
False Positive Identification of Pesticides in Food Using the European Standard Method and LC-MS/MS Determination: Examples and Solutions from Routine Applications
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents, Instruments and Samples
2.2. Sample Preparation
2.3. LC-MS/MS Separation for Screening
2.4. LC-MS/MS Separation for Confirmation
2.5. Standard Method Verification
2.5.1. Screening
2.5.2. Confirmation
3. Results and Discussion
3.1. Method Evaluation
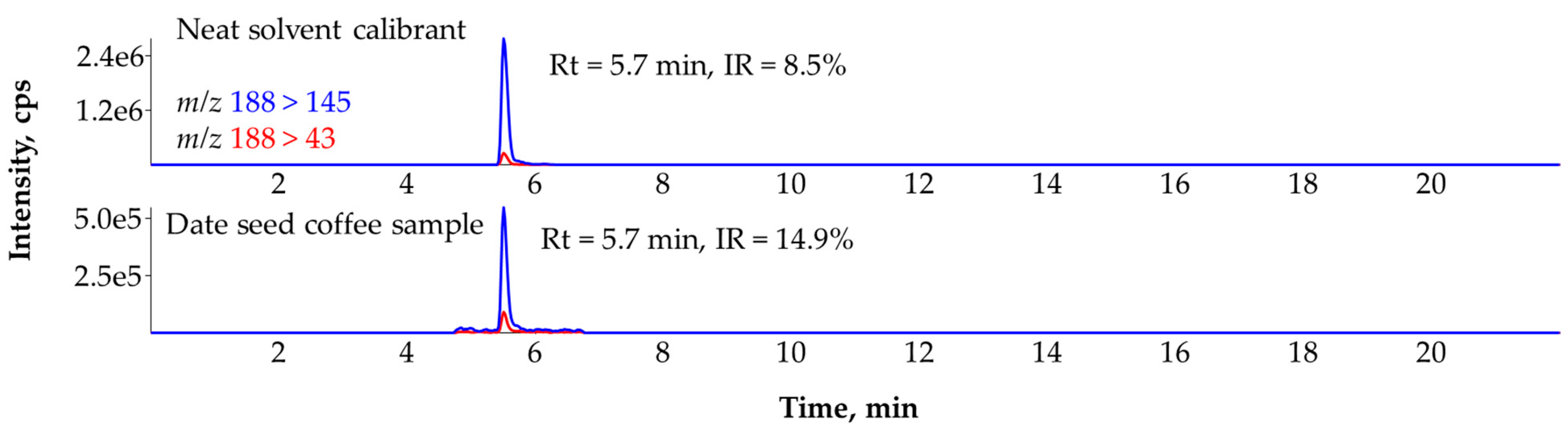
3.2. Ruling Out the False Identification of Atrazine-Desethyl in a Date Seed Coffee (Coffea Phoenix dactylifera) Sample with the Help of IR
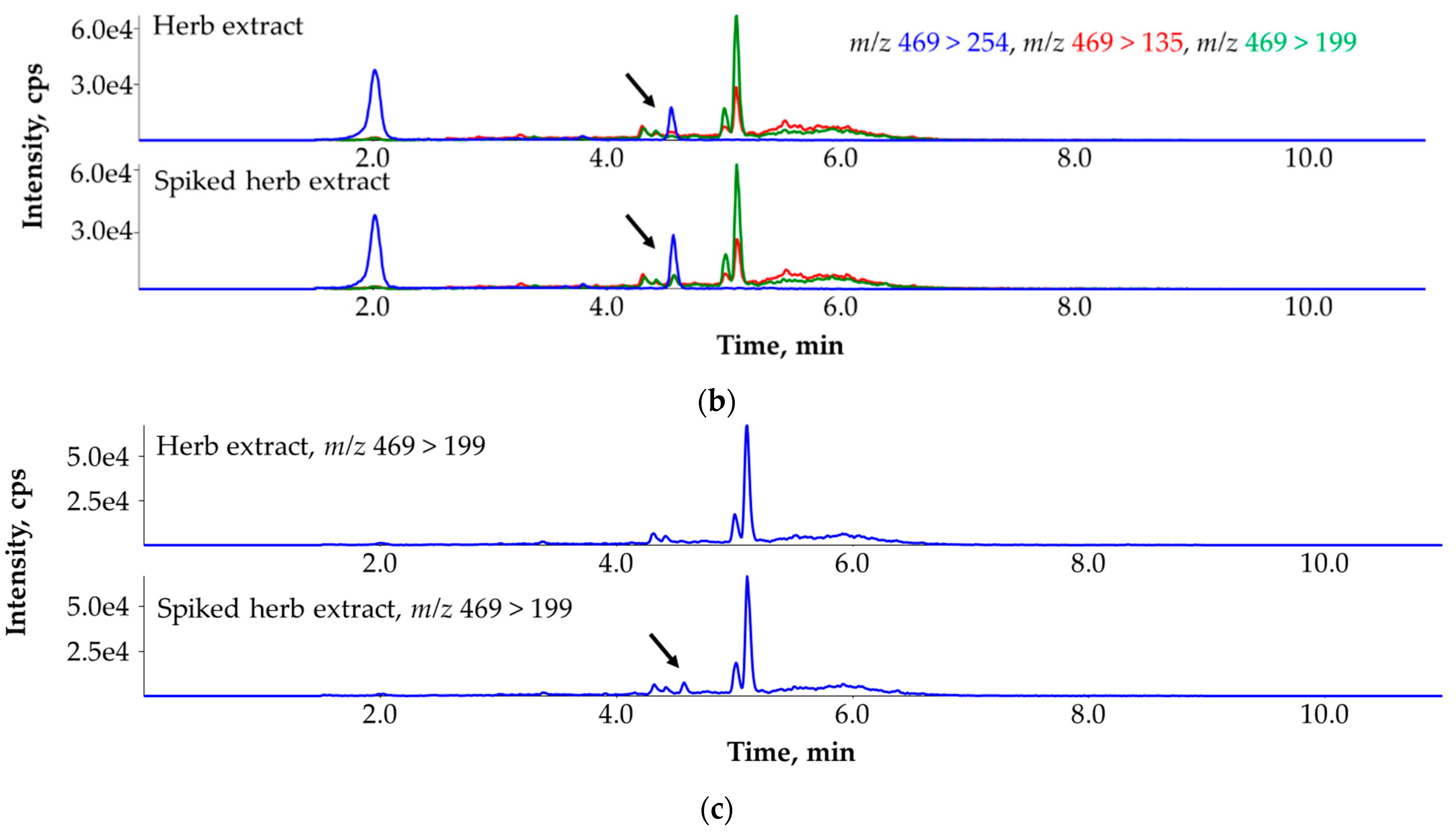
3.3. Ruling Out the False Identification of Primisulfuron-Methyl in a Herb Extract Sample with the Help of the Third MRM Transition
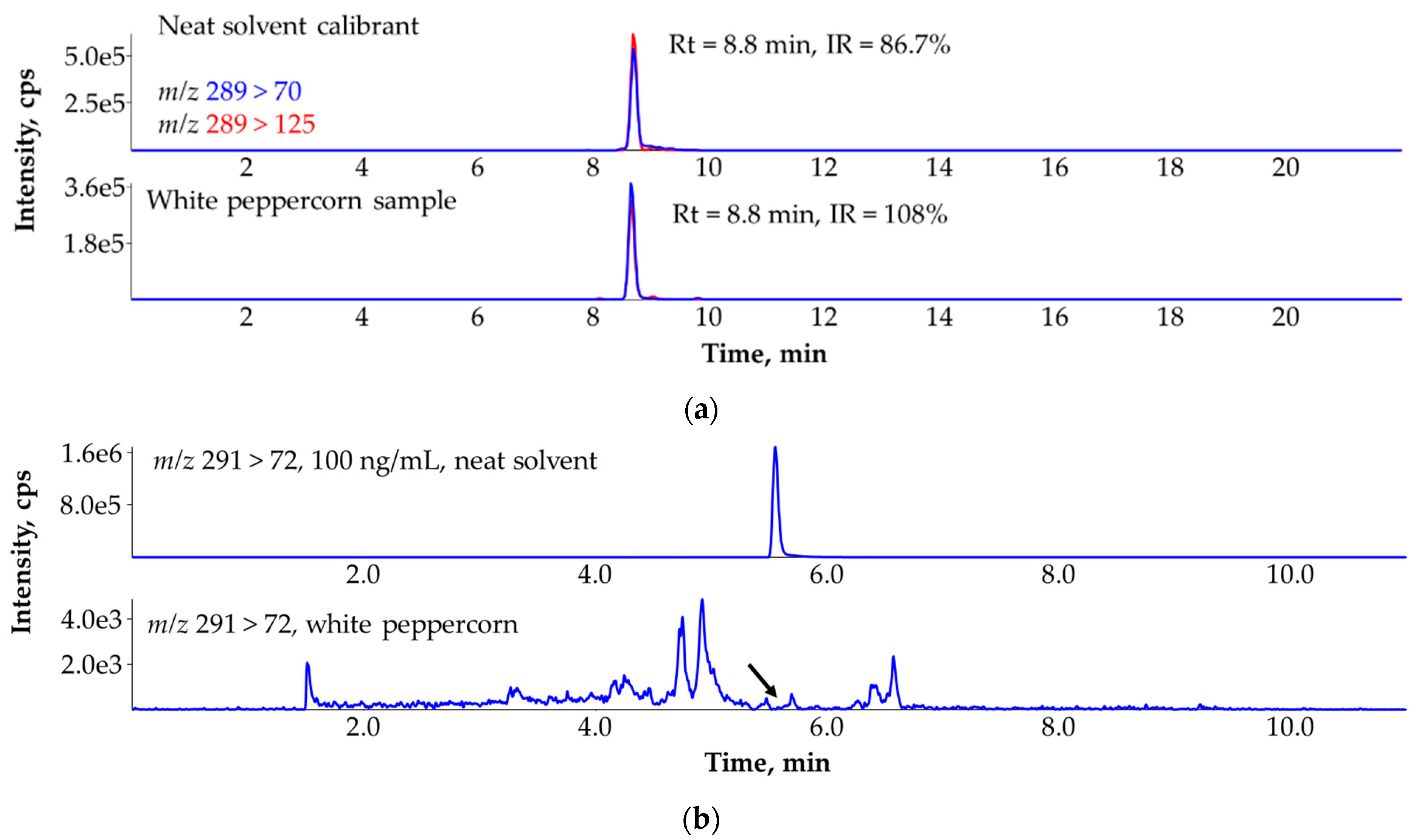
3.4. Ruling Out the False Identification of Myclobutanil in White Peppercorn (Piper nigrum) Sample with the Help of the A + 2 Isotopologue Based MRM Transition
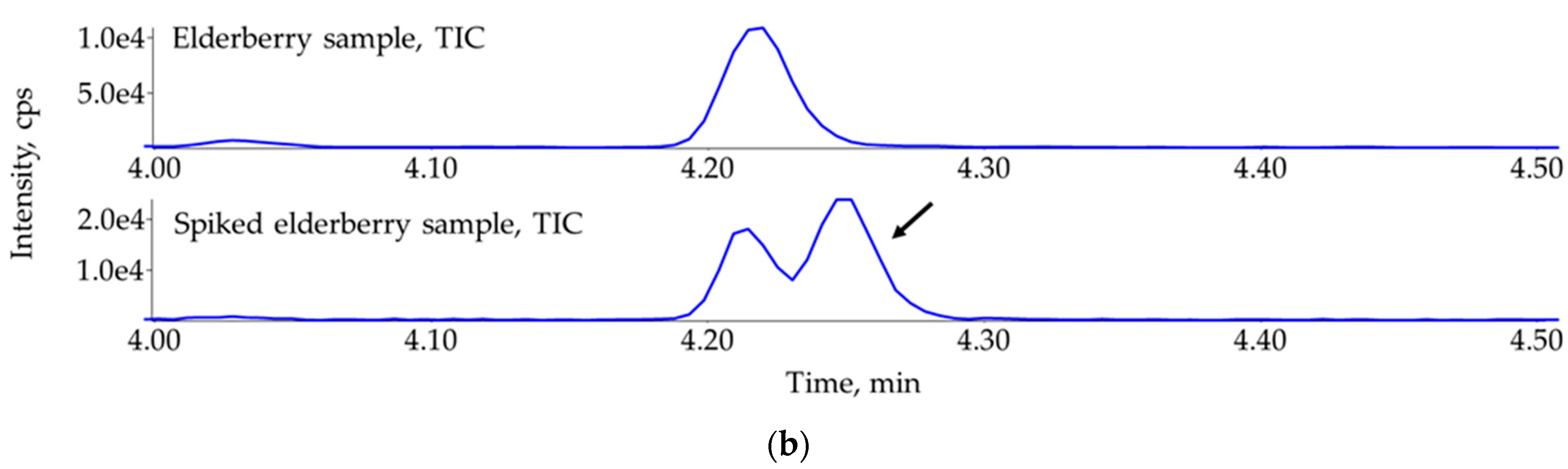
3.5. Ruling Out the False Identification of Propham in Elderberry (Sambucus nigra) Sample with the Help of Standard Addition
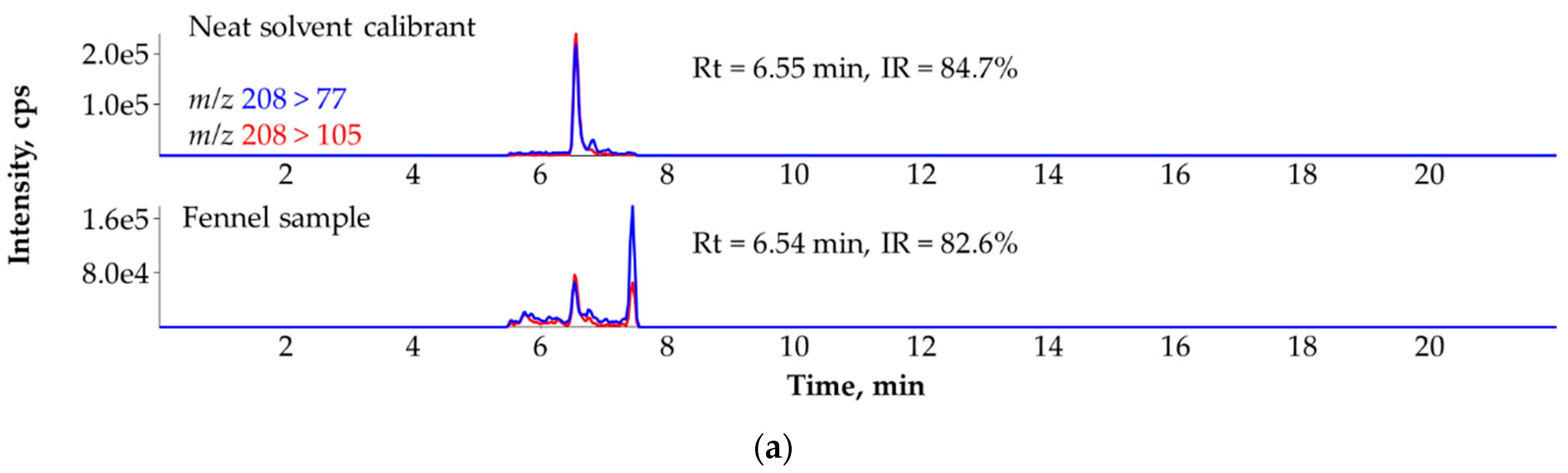
3.6. Ruling Out the False Identification of Quinoclamine in Fennel (Foeniculum vulgare) with the Help of Standard Addition and the Third MRM Transition
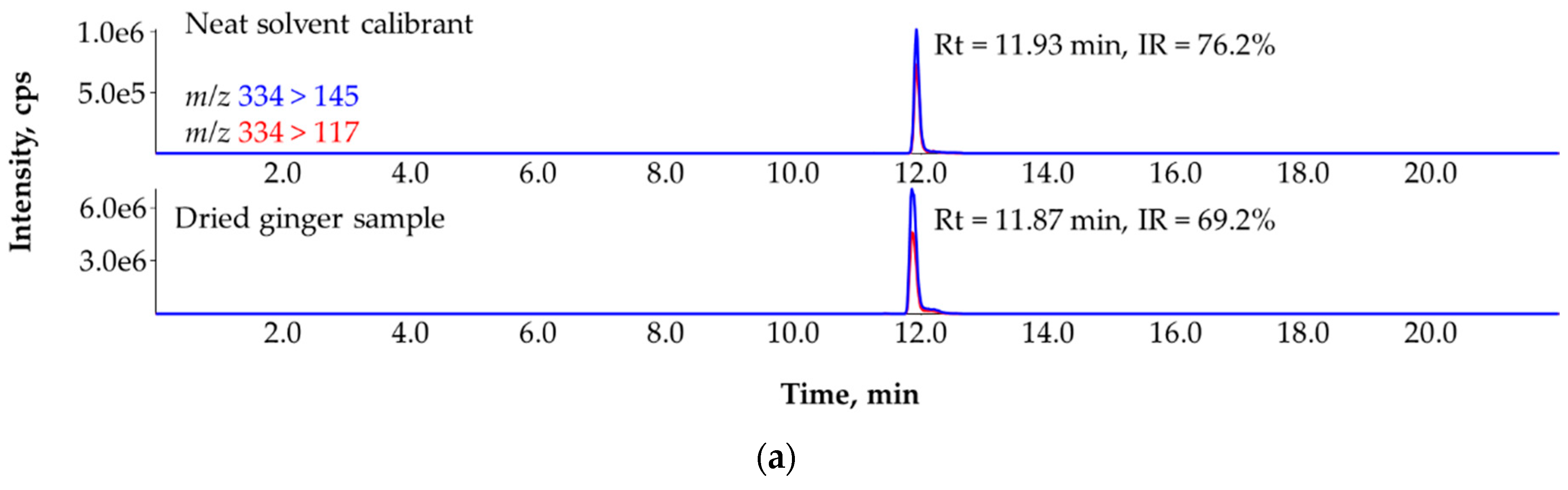
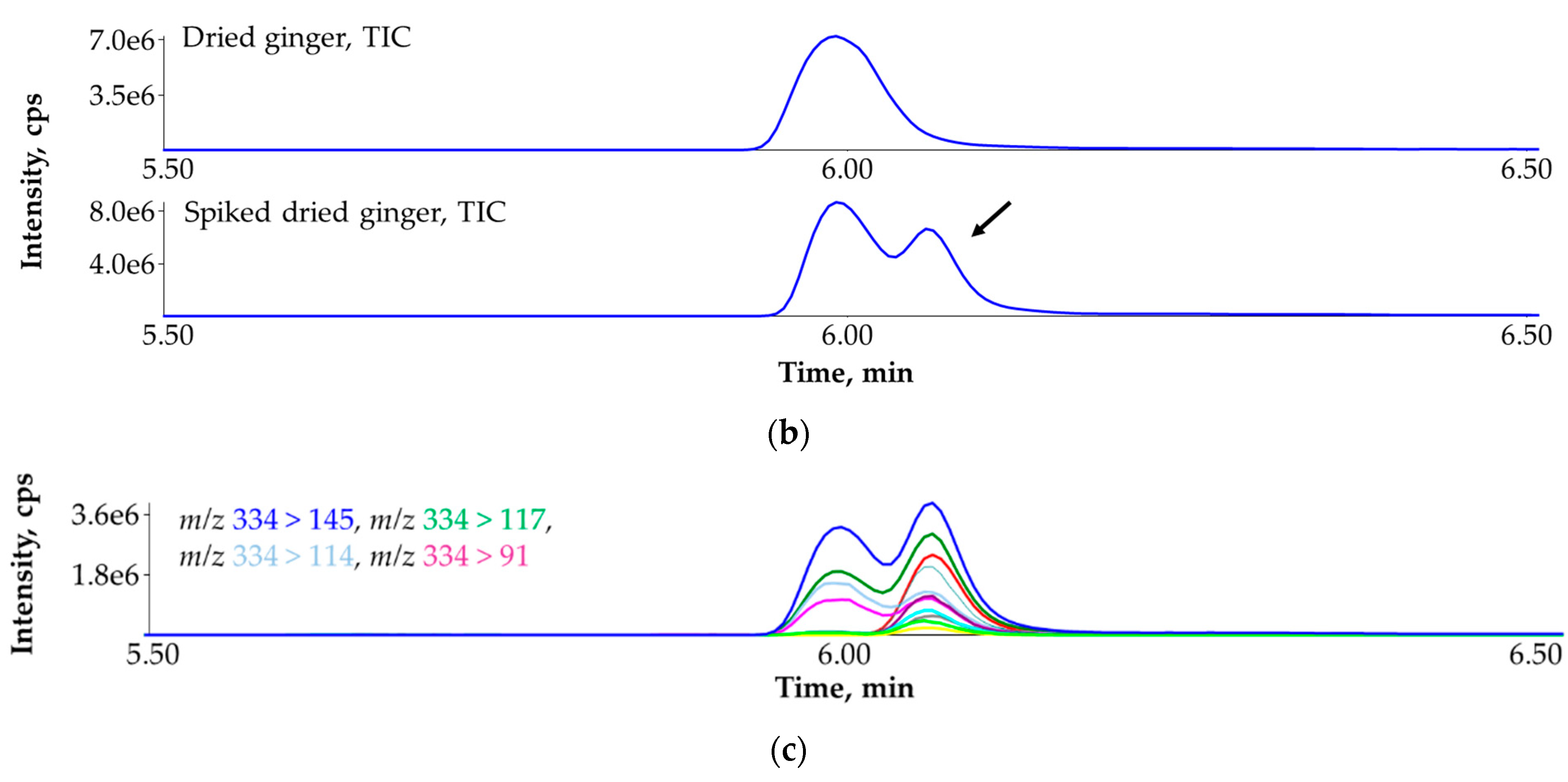
3.7. Ruling Out the False Identification of Tebufenpyrad in Dried Ginger (Zingiber officinale) with the Help of Standard Addition and More MRM Transitions
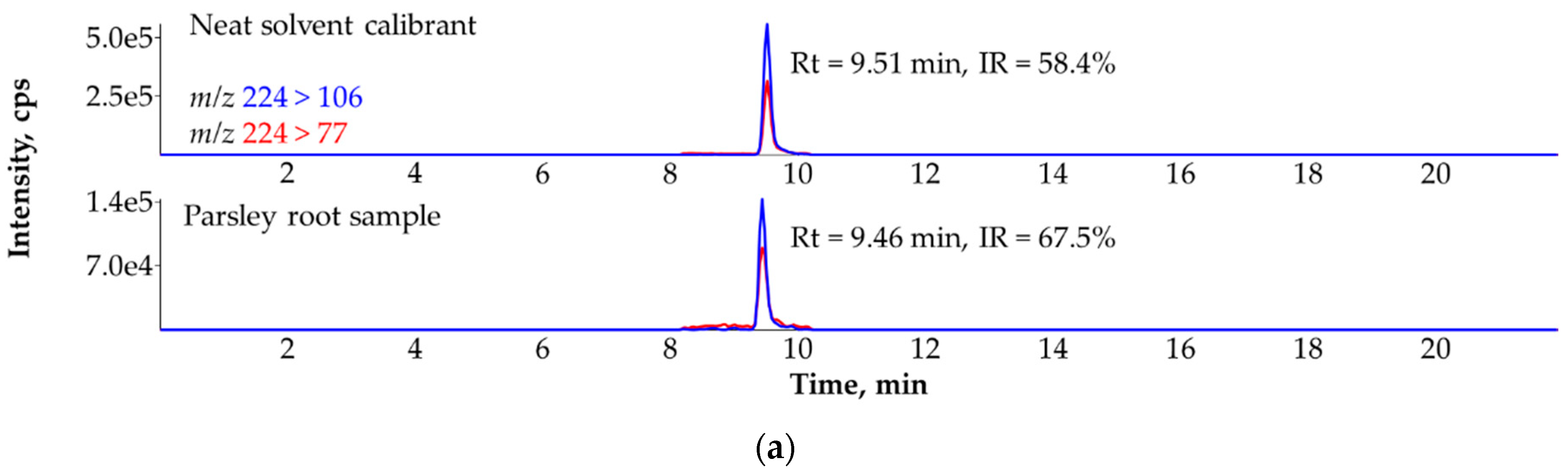
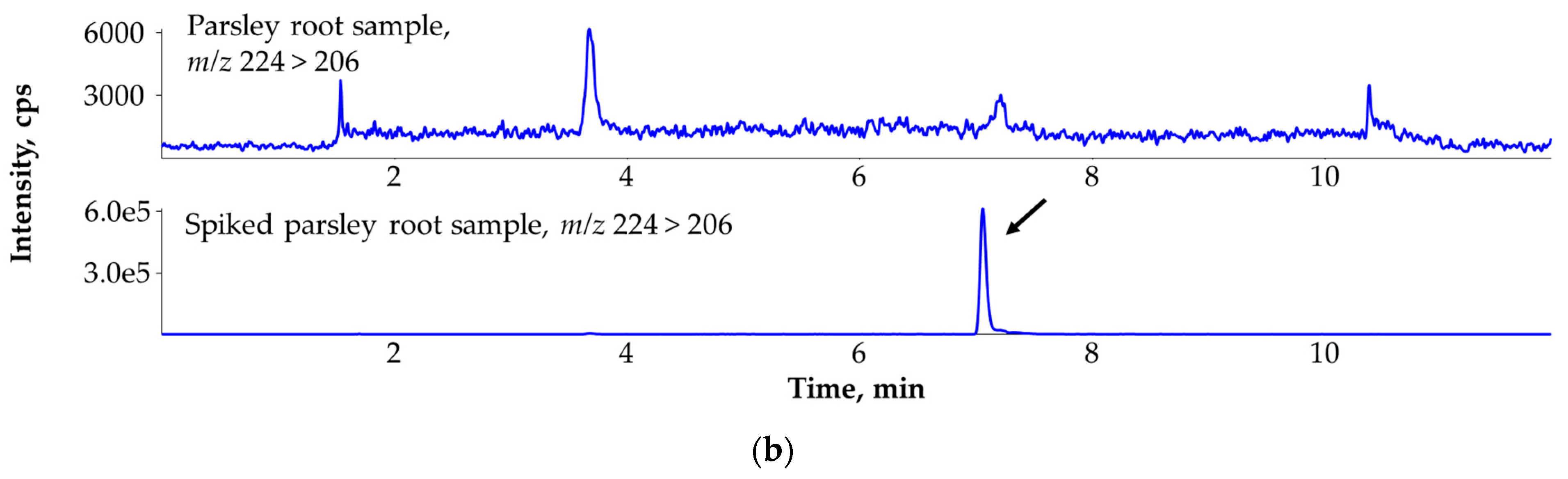
3.8. Ruling Out the False Identification of Mepanipyrim in Parsley Root (Petroselinum crispum) with the Help of the Third MRM Transition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Narenderan, S.T.; Meyyanathan, S.N.; Babu, B. Review of pesticide residue analysis in fruits and vegetables. Pre-treatment, extraction and detection techniques. Int. Food Res. J. 2020, 133, 109141. [Google Scholar] [CrossRef] [PubMed]
- Wahab, S.; Muzammil, K.; Nasir, N.; Khan, M.S.; Ahmad, M.F.; Khalid, M.; Ahmad, W.; Dawria, A.; Reddy, L.K.V.; Busayli, A.M. Advancement and New Trends in Analysis of Pesticide Residues in Food: A Comprehensive Review. Plants 2022, 11, 1106. [Google Scholar] [CrossRef] [PubMed]
- Schleiffer, M.; Speiser, B. Presence of pesticides in the environment, transition into organic food, and implications for quality assurance along the European organic food chain—A review. Environ. Pollut. 2022, 313, 120116. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.J.; McIntosh, L.J.; Mink, P.J.; Jurek, A.M.; Li, A.A. Pesticide exposure and neurodevelopmental outcomes: Review of the epidemiologic and animal studies. J. Toxicol. Environ. Health B Crit. Rev. 2013, 16, 127–283. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Nurul Amin, S.M.; Aminur Rahman, M.; Juraimi, A.S.; Uddin, K.; Brown, K.L.; Arshad, A. Chronic effects of organic pesticides on the aquatic environment and human health: A review. Environ. Nanotechnol. Monit. Manag. 2022, 18, 100740. [Google Scholar] [CrossRef]
- O’Driscoll, H.J.; Siggins, A.; Healy, M.G.; McGinley, J.; Mellander, P.-E.; Morrison, L.; Ryan, P.C. A risk ranking of pesticides in Irish drinking water considering chronic health effects. Sci. Total Environ. 2022, 829, 154532. [Google Scholar] [CrossRef] [PubMed]
- Regulation (EC) No. 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC. Available online: http://data.europa.eu/eli/reg/2005/396/2016-05-13 (accessed on 18 September 2022).
- Pesticide Residue Monitoring Program, Fiscal Year 2020 Pesticide Report, U.S. Food and Drug Administration. Available online: https://www.fda.gov/media/160464/download (accessed on 18 September 2022).
- Pesticide Evaluations: Overview and Procedure. Available online: https://www.efsa.europa.eu/en/applications/pesticides (accessed on 18 September 2022).
- Xie, Y.; Wu, X.; Song, Y.; Sun, Y.; Tong, K.; Yu, X.; Fan, C.; Chen, H. Screening of 258 Pesticide Residues in Silage Using Modified QuEChERS with Liquid- and Gas Chromatography-Quadrupole/Orbitrap Mass Spectrometry. Agriculture 2022, 12, 1231. [Google Scholar] [CrossRef]
- Ghosh, R.K.; Khan, Z.S.; Banerjee, K.; Reddy, D.D.; Johnson, N.; Ray, D.P. Elucidation of False Detection of Pesticides During Residue Analysis in Indian Tobacco by Multidimensional GC-MS. J. AOAC Int. 2020, 103, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.-W.; Park, M.-G.; Jeon, H.-J.; Moon, J.-K.; Lee, S.-E. Analysis of Multiresidue Pesticides in Agricultural Paddy Soils Near Industrial Areas in Korea by GC–MS/MS and LC–MS/MS Using QuEChERS Extraction with dSPE Clean-Up. Appl. Sci. 2021, 11, 8415. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, Z.; Huang, S.; Wang, J. Non-targeted screening of pesticides for food analysis using liquid chromatography high-resolution mass spectrometry-a review. Food Addit. Contam Part A 2020, 37, 1180–1201. [Google Scholar] [CrossRef] [PubMed]
- Kalogiouri, N.P.; Papadakis, E.-N.; Maggalou, M.G.; Karaoglanidis, G.S.; Samanidou, V.F.; Menkissoglu-Spiroudi, U. Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS. Appl. Sci. 2021, 11, 10931. [Google Scholar] [CrossRef]
- Jang, S.; Seo, H.; Kim, H.; Kim, H.; Ahn, J.; Cho, H.; Hong, S.; Lee, S.; Na, T. Development of a Quantitative Method for Detection of Multiclass Veterinary Drugs in Feed Using Modified QuPPe Extraction and LC–MS/MS. Molecules 2022, 27, 4483. [Google Scholar] [CrossRef] [PubMed]
- Tankiewicz, M.; Berg, A. Improvement of the QuEChERS method coupled with GC–MS/MS for the determination of pesticide residues in fresh fruit and vegetables. Microchem. J. 2022, 181, 107794. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Li, M.; Li, X.; Zhang, Q.; Li, H. Emulsification/demulsification method coupled to GC–MS/MS for analysis of multiclass pesticide residues in edible oils. Food Chem. 2022, 379, 132098. [Google Scholar] [CrossRef] [PubMed]
- BS EN 15662:2018; Foods of Plant Origin. Multimethod for the Determination of Pesticide Residues Using GC- and LC-Based Analysis Following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE. Modular QuEChERS-Method. 2018. Available online: https://www.en-standard.eu/bs-en-15662-2018-foods-of-plant-origin-multimethod-for-the-determination-of-pesticide-residues-using-gc-and-lc-based-analysis-following-acetonitrile-extraction-partitioning-and-clean-up-by-dispersive-spe-modular-quechers-method/ (accessed on 18 September 2022).
- Main Changes Introduced in Document N° SANTE/11312/2021 with Respect to the Previous Version (Document N° SANTE 12682/2019). Available online: https://food.ec.europa.eu/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf (accessed on 18 September 2022).
- Malato, O.; Lozano, A.; Mezcua, M.; Agüera, A.; Fernandez-Alba, A.R. Benefits and pitfalls of the application of screening methods for the analysis of pesticide residues in fruits and vegetables. J. Chromatogr. A 2011, 1218, 7615–7626. [Google Scholar] [CrossRef] [PubMed]
- Mol, H.G.J.; Zomer, P.; de Koning, M. Qualitative aspects and validation of a screening method for pesticides in vegetables and fruits based on liquid chromatography coupled to full scan high resolution (Orbitrap) mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 2891–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schürmann, A.; Dvorak, V.; Crüzer, C.; Butcher, P.; Kaufmann, A. False-positive liquid chromatography/tandem mass spectrometric confirmation of sebuthylazine residues using the identification points system according to EU directive 2002/657/EC due to a biogenic insecticide in tarragon. Rapid Commun. Mass Spectrom. 2009, 23, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Mol Hans, G.J.; Zomer, P.; López, M.G.; Fussell, R.J.; Scholten, J.; de Kok, A.; Wolheim, A.; Anastassiades, M.; Lozano, A.; Alba, A.F. Identification in residue analysis based on liquid chromatography with tandem mass spectrometry: Experimental evidence to update performance criteria. Anal. Chim. Acta 2015, 873, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Tölgyesi, Á.; Simon, A.; Bálint, M.; Ma, X.; Sharma, V.K. An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry. Molecules 2022, 27, 1872. [Google Scholar] [CrossRef] [PubMed]
- EU Pesticides Database. Available online: https://ec.europa.eu/food/plants/pesticides/eu-pesticides-database_hu (accessed on 18 September 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Benalaxyl | 68.8 | 71.5 | 0.2 |
| Boscalid | 96.1 | 88.9 | −0.3 |
| Diazinon | 19.5 | 20.3 | 0.2 |
| Metolachlor (sum of constituent isomers including S-metolachlor) | 91.2 | 94.8 | 0.2 |
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Buprofezin | 42.8 | 53.0 | 1.1 |
| Dimethoate | 152 | 166 | 0.4 |
| Hexythiazox | 54.6 | 63.0 | 0.7 |
| Methamidophos | 71.8 | 84.0 | 0.8 |
| Methomyl | 64.5 | 64.0 | 0.0 |
| Phenthoate | 87.2 | 98.0 | 0.6 |
| Pyrimethanil | 109 | 128 | 0.8 |
| Quinalphos | 72.8 | 84.0 | 0.7 |
| Tebuconazole | 137 | 162 | 0.8 |
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Dinotefuran | 76.2 | 83.1 | 0.4 |
| Kresoxim-methyl | 36.2 | 53.4 | 2.2 |
| Omethoate | 117 | 143 | 1.0 |
| Pendimethalin | 41.1 | 58.6 | 1.9 |
| Proquinazid | 82.7 | 101 | 1.0 |
| Tebufenpyrad | 59.1 | 74.9 | 1.2 |
| Thiacloprid | 75.9 | 80.1 | 0.3 |
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Azoxystrobin | 36.5 | 43.3 | 0.8 |
| Carbendazim | 46.2 | 51.3 | 0.5 |
| Carbofuran | 62.4 | 69.6 | 0.5 |
| Clothianidin | 77.0 | 83.8 | 0.4 |
| Cyprodinil | 42.7 | 45.9 | 0.3 |
| Dimethoate | 57.1 | 62.8 | 0.5 |
| Dimethomorph (sum of isomers) | 87.8 | 88.6 | 0.0 |
| Emamectin (as emamectin benzoate B1a, expressed as emamectin) | 61.1 | 72.3 | 0.8 |
| Monocrotophos | 47.5 | 50.9 | 0.3 |
| Omethoate | 78.6 | 83.7 | 0.3 |
| Tetraconazole | 75.5 | 82.7 | 0.4 |
| Trifloxystrobin | 112 | 118 | 0.3 |
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Atrazine | 39.3 | 47.2 | 0.77 |
| Azoxystrobin | 172 | 192 | 0.48 |
| Bocalid | 50.2 | 80.9 | 2.23 |
| Cumaphos | 51.4 | 52.8 | 0.11 |
| Linuron | 75.3 | 75.1 | −0.01 |
| Pyrimethanil | 70.5 | 47.9 | 0.71 |
| Triadimefon | 96.9 | 99.8 | 0.13 |
| Component | Assigned Value (µg/kg) | Result (µg/kg) | z-Score |
|---|---|---|---|
| Malathion | 58.0 | 49.1 | −0.7 |
| Phosmet | 51.3 | 52.6 | 0.1 |
| Prochloraz | 58.2 | 63.7 | 0.4 |
| Propoxur | 78.0 | 71.0 | −0.4 |
| Tetraconazole | 76.0 | 70.8 | −0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, E.; Bálint, M.; Tölgyesi, Á. False Positive Identification of Pesticides in Food Using the European Standard Method and LC-MS/MS Determination: Examples and Solutions from Routine Applications. Appl. Sci. 2022, 12, 12005. https://doi.org/10.3390/app122312005
Tóth E, Bálint M, Tölgyesi Á. False Positive Identification of Pesticides in Food Using the European Standard Method and LC-MS/MS Determination: Examples and Solutions from Routine Applications. Applied Sciences. 2022; 12(23):12005. https://doi.org/10.3390/app122312005
Chicago/Turabian StyleTóth, Edgár, Mária Bálint, and Ádám Tölgyesi. 2022. "False Positive Identification of Pesticides in Food Using the European Standard Method and LC-MS/MS Determination: Examples and Solutions from Routine Applications" Applied Sciences 12, no. 23: 12005. https://doi.org/10.3390/app122312005