
Molecular Dynamics Study of Interfacial Properties for Crude Oil with Pure and Impure CH4


Abstract
:1. Introduction
2. Methodology
2.1. Molecular Model
2.2. Force Field
2.3. Simulation Details
3. Results and Discussion
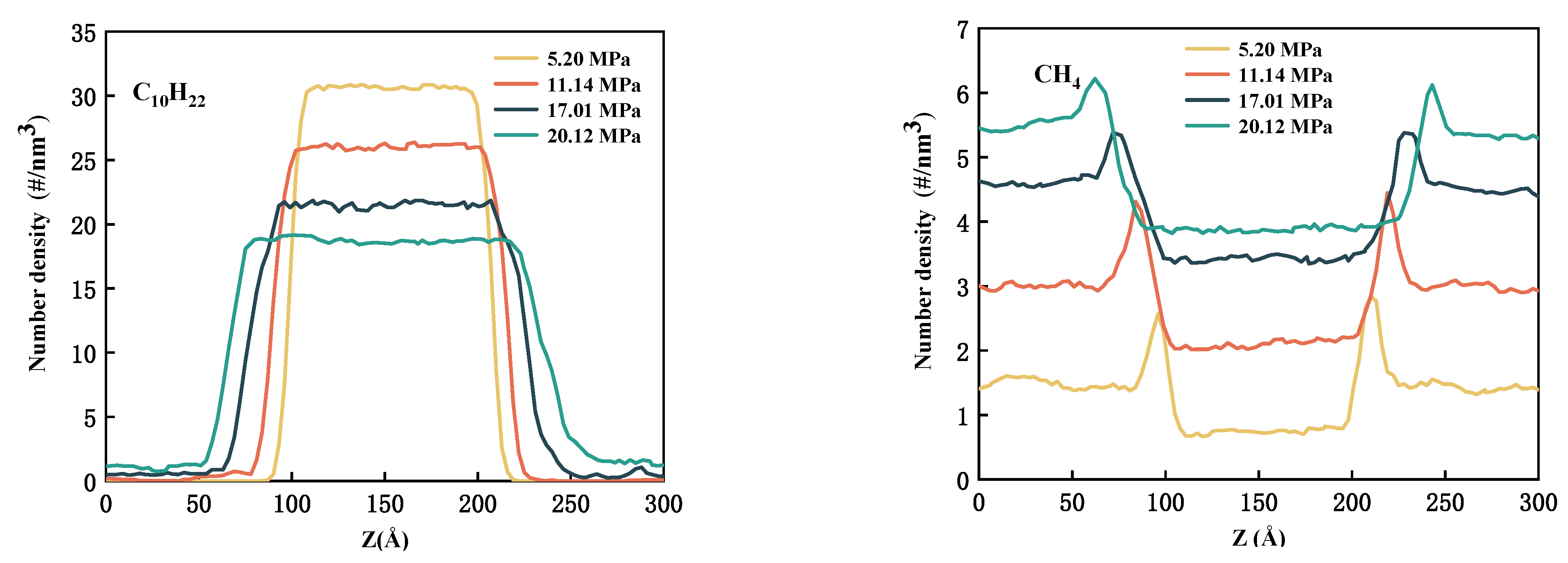
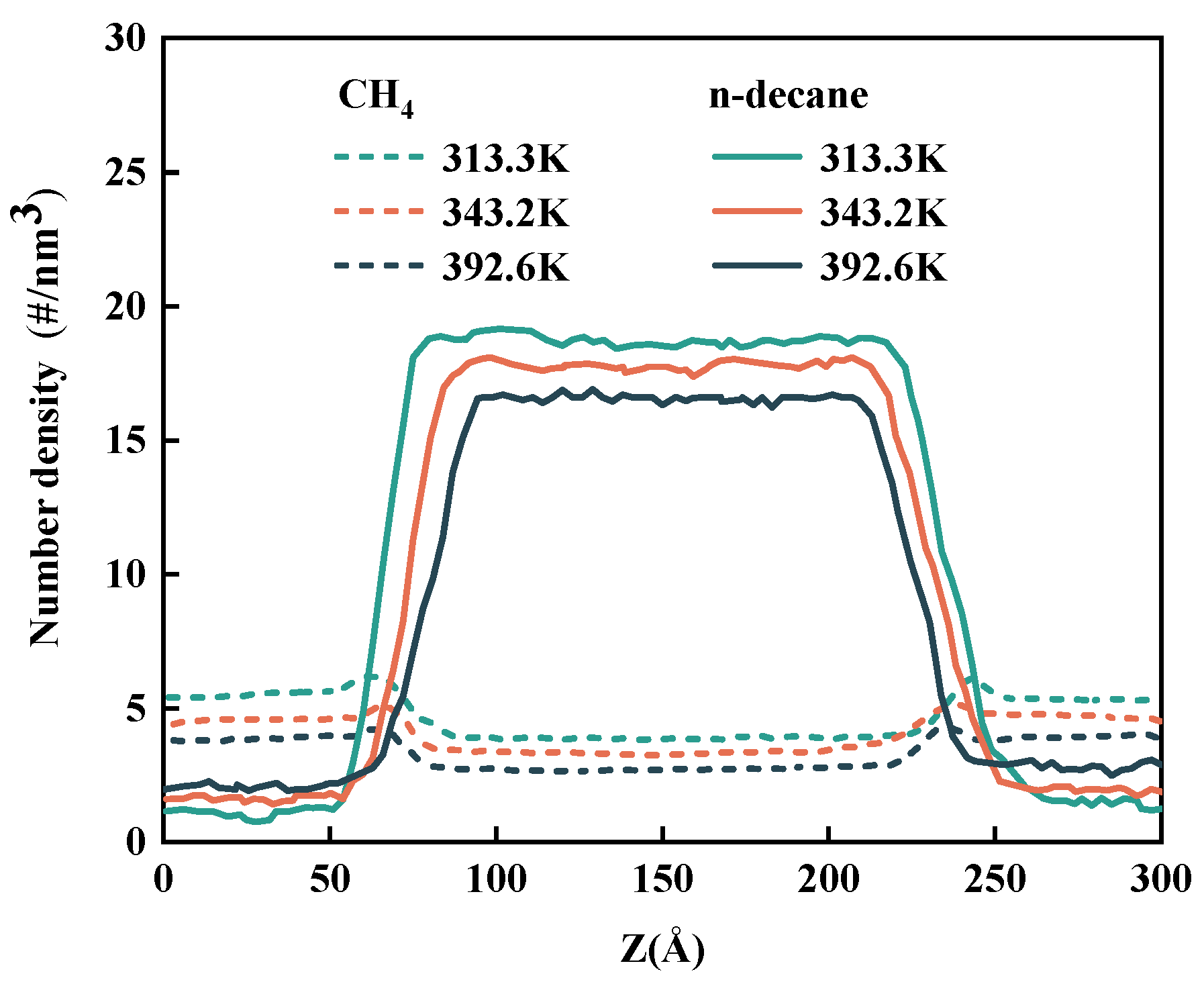
3.1. Density Profile
3.2. Interfacial Tension
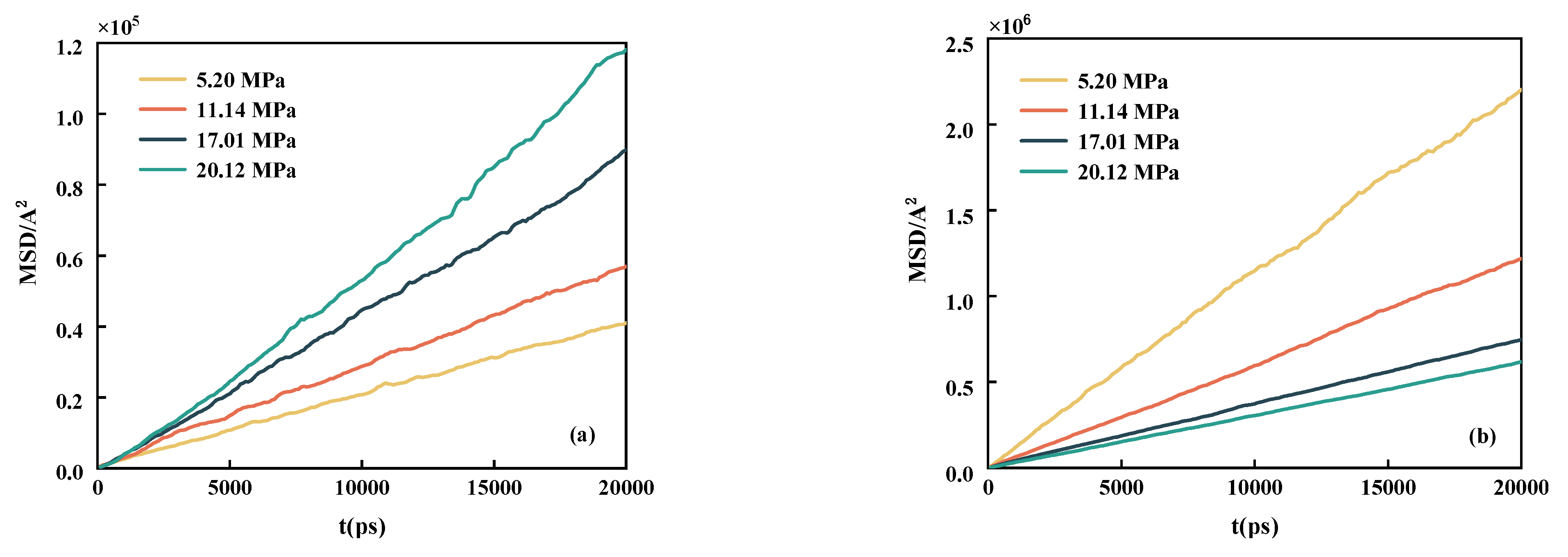
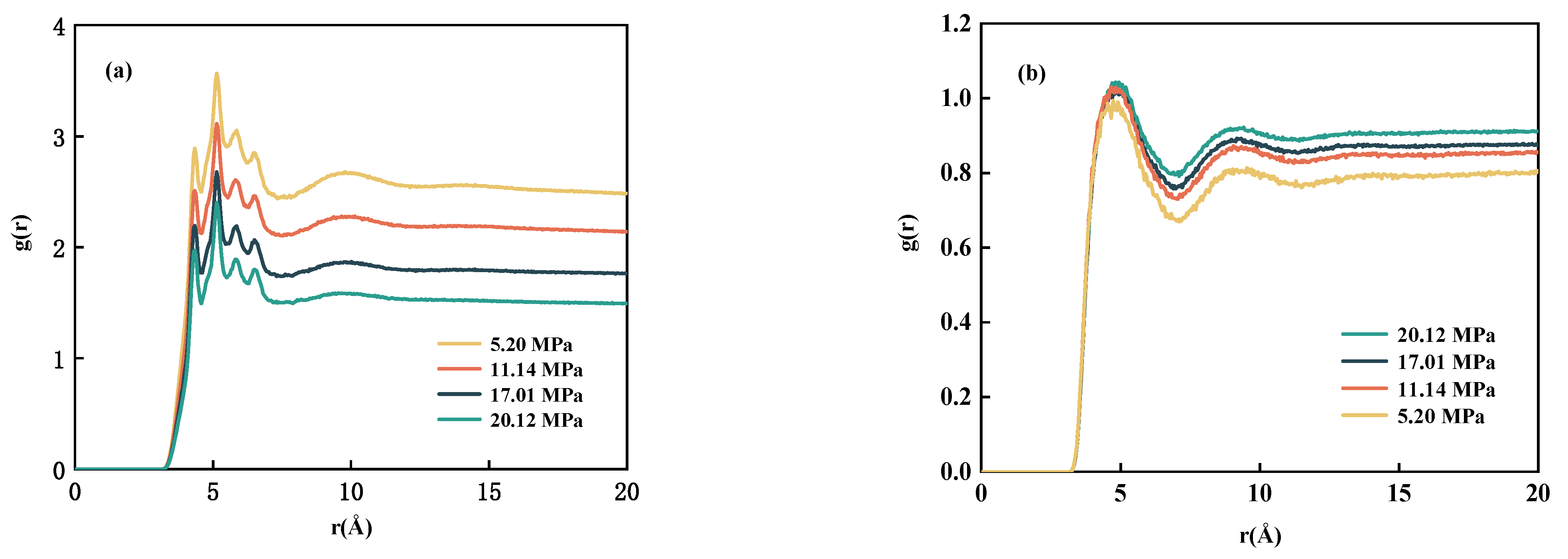
3.3. Diffusion Properties
4. Conclusions
- With rising pressure and falling temperature, there will be more CH4 enrichment at the interface. It is worth noting that after adding CO2 to CH4, the accumulation of CO2 at the interface is greater than that of CH4.
- Under low pressure, the IFT of the CH4+n-decane system decreases dramatically as temperature rises. The IFT remains unchanged as the temperature rises when pressure is high.
- The addition of CO2 can lower the IFT of the CH4+n-decane system. The IFT is affected in the opposite way by the addition of n-hexane and n-nonadecane, with the former having a lower IFT and the latter having a greater IFT.
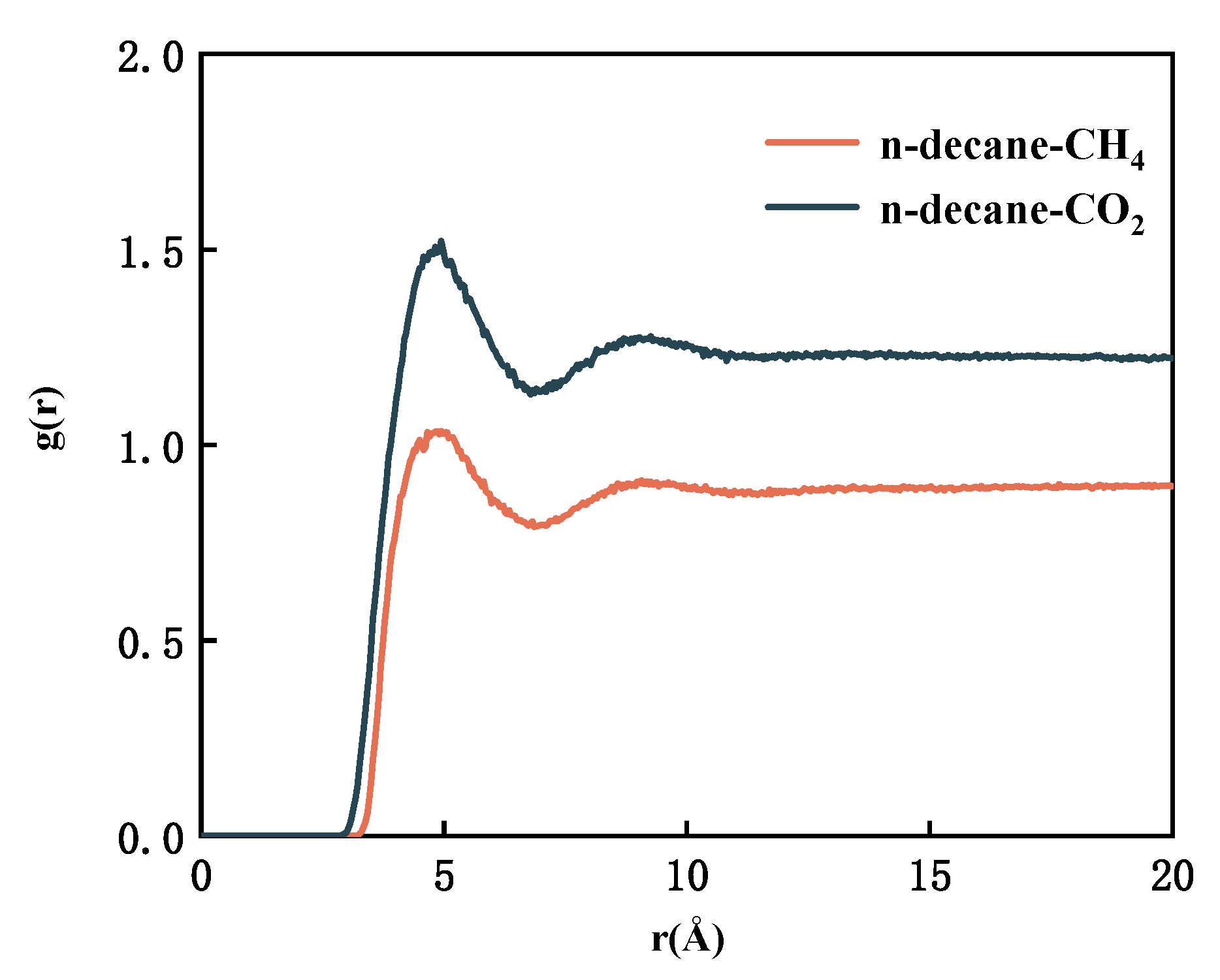
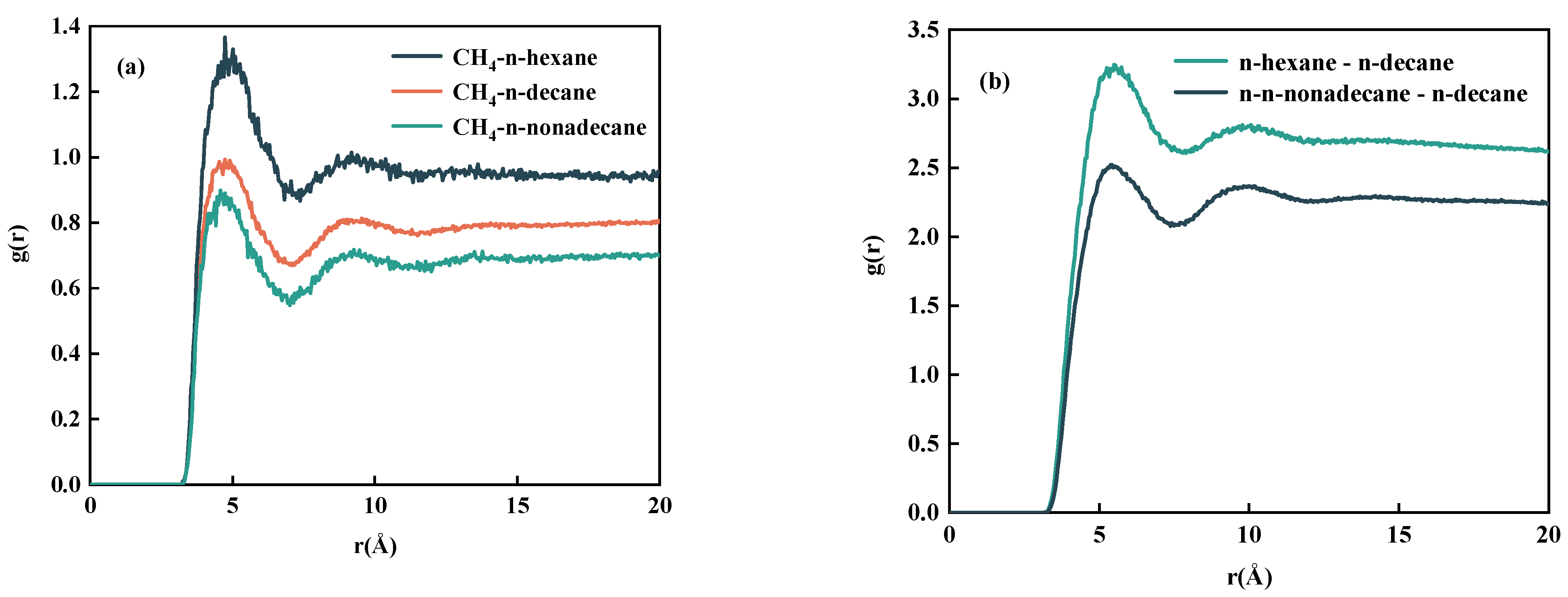
- By studying the diffusion properties, it was discovered that CH4 can weak the interaction between n-decane, thereby increasing its diffusion ability. One significant finding is that the interaction between n-decane and CO2 is significantly more potent than that of CH4. Simultaneously, systems containing lighter components are more easily aggregated around CH4 and dissolved in CH4.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wankui, G.; Guangzhi, L. Gas Injection Enhanced Recovery Technology; Petroleum Industry Press: Beijing, China, 2003; pp. 2–7. [Google Scholar]
- Baidakov, V.G.; Khotienkova, M.N. Capillary constant and surface tension of propane (R-290) with helium dissolved in it. Int. J. Refrig. 2019, 98, 261–265. [Google Scholar] [CrossRef]
- Fukuta, M.; Sumiyama, J.; Motozawa, M.; Yanagisawa, T. Surface tension measurement of oil/refrigerant mixture by maximum bubble pressure method. Int. J. Refrig. 2017, 73, 125–133. [Google Scholar] [CrossRef]
- Fainerman, V.B.; Miller, R.; Joos, P. The measurement of dynamic surface tension by the maximum bubble pressure method. Colloid Polym. Sci. 1994, 272, 731–739. [Google Scholar] [CrossRef]
- Andreas, J.M.; Hauser, E.A.; Tucker, W.B. Boundary tension by pendant drops. J. Phys. Chem. 1938, 42, 1001–1019. [Google Scholar] [CrossRef]
- Wakayama, N.I. Effect of a decreasing magnetic field on the flow of nitrogen gas. Chem. Phys. Lett. 1991, 185, 449–451. [Google Scholar] [CrossRef]
- Chapela, G.A.; Martínez-Casas, S.E.; Varea, C. Square well orthobaric densities via spinodal decomposition. J. Chem. Phys. 1987, 86, 5683–5688. [Google Scholar] [CrossRef]
- Chapela, G.A.; Saville, G.; Thompson, S.M.; Rowlinson, J.S. Computer simulation of a gas–liquid surface. Part 1. J. Chem. Soc. Faraday Trans. 1977, 73, 1133–1144. [Google Scholar] [CrossRef]
- Nijmeijer, M.J.P.; Bakker, A.F.; Bruin, C.; Sikkenk, J.H. A molecular dynamics simulation of the Lennard-Jones liquid–vapor interface. J. Chem. Phys. 1988, 89, 3789–3792. [Google Scholar] [CrossRef]
- Thompson, S.M.; Gubbins, K.E.; Walton, J.P.R.B.; Chantry, R.A.R.; Rowlinson, J.S. A molecular dynamics study of liquid drops. J. Chem. Phys. 1984, 81, 530–542. [Google Scholar] [CrossRef]
- Mecke, M.; Winkelmann, J.; Fischer, J. Molecular dynamics simulation of the liquid-vapor interface: The Lennard-Jones fluid. J. Chem. Phys. 1997, 107, 9264–9270. [Google Scholar] [CrossRef]
- Bhansali, A.P.; Bayazitoglu, Y.; Maruyama, S. Molecular dynamics simulation of an evaporating sodium droplet. Int. J. Therm. Sci. 1999, 38, 66–74. [Google Scholar] [CrossRef]
- Trokhymchuk, A.; Alejandre, J. Computer simulations of liquid/vapor interface in Lennard-Jones fluids: Some questions and answers. J. Chem. Phys. 1999, 111, 8510–8523. [Google Scholar] [CrossRef]
- Mecke, M.; Winkelmann, J.; Fischer, J. Molecular dynamics simulation of the liquid-vapor interface: Binary mixtures of Lennard-Jones fluids. J. Chem. Phys. 1999, 110, 1188–1194. [Google Scholar] [CrossRef]
- Frezzotti, A. Boundary conditions at the vapor-liquid interface. Phys. Fluids 2011, 23, 030609. [Google Scholar] [CrossRef]
- Chilukoti, H.K.; Kikugawa, G.; Ohara, T. A molecular dynamics study on transport properties and structure at the liquid-vapor interfaces of alkanes. Int. J. Heat Mass Tran. 2013, 59, 144–154. [Google Scholar] [CrossRef]
- Weinaug, C.F.; Katz, D.L. Surface Tensions of Methane-Propane Mixtures. Ind. Eng. Chem. 1943, 35, 239–246. [Google Scholar] [CrossRef]
- Amin, R.; Smith, T.N. Interfacial tension and spreading coefficient under reservoir conditions. Fluid Phase Equilibria 1998, 142, 231–241. [Google Scholar] [CrossRef]
- Miqueu, C.; Mendiboure, B.; Graciaa, C.; Lachaise, J. Modeling of the surface tension of binary and ternary mixtures with the gradient theory of fluid interfaces. Fluid Phase Equilibria 2004, 218, 189–203. [Google Scholar] [CrossRef]
- Ramírez-Verduzco, L.F.; Romero-Martínez, A.; Trejo, A. Prediction of the surface tension, surface concentration, and the relative Gibbs adsorption isotherm of binary liquid systems. Fluid Phase Equilibria 2006, 246, 119–130. [Google Scholar] [CrossRef]
- Pereira, L.M.C.; Chapoy, A.; Burgass, R.; Tohidi, B. Measurement and modeling of high pressure density and interfacial tension of (gas + n -alkane) binary mixtures. J. Chem. Thermodyn. 2016, 97, 55–69. [Google Scholar] [CrossRef]
- Choudhary, N.; Narayanan Nair, A.K.; Che Ruslan, M.F.A.; Sun, S. Bulk and interfacial properties of decane in the presence of carbon dioxide, methane, and their mixture. Sci. Rep. 2019, 9, 19784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, T.; Lenahan, F.D.; Kerscher, M.; Jander, J.H.; Rausch, M.H.; Koller, T.M. Viscosity and Interfacial Tension of Binary Mixtures of n-Hexadecane with Dissolved Gases Using Surface Light Scattering and Equilibrium Molecular Dynamics Simulations. J. Chem. Eng. Data 2021, 66, 3205–3218. [Google Scholar] [CrossRef]
- De Lara, L.S.; Michelon, M.F.; Miranda, C.R. Molecular Dynamics Studies of Fluid/Oil Interfaces for Improved Oil Recovery Processes. J. Phys. Chem. B 2012, 116, 14667–14676. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.K.; Escobedo, F.A.; de Pablo, J.J. On the simulation of vapor-liquid equilibria for alkanes. J. Chem. Phys. 1998, 108, 9905–9911. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Zhu, A.; Zhang, X.; Liu, Q.; Zhang, Q. A Fully Flexible Potential Model for Carbon Dioxide. Chin. J. Chem. Eng. 2009, 17, 268–272. [Google Scholar]
- Stanishneva-Konovalova, T.B.; Sokolova, O.S. Molecular dynamics simulations of negatively charged DPPC/DPPI lipid bilayers at two levels of resolution. Comput. Theor. Chem. 2015, 1058, 61–66. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Hoover, W.G.; Ladd, A.J.C.; Moran, B. High-Strain-Rate Plastic Flow Studied via Nonequilibrium Molecular Dynamics. Phys. Rev. Lett. 1982, 48, 1818–1820. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Pollock, E.L.; Glosli, J. Comments on P3M, FMM, and the Ewald method for large periodic Coulombic systems. Comput. Phys. Commun. 1996, 95, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Müller, E.A.; Mejía, A. Interfacial properties of selected binary mixtures containing n-alkanes. Fluid Phase Equilibria 2009, 282, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Bresme, F.; Chacón, E.; Tarazona, P.; Tay, K. Intrinsic Structure of Hydrophobic Surfaces: The Oil-Water Interface. Phys. Rev. Lett. 2008, 101, 056102. [Google Scholar] [CrossRef] [Green Version]
- Bresme, F.; Chacón, E.; Tarazona, P. Force-field dependence on the interfacial structure of oil–water interfaces. Mol. Phys. 2010, 108, 1887–1898. [Google Scholar] [CrossRef]
- Zhang, Y.; Feller, S.E.; Brooks, B.R.; Pastor, R.W. Computer simulation of liquid/liquid interfaces. I. Theory and application to octane/water. J. Chem. Phys. 1995, 103, 10252–10266. [Google Scholar] [CrossRef]
- Smith, F.T. Atomic Distortion and the Combining Rule for Repulsive Potentials. Phys. Rev. A 1972, 5, 1708–1713. [Google Scholar] [CrossRef]
- Mo, Z.; Zhu, X.; Wang, G.; Han, W.; Guo, R. Molecular dynamics simulation on the interaction between single-walled carbon nanotubes and binaphthyl core-based chiral phenylene dendrimers. J. Mater. Res. 2014, 29, 2156–2161. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular | Atom | σ (nm) | εij (KJ·mol−1) | qi (e) | Model |
|---|---|---|---|---|---|
| n-decane | C(CH3) | 0.3910 | 0.8647 | 0 | NERD [26] |
| C(CH2) | 0.3930 | 0.3808 | 0 | ||
| CH4 | C(CH4) | 0.3730 | 1.2300 | 0 | TraPPE-UA [27] |
| CO2 | C | 0.2800 | 0.23397 | +0.6512 | ZHU [28] |
| O | 0.3028 | 0.66824 | −0.3256 |
| Stretch | r0 (nm) | (KJ·mol−1·nm−2) | ||
|---|---|---|---|---|
| CHx-CHy | 0.1540 | 80,235.028 | ||
| C-O | 0.1162 | 60,000 | ||
| Bend | θ0 (°) | (KJ·mol−1·rad−2) | ||
| CHx-CH2-CHy | 114.0 | 519.657 | ||
| O-C-O | 180 | 110 | ||
| Torsion | V0 (KJ·mol−1) | V1 (KJ·mol−1) | V2 (KJ·mol−1) | V3 (KJ·mol−1) |
| CHx-CH2-CH2-CHy | 0 | 5.9038 | −1.1339 | 13.159 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Ma, X.; Xu, H.; Li, W.; Qian, S.; Wang, Z.; Liu, Z.; Lei, G. Molecular Dynamics Study of Interfacial Properties for Crude Oil with Pure and Impure CH4. Appl. Sci. 2022, 12, 12239. https://doi.org/10.3390/app122312239
Dong Z, Ma X, Xu H, Li W, Qian S, Wang Z, Liu Z, Lei G. Molecular Dynamics Study of Interfacial Properties for Crude Oil with Pure and Impure CH4. Applied Sciences. 2022; 12(23):12239. https://doi.org/10.3390/app122312239
Chicago/Turabian StyleDong, Zhenzhen, Xinle Ma, Haobin Xu, Weirong Li, Shihao Qian, Zhengbo Wang, Zhaoxia Liu, and Gang Lei. 2022. "Molecular Dynamics Study of Interfacial Properties for Crude Oil with Pure and Impure CH4" Applied Sciences 12, no. 23: 12239. https://doi.org/10.3390/app122312239
APA StyleDong, Z., Ma, X., Xu, H., Li, W., Qian, S., Wang, Z., Liu, Z., & Lei, G. (2022). Molecular Dynamics Study of Interfacial Properties for Crude Oil with Pure and Impure CH4. Applied Sciences, 12(23), 12239. https://doi.org/10.3390/app122312239