. Apidologie 1992, 23, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Fries, I.; Lindström, A.; Korpela, S. Vertical transmission of American foulbrood (Paenibacillus larvae) in honey bees (Apis mellifera). Vet. Microbiol. 2006, 114, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Lindström, A.; Fries, I. Sampling of adult bees for detection of American foulbrood (Paenibacillus larvae subsp. larvae) spores in honey bee (Apis mellifera) colonies. J. Apic. Res. 2005, 44, 82–86. [Google Scholar] [CrossRef]
- Von der Ohe, W.; Dustmann, J.H. Efficient prophylactic measures against American foulbrood by bacteriological analysis of honey for spore contamination. Am. Bee J. 1997, 137, 603–606. Available online: https://agris.fao.org/agris-search/search.do?recordID=US9748622 (accessed on 27 September 2022).
- De Graaf, D.C.; Alippi, A.M.; Antúnez, K.; Aronstein, K.A.; Budge, G.; De Koker, D.; De Smet, L.; Dingman, D.W.; Evans, J.D.; Foster, L.J.; et al. Standard methods for American foulbrood research. J. Apic. Res. 2013, 52, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Bassi, S.; Galletti, G.; Carpana, E.; Palminteri, S.; Bosi, F.; Loglio, G.; Carra, E. Powdered Sugar Examination as a Tool for the Assessment of Paenibacillus larvae Infection Levels in Honey Bee Colonies. Front. Vet. Sci. 2022, 9, 853707. [Google Scholar] [CrossRef]
- Govan, V.A.; Allsopp, M.H.; Davison, S. A PCR Detection Method for Rapid Identification of Paenibacillus larvae. Appl. Environ. Microbiol. 1999, 65, 2243–2245. [Google Scholar] [CrossRef] [Green Version]
- Dobbelaere, W.; de Graaf, D.C.; Peeters, J.E. Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp. larvae) using a 16S rRNA gene based PCR. Apidologie 2001, 32, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Piccini, C.; D’ Alessandro, B.; Antunez, K.; Zunino, P. Detection of Paenibacillus larvae subspecies larvae spores in naturally infected bee larvae and artificially contaminated honey by PCR. World J. Microbiol. Biotechnol. 2002, 18, 761–765. [Google Scholar] [CrossRef]
- Bakonyi, T.; Derakhshifar, I.; Grabensteiner, E.; Nowotny, N. Development and Evaluation of PCR Assays for the Detection of Paenibacillus larvae in Honey Samples: Comparison with Isolation and Biochemical Characterization. Appl. Environ. Microbiol. 2003, 69, 1680–1686. [Google Scholar] [CrossRef] [Green Version]
- Lauro, F.M.; Favaretto, M.; Covolo, L.; Rassu, M.; Bertoloni, G. Rapid detection of Paenibacillus larvae from honey and hive samples with a novel nested PCR protocol. Int. J. Food Microbiol. 2002, 81, 195–201. [Google Scholar] [CrossRef]
- Kilwinski, J.; Peters, M.; Ashiralieva, A.; Genersch, E. Proposal to reclassify Paenibacillus larvae subsp. pulvifaciens DSM 3615 (ATCC 49843) as Paenibacillus larvae subsp. larvae. Results of a comparative biochemical and genetic study. Vet. Microbiol. 2004, 104, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Ryba, S.; Titera, D.; Haklova, M.; Stopka, P. A PCR method of detecting American Foulbrood (Paenibacillus larvae) in winter beehive wax debris. Vet. Microbiol. 2009, 139, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Bassi, S.; Carra, E.; Carpana, E.; Paganelli, G.L.; Pongolini, S. A scientific note on the detection of spores ofPaenibacillus larvaein naturally and artificially contaminated honey: Comparison of cultural and molecular methods. Apidologie 2010, 41, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-H.; Lee, D.-B.; Lee, D.-W.; Kim, E.-H.; Yoon, B.-S. Ultra-rapid real-time PCR for the detection of Paenibacillus larvae, the causative agent of American Foulbrood (AFB). J. Invertebr. Pathol. 2008, 99, 8–13. [Google Scholar] [CrossRef]
- Chagas, S.S.; Vaucher, R.A.; Brandelli, A. Detection of Paenibacillus larvae by real-time PCR. Acta Sci. Vet. 2010, 38, 251–256. Available online: https://www.cabi.org/isc/FullTextPDF/2010/20103356040.pdf (accessed on 30 July 2021). [CrossRef]
- Martínez, J.; Simon, V.; González, B.; Conget, P. A real-time PCR-based strategy for the detection of Paenibacillus larvae vegetative cells and spores to improve the diagnosis and the screening of American foulbrood. Lett. Appl. Microbiol. 2010, 50, 603–610. [Google Scholar] [CrossRef]
- Quintana, S.; Fernández, N.J.; Pagnuco, I.; Medici, S.; Eguaras, M.J.; Gende, L.B. Report of a Real-Time pcr Assay for Paenibacillus Larvae dna Detection from Spores of Scale Samples. Revista Argentina de Producción Animal. 2017; Volume 37, pp. 83–88. Available online: https://ri.conicet.gov.ar/bitstream/handle/11336/55325/CONICET_Digital_Nro.1e426611-2f4b-49f3-a681-e75bc46f6925_A.pdf?sequence=2&isAllowed=y (accessed on 27 September 2022).
- Dainat, B.; Grossar, D.; Ecoffey, B.; Haldemann, C. Triplex real-time PCR method for the qualitative detection of European and American foulbrood in honeybee. J. Microbiol. Methods 2018, 146, 61–63. [Google Scholar] [CrossRef]
- Rossi, F.; Amadoro, C.; Ruberto, A.; Ricchiuti, L. Evaluation of Quantitative PCR (qPCR) Paenibacillus larvae Targeted Assays and Definition of Optimal Conditions for Its Detection/Quantification in Honey and Hive Debris. Insects 2018, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Beims, H.; Janke, M.; Von der Ohe, W.; Steinert, M. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Vet. J. 2020, 10, 53–58. [Google Scholar] [CrossRef]
- Kušar, D.; Papić, B.; Zajc, U.; Zdovc, I.; Golob, M.; Žvokelj, L.; Knific, T.; Avberšek, J.; Ocepek, M.; Ocepek, M.P. Novel TaqMan PCR Assay for the Quantification of Paenibacillus larvae Spores in Bee-Related Samples. Insects 2021, 12, 1034. [Google Scholar] [CrossRef] [PubMed]
- Riviere, M.-P.; Ribière, M.; Chauzat, M.-P. Recent molecular biology methods for foulbrood and nosemosis diagnosis. Rev. Sci. Tech. 2013, 32, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Ågren, J.; Schäfer, M.O.; Forsgren, E. Using whole genome sequencing to study American foulbrood epidemiology in honeybees. PLoS ONE 2017, 12, e0187924. [Google Scholar] [CrossRef] [PubMed]
- Djukic, M.; Brzuszkiewicz, E.; Fünfhaus, A.; Voss, J.; Gollnow, K.; Poppinga, L.; Liesegang, H.; Garcia-Gonzalez, E.; Genersch, E.; Daniel, R. How to Kill the Honey Bee Larva: Genomic Potential and Virulence Mechanisms of Paenibacillus larvae. PLoS ONE 2014, 9, e90914. [Google Scholar] [CrossRef]
- Genersch, E.; Otten, C. The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie 2003, 34, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Nordström, S.; Fries, I. A comparison of media and cultural conditions for identification of Bacillus larvae in honey. J. Apic. Res. 1995, 34, 97–103. [Google Scholar] [CrossRef]
- Dingman, D.W.; Stahly, D.P. Medium Promoting Sporulation of Bacillus larvae and Metabolism of Medium Components. Appl. Environ. Microbiol. 1983, 46, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Forsgren, E.; Stevanovic, J.; Fries, I. Variability in germination and in temperature and storage resistance among Paenibacillus larvae genotypes. Vet. Microbiol. 2008, 129, 342–349. [Google Scholar] [CrossRef]
- Crudele, S.; Ricchiuti, L.; Ruberto, A.; Rossi, F. Quantitative PCR (qPCR) vs culture-dependent detection to assess honey contamination by Paenibacillus larvae. J. Apic. Res. 2019, 59, 218–222. [Google Scholar] [CrossRef]
- Antúnez, K.; D’Alessandro, B.; Piccini, C.; Corbella, E.; Zunino, P. Paenibacillus larvae larvae spores in honey samples from Uruguay: A nationwide survey. J. Invertebr. Pathol. 2004, 86, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Ribani, A.; Utzeri, V.J.; Taurisano, V.; Fontanesi, L. Honey as a Source of Environmental DNA for the Detection and Monitoring of Honey Bee Pathogens and Parasites. Vet. Sci. 2020, 7, 113. [Google Scholar] [CrossRef]
- Forsgren, E.; Laugen, A.T. Prognostic value of using bee and hive debris samples for the detection of American foulbrood disease in honey bee colonies. Apidologie 2013, 45, 10–20. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Culture (+) | Culture (−) | ||
|---|---|---|---|
| RT-PCR (+) | 51 | 18 | 69 |
| RT-PCR (−) | 6 | 30 | 36 |
| 57 | 48 | Tot. 105 |
| Group A | Group B (n = 45) | ||||||
|---|---|---|---|---|---|---|---|
| Sub-Group A1 (n = 15) | Sub-Group A2 (n = 45) | ||||||
| Classes of Contamination | Culture Method | qPCR | Culture Method | qPCR | Culture Method | qPCR | |
| (CFU/g–Spore/g) | |||||||
| <20 | 0 | 0 | 11 (24%) | 8 (18%) | 37 (82%) | 28 (62%) | |
| 20–100 | 0 | 0 | 16 (36%) | 5 (11%) | 6 (13%) | 8 (18%) | |
| 101–1000 | 0 | 0 | 13 (29%) | 18 (40%) | 2 (4%) | 9 (20%) | |
| 1001–10,000 | 0 | 0 | 4 (9%) | 8 (18%) | 0 | 0 | |
| 10,001–100,000 | 3 (20%) | 0 | 1 (2%) | 6 (13%) | 0 | 0 | |
| 100,001–1,000,000 | 8 (53%) | 5 (33%) | 0 | 0 | 0 | 0 | |
| >1,000,001 | 4 (27%) | 10 (67%) | 0 | 0 | 0 | 0 | |
| Total | 15 | 15 | 45 | 45 | 45 | 45 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carra, E.; Galletti, G.; Carpana, E.; Bergamini, F.; Loglio, G.; Bosi, F.; Palminteri, S.; Bassi, S. A Probe-Based qPCR Method, Targeting 16S rRNA Gene, for the Quantification of Paenibacillus larvae Spores in Powdered Sugar Samples. Appl. Sci. 2022, 12, 9895. https://doi.org/10.3390/app12199895
Carra E, Galletti G, Carpana E, Bergamini F, Loglio G, Bosi F, Palminteri S, Bassi S. A Probe-Based qPCR Method, Targeting 16S rRNA Gene, for the Quantification of Paenibacillus larvae Spores in Powdered Sugar Samples. Applied Sciences. 2022; 12(19):9895. https://doi.org/10.3390/app12199895
Chicago/Turabian StyleCarra, Elena, Giorgio Galletti, Emanuele Carpana, Federica Bergamini, Giulio Loglio, Filippo Bosi, Stefano Palminteri, and Stefano Bassi. 2022. "A Probe-Based qPCR Method, Targeting 16S rRNA Gene, for the Quantification of Paenibacillus larvae Spores in Powdered Sugar Samples" Applied Sciences 12, no. 19: 9895. https://doi.org/10.3390/app12199895