
Comparative Analysis of Grapevine Epiphytic Microbiomes among Different Varieties, Tissues, and Developmental Stages in the Same Terroir



Abstract
:1. Introduction
2. Materials and Methods
2.1. Vineyard, Vine Cultivars and Sampling
2.2. DNA Extraction
2.3. High-Throughput Sequencing and Statistical Analysis
3. Results
3.1. Diversity Assessment among Phenological Stages
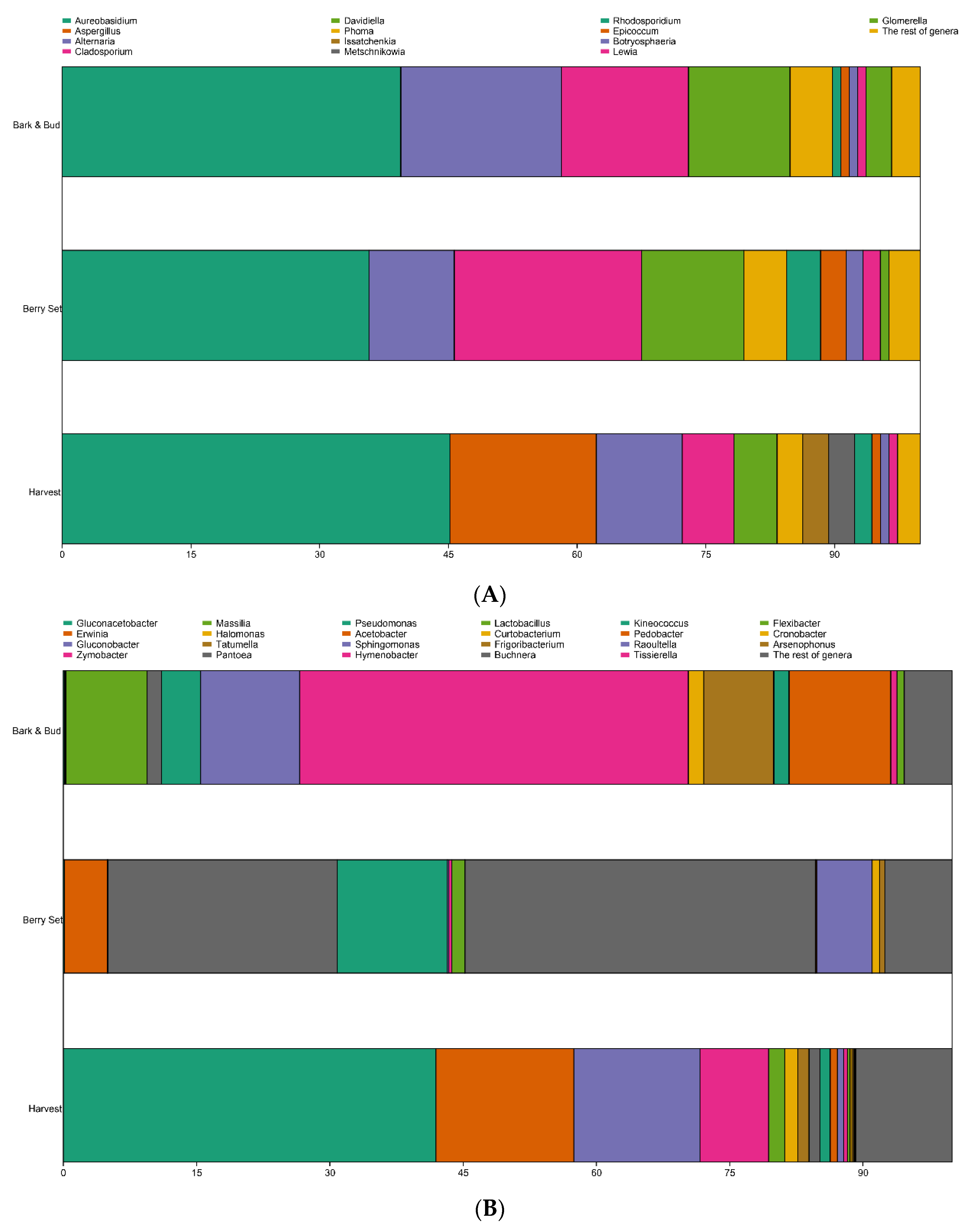
3.2. Microbiome Varies According to Grapevine Phenological Stage
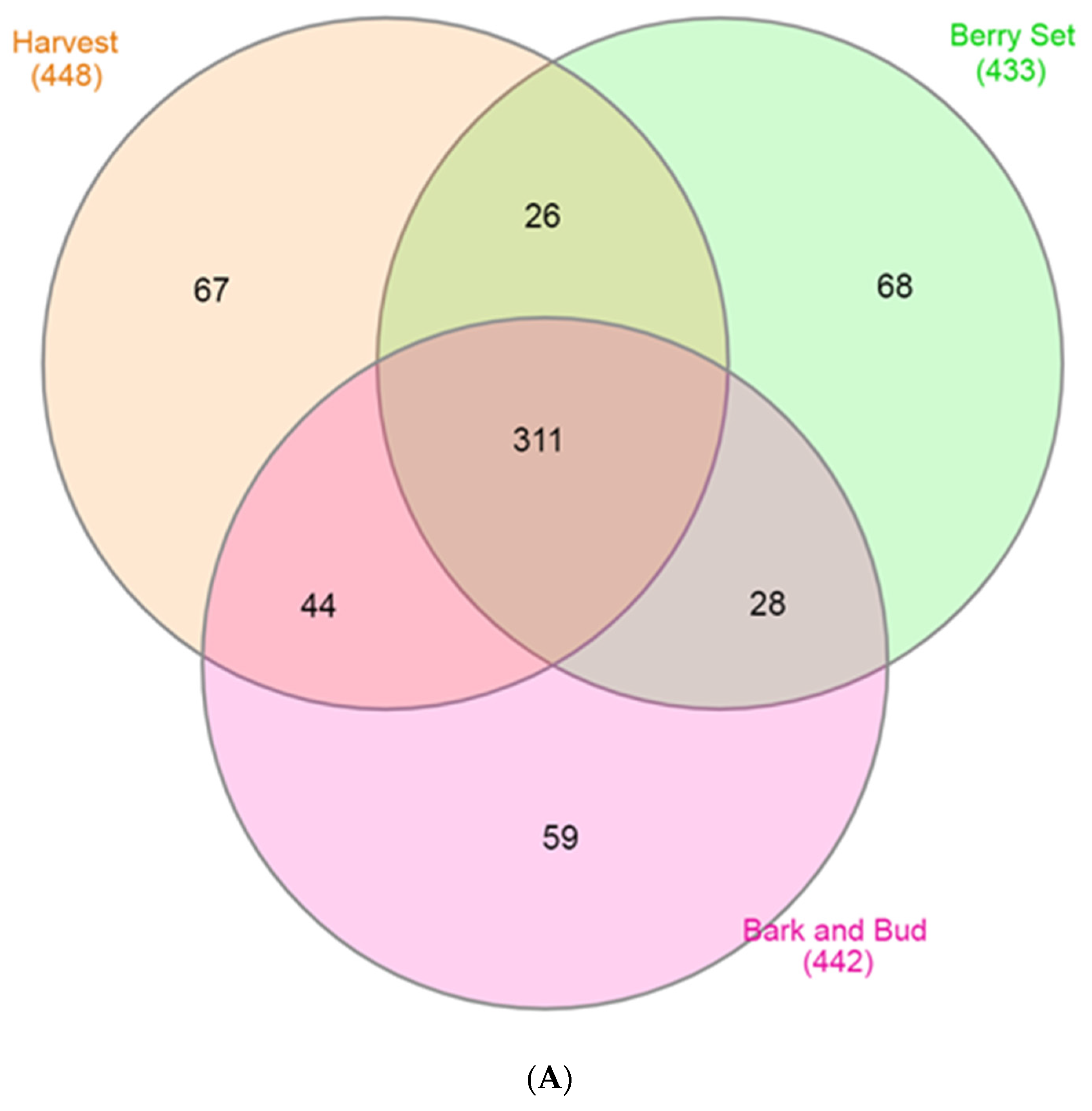
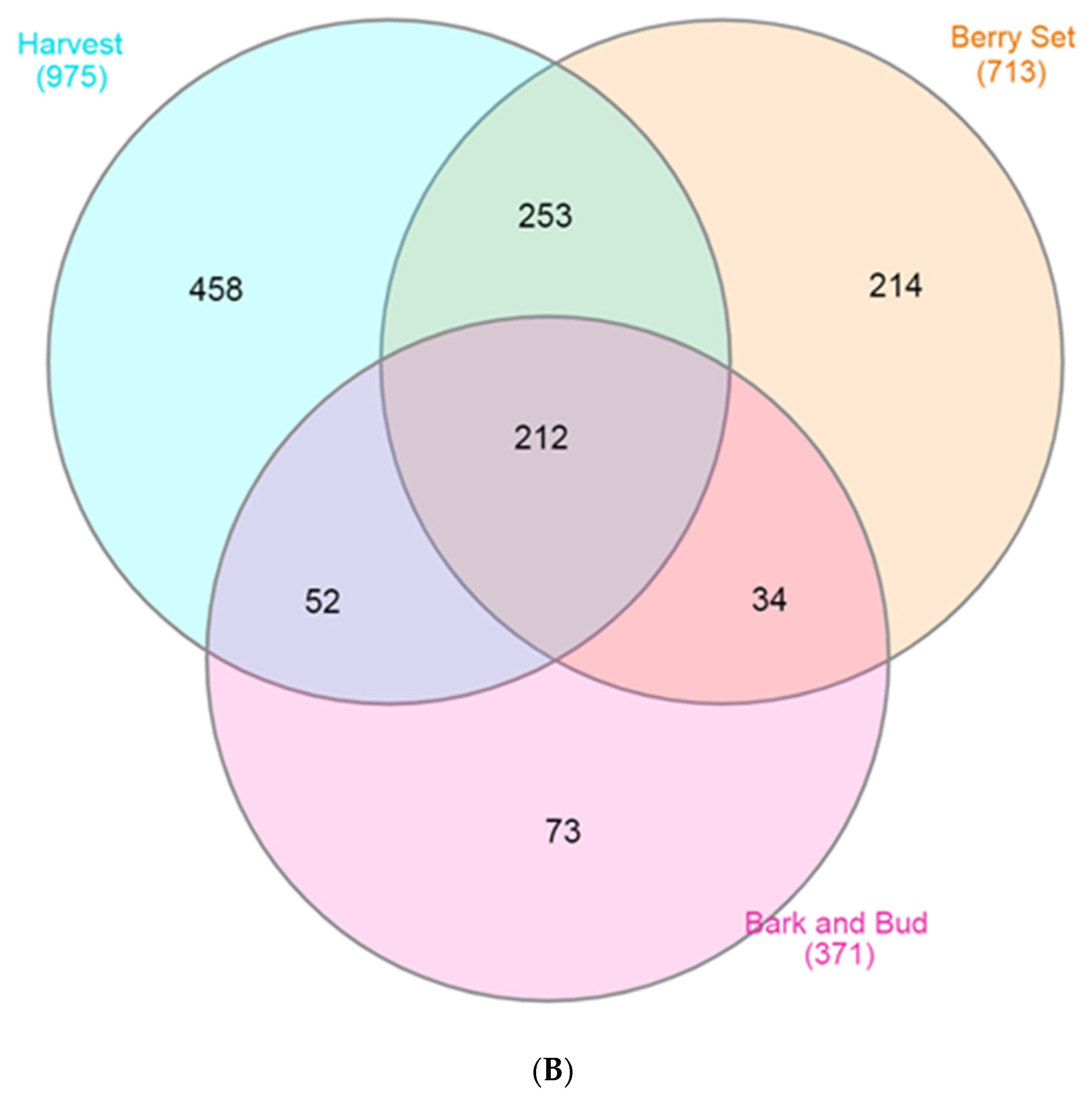
3.3. Core and Unique Microbiome Vary among the Cultivars in each Phenological Stage
3.4. Temporal Dynamics of Grapevine Microbial Communities through Phenological Stages
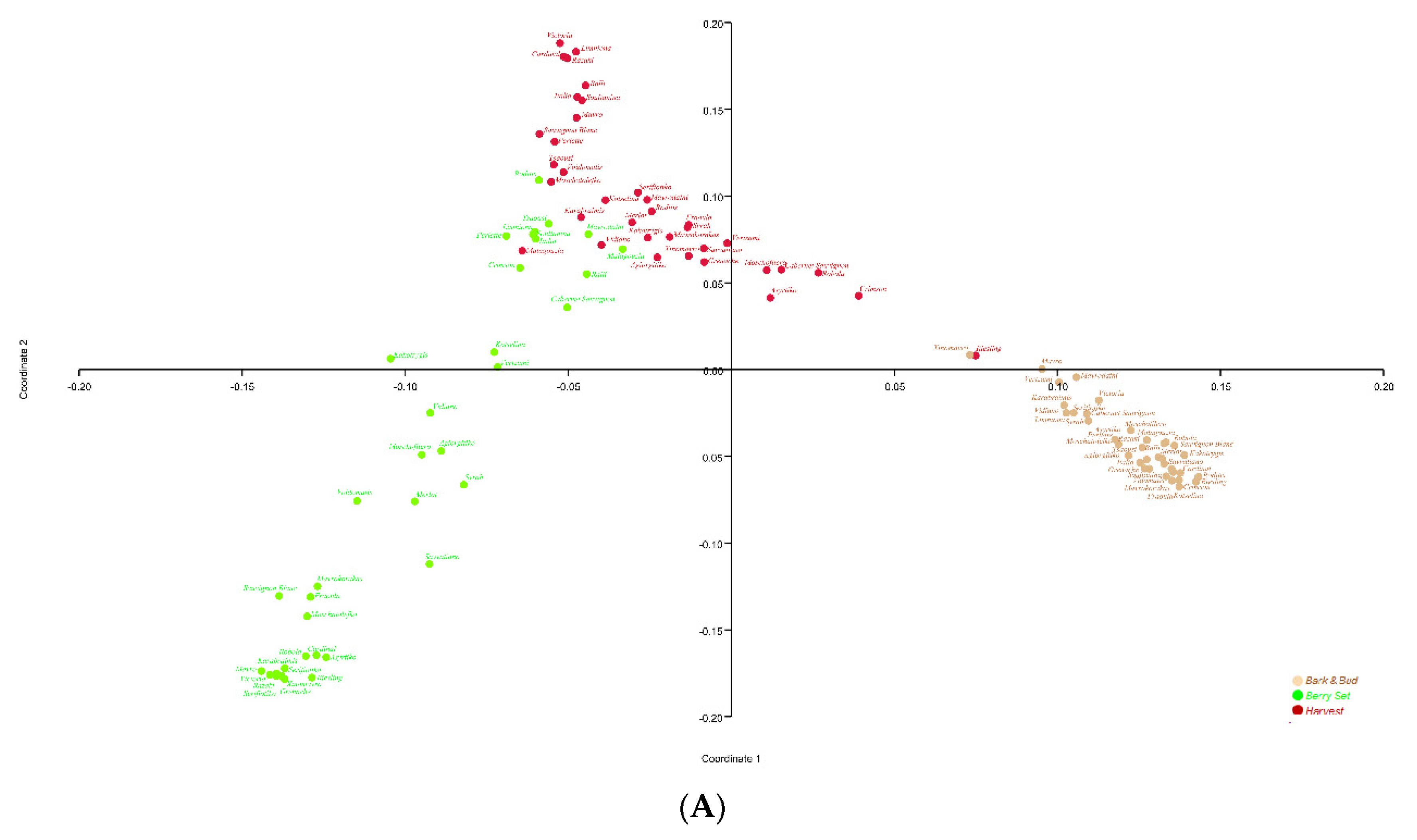
3.5. Grapevine Genotypes ShapeMicrobial Communities under the Phenological Stage Influence
3.6. Microbial Diversity among Cultivars at the different Phenological Stages
3.7. Assessment of Vine Microbiome of Economic Importance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OIV; International Organisation of Vine and Wine. 2019 Statistical Report on World Vitiviniculture; OIV: Ensenada, Mexico, 2019; p. 23. [Google Scholar]
- Poni, S.; Gatti, M.; Palliotti, A.; Dai, Z.; Duchêne, E.; Truong, T.T.; Ferrara, G.; Matarrese, A.M.S.; Gallotta, A.; Bellincontro, A.; et al. Grapevine quality: A multiple choice issue. Sci. Hortic. 2018, 234, 445–462. [Google Scholar] [CrossRef] [Green Version]
- Döring, J.; Frisch, M.; Tittmann, S.; Stoll, M.; Kauer, R. Growth, yield and fruit quality of grapevines under organic and biodynamic management. PLoS ONE 2015, 10, e0138445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.F.; Hall, M.E.; Harris, Z.N.; Kwasniewski, M.T.; Miller, A.J. Grapevine Microbiota Reflect Diversity among Compartments and Complex Interactions within and among Root and Shoot Systems. Microorganisms 2021, 9, 92. [Google Scholar] [CrossRef]
- Carbone, M.J.; Alaniz, S.; Mondino, P.; Gelabert, M.; Eichmeier, A.; Tekielska, D.; Bujanda, R.; Gramaje, D. Drought influences fungal community dynamics in the grapevine rhizosphere and root microbiome. J. Fungi 2021, 7, 686. [Google Scholar] [CrossRef]
- Bernardo, S.; Dinis, L.T.; Machado, N.; Moutinho-Pereira, J. Grapevine abiotic stress assessment and search for sustainable adaptation strategies in Mediterranean-like climates. A review. Agron. Sustain. Dev. 2018, 38, 66. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Van Der Lelie, D.; Zarraonaindia, I. Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. USA 2014, 111, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.N.; Kluepfel, D.A.; Strauss, S.L.; Bokulich, N.A.; Cantu, D.; Steenwerth, K.L. Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: Differentiation by geographic features. Soil Biol. Biochem. 2015, 91, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Zarraonaindia, I.; Gilbert, J.A. Understanding grapevine-microbiome interactions: Implications for viticulture industry. Microb. Cell 2015, 2, 171–173. [Google Scholar] [CrossRef]
- Portillo, C.; Mas, A. Analysis of microbial diversity and dynamics during wine fermentation of Grenache grape variety by high-throughput barcoding sequencing. LWT-Food Sci. Technol. 2016, 72, 317–321. [Google Scholar] [CrossRef]
- Morrison-Whittle, P.; Lee, S.A.; Goddard, M.R. Fungal communities are differentially affected by conventional and biodynamic agricultural management approaches in vineyard ecosystems. Agric. Ecosyst. Environ. 2017, 246, 306–313. [Google Scholar] [CrossRef]
- Liu, D.; Howell, K. Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 2020, 23, 1842–1857. [Google Scholar] [CrossRef] [PubMed]
- Tello, J.; Torres-Pérez, R.; Grimplet, J.; Carbonell-Bejerano, P.; Martínez-Zapater, J.M.; Ibáñez, J. Polymorphisms and minihaplotypes in the VvNAC26 gene associate with berry size variation in grapevine. BMC Plant Biol. 2015, 15, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armijo, G.; Schlechter, R.; Agurto, M.; Muñoz, D.; Nuñez, C.; Arce-Johnson, P. Grapevine Pathogenic Microorganisms: Understanding Infection Strategies and Host Response Scenarios. Front. Plant Sci. 2016, 7, 382. [Google Scholar] [CrossRef] [Green Version]
- Patanita, M.; Albuquerque, A.; Campos, M.D.; Materatski, P.; Varanda, C.M.R.; Ribeiro, J.A.; Félix, M.D.R. Metagenomic Assessment Unravels Fungal Microbiota Associated to Grapevine Trunk Diseases. Horticulturae 2022, 8, 288. [Google Scholar] [CrossRef]
- Awad, M.; Giannopoulos, G.; Mylona, P.V.; Polidoros, A.N. Genotype may influence bacterial diversity in bark and bud of Vitis vinifera cultivars grown under the same environment. Appl. Sci. 2020, 10, 8405. [Google Scholar] [CrossRef]
- Awad, M. Genetic Diversity in V. vinifera and Its Impact on Hosted Microbiota. Ph.D. Thesis, Aristotle University of Thessaloniki, Thessaloniki, Greece, 2020. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, X.; Zhong, Q.; Huang, Z.; Bai, Z. Relations among epiphytic microbial communities from soil, leaves and grapes of the grapevine. Front. Life Sci. 2017, 10, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Salvetti, E.; Campanaro, S.; Campedelli, I.; Fracchetti, F.; Gobbi, A.; Tornielli, G.B.; Torriani, S.; Felis, G.E. Whole-metagenome-sequencing-based community profiles of Vitis vinifera L. cv. Corvina berries withered in two post-harvest conditions. Front. Microbiol. 2016, 7, 937. [Google Scholar] [CrossRef] [Green Version]
- Martins, G.; Lauga, B.; Miot-Sertier, C.; Mercier, A.; Lonvaud, A.; Soulas, M.L.; Soulas, G.; Masneuf-Pomarède, I. Characterization of Epiphytic Bacterial Communities from Grapes, Leaves, Bark and Soil of Grapevine Plants Grown, and Their Relations. PLoS ONE 2013, 8, e73013. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, X.; Zhong, Q.; Zhuang, X.; Bai, Z. Microbial Community Analyses Associated with Nine Varieties of Wine Grape Carposphere Based on High-Throughput Sequencing. Microorganisms 2019, 7, 668. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Santoni, S.; This, P.; Péros, J.-P. Genotype-Environment Interaction Shapes the Microbial Assemblage in Grapevine’s Phyllosphere and Carposphere: An NGS Approach. Microorganisms 2018, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Vitulo, N.; Lemos, W.J.F.; Calgaro, M.; Confalone, M.; Felis, G.E.; Zapparoli, G.; Nardi, T. Bark and grape microbiome of Vitis vinifera: Influence of geographic patterns and agronomic management on bacterial diversity. Front. Microbiol. 2019, 9, 3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.J.; Wu, Y.; Yan, Y.Z.; Zou, W.; Xue, J.; Ma, W.R.; Wang, W.; Tian, G.; Wang, L.Y. High-throughput sequencing of microbial community diversity in soil, grapes, leaves, grape juice and wine of grapevine from China. PLoS ONE 2018, 13, e0193097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faist, H.; Keller, A.; Hentschel, U.; Deeken, R. Grapevine (Vitis vinifera) crown galls host distinct microbiota. Appl. Environ. Microbiol. 2016, 82, 5542–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, E.; Moreira, L.; Rivera, C. Confirmation of Xylella fastidiosa infecting grapes Vitis vinifera in Costa Rica. Trop. Plant Pathol. 2008, 33, 444–448. [Google Scholar] [CrossRef]
- Zheng, C.; Choquer, M.; Zhang, B.; Ge, H.; Hu, S.; Ma, H.; Chen, S. LongSAGE gene-expression profiling of Botrytis cinerea germination suppressed by resveratrol, the major grapevine phytoalexin. Fungal Biol. 2011, 115, 815–832. [Google Scholar] [CrossRef]
- Brewer, M.T.; Milgroom, M.G. Phylogeography and population structure of the grape powdery mildew fungus, Erysiphe necator, from diverse Vitis species. BMC Evol. Biol. 2010, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Gessler, C.; Pertot, I.; Perazzolli, M. Plasmopara viticola: A review of knowledge on downy mildew of grapevine and effective disease management. Phytopathol. Mediterr. 2011, 50, 3–44. [Google Scholar] [CrossRef]
- González-Alonso, I.; Walker, M.E.; Pascual-Vallejo, M.E.; Naharro-Carrasco, G.; Jiranek, V. Capturing yeast associated with grapes and spontaneous fermentations of the Negro Saurí minority variety from an experimental vineyard near León. Sci. Rep. 2021, 11, 3748. [Google Scholar] [CrossRef]
- Ganucci, D.; Guerrini, S.; Mangani, S.; Vincenzini, M.; Granchi, L. Quantifying the effects of ethanol and temperature on the fitness advantage of predominant Saccharomyces cerevisiae strains occurring in spontaneous wine fermentations. Front. Microbiol. 2018, 9, 1563. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.G.; Aplin, J.J. Application of Cool Fermentation Temperatures to Encourage Non-Saccharomyces Yeasts to Yield Lower Ethanol Concentrations in Wines. Fermentation 2022, 8, 421. [Google Scholar] [CrossRef]
- Mezzasalma, V.; Sandionigi, A.; Bruni, I.; Bruno, A.; Lovicu, G.; Casiraghi, M.; Labra, M. Grape microbiome as a reliable and persistent signature of field origin and environmental conditions in Cannonau wine production. PLoS ONE 2017, 12, e0184615. [Google Scholar] [CrossRef] [PubMed]
- Morrison-Whittle, P.; Goddard, M.R. From vineyard to winery: A source map of microbial diversity driving wine fermentation. Environ. Microbiol. 2018, 20, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, H.H.; du Toit, M.; Setati, M.E. The grapevine and wine microbiome: Insights from high-throughput amplicon sequencing. Front. Microbiol. 2017, 8, 820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, R.G.; Steenwerth, K.L.; Mills, D.A.; Cantu, D.; Bokulich, N.A. Sources and Assembly of Microbial Communities in Vineyards as a Functional Component of Winegrowing. Front. Microbiol. 2021, 12, 673810. [Google Scholar] [CrossRef] [PubMed]
- Marasco, R.; Rolli, E.; Fusi, M.; Michoud, G.; Daffonchio, D. Grapevine rootstocks shape underground bacterial microbiome and networking but not potential functionality. Microbiome 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriaut, R.; Martins, G.; Dewasme, C.; Mary, S.; Darrieutort, G.; Ballestra, P.; Marguerit, E.; Vivin, P.; Ollat, N.; Masneuf-Pomarède, I.; et al. Grapevine decline is associated with difference in soil microbial composition and activity. OENO One 2021, 55, 67–84. [Google Scholar] [CrossRef]
- Gan, H.M.; Szegedi, E.; Fersi, R.; Chebil, S.; Kovács, L.; Kawaguchi, A.; Hudson, A.O.; Burr, T.J.; Savka, M.A. Insight into the microbial co-occurrence and diversity of 73 grapevine (Vitis vinifera) crown galls collected across the northern hemisphere. Front. Microbiol. 2019, 10, 1896. [Google Scholar] [CrossRef] [Green Version]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Compant, S.; Mitter, B.; Colli-Mull, J.G.; Gangl, H.; Sessitsch, A. Endophytes of grapevine flowers, berries, and seeds: Identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb. Ecol. 2011, 62, 188–197. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server-A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.; Ryan, P. PAST: Paleontological statistics software package for education education and data analysis. Palaeontol. Electron. 2015, 1, 1–9. [Google Scholar]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, E.T.; Singh, R.P.; Guo, C.; Shang, Y.; Chen, J.; Liu, C. Grape berry surface bacterial microbiome: Impact from the varieties and clones in the same vineyard from central China. J. Appl. Microbiol. 2019, 126, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Siegwald, L.; Touzet, H.; Lemoine, Y.; Hot, D.; Audebert, C.; Caboche, S. Assessment of common and emerging bioinformatics pipelines for targeted metagenomics. PLoS ONE 2017, 12, e0169563. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, L.; Fan, X.; Jiang, J.; Zheng, X.-b.; Sun, H.; Chonghuai, L. Genome-wide assessment of population structure, linkage disequilibrium and resistant QTLs in Chinese wild grapevine. Sci. Hortic. 2017, 215, 59–64. [Google Scholar] [CrossRef]
- Liu, D.; Chen, Q.; Zhang, P.; Chen, D.; Howell, K.S. Vineyard ecosystems are structured and distinguished by fungal communities impacting the flavour and quality of wine. bioRxiv 2020, 1–52. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, I.; Cavalieri, D. Metagenomic approaches to investigate the contribution of the vineyard environment to the quality of wine fermentation: Potentials and difficulties. Front. Microbiol. 2018, 9, 991. [Google Scholar] [CrossRef]
- Pinto, C.; Pinho, D.; Cardoso, R.; Custódio, V.; Fernandes, J.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Wine fermentation microbiome: A landscape from different Portuguese wine appellations. Front. Microbiol. 2015, 6, 905. [Google Scholar] [CrossRef]
- Hendgen, M.; Hoppe, B.; Döring, J.; Friedel, M.; Kauer, R.; Frisch, M.; Dahl, A.; Kellner, H. Effects of different management regimes on microbial biodiversity in vineyard soils. Sci. Rep. 2018, 8, 9393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chu, Z.; Ren, M.; Jia, R.; Zhao, C.; Fei, D.; Su, H.; Fan, X.; Zhang, X.; Li, Y.; et al. Identification of anthocyanin composition and functional analysis of an anthocyanin activator in Solanum nigrum fruits. Molecules 2017, 22, 876. [Google Scholar] [CrossRef] [PubMed]
- Canfora, L.; Vendramin, E.; Florio, A.; Benedetti, A.; Felici, B.; Tarricone, L. Vineyard microbiome variations during di ff erent fertilisation practices revealed by 16s rRNA gene sequencing. Appl. Soil Ecol. 2018, 125, 71–80. [Google Scholar] [CrossRef]
- Mezzasalma, V.; Sandionigi, A.; Guzzetti, L.; Galimberti, A.; Grando, M.S.; Tardaguila, J.; Labra, M. Geographical and cultivar features differentiate grape microbiota in Northern Italy and Spain vineyards. Front. Microbiol. 2018, 9, 946. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.Y.; Vanden Heuvel, J.; Bell, T.H.; Panke-Buisse, K.; Kao-Kniffin, J. Vineyard under-vine floor management alters soil microbial composition, while the fruit microbiome shows no corresponding shifts. Sci. Rep. 2018, 8, 11039. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Liu, W.; Li, X.; Tan, W.; Zhao, Q.; Wang, M.; Ren, R.; Ma, X.; Tang, X. Genetic relationships of 34 grapevine varieties and construction of molecular fingerprints by SSR markers. Biotechnol. Biotechnol. Equip. 2018, 32, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, A.; Kyrkou, I.; Filippi, E.; Ellegaard-Jensen, L.; Hansen, L.H. Seasonal epiphytic microbial dynamics on grapevine leaves under biocontrol and copper fungicide treatments. Sci. Rep. 2020, 10, 681. [Google Scholar] [CrossRef] [Green Version]
- Leveau, J.H.J.; Tech, J.J. Grapevine microbiomics: Bacterial diversity on grape leaves and berries revealed by high-throughput sequence analysis of 16S rRNA amplicons. Acta Hortic. 2011, 905, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Sánchez, R.; Castañeda, L.E.; Godoy, K.; Barbosa, O. Is microbial terroir related to geographic distance between vineyards? Environ. Microbiol. Rep. 2017, 9, 742–749. [Google Scholar] [CrossRef]
- Briceño, E.X.; Latorre, B.A. Characterization of cladosporium rot in grapevines, a problem of growing importance in Chile. Plant Dis. 2008, 92, 1635–1642. [Google Scholar] [CrossRef]
- Latorre, B.A.; Briceño, E.X.; Torres, R. Increase in Cladosporium spp. populations and rot of wine grapes associated with leaf removal. Crop Prot. 2011, 30, 52–56. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | |||||||||
| Phenological Stage | n | OTUs Withsingletons | OTUs without Singletons | Individuals (Classfied) | Individuals (Unclassfied) | Simpson | Shannon | Chao1 | Good’s Coverage |
| Bark and bud | 36 | 628 | 371 | 177,834 | 721,620 | 0.710 | 1.91 | 192.2 | 0.988 |
| Berry Set | 36 | 1157 | 713 | 262,982 | 239,866 | 0.591 | 1.76 | 301.2 | 0.979 |
| Harvest | 36 | 1450 | 975 | 349,639 | 236,577 | 0.690 | 2.35 | 339.3 | 0.978 |
| Total | 108 | 1961 | 1296 | 790,455 | 1,198,063 | ||||
| B | |||||||||
| Phenological Stage | n | OTUs Withsingletons | OTUs without Singletons | Classfied Individuals | Unclassfied Individuals | Simpson | Shannon | Chao1 | Good’s Coverage |
| Bark and bud | 36 | 579 | 422 | 549,446 | - | 0.679 | 1.53 | 172.2 | 0.994 |
| Berry Set | 36 | 561 | 433 | 763,410 | - | 0.691 | 1.62 | 214.3 | 0.994 |
| Harvest | 36 | 568 | 448 | 731,220 | - | 0.668 | 1.57 | 174.3 | 0.995 |
| Total | 108 | 789 | 603 | 2,044,076 | - | ||||
| A | |||||
| Phyllum | Family | Genus | Harvest | Berry Set | Bark and Bud |
| Ascomycota | Dothioraceae | Aureobasidium | 45.35 | 35.74 | 39.80 |
| Ascomycota | Trichocomaceae | Aspergillus | 17.39 | 0.10 | 0.02 |
| Ascomycota | Pleosporaceae | Alternaria | 10.31 | 10.07 | 18.54 |
| Ascomycota | Davidiellaceae | Cladosporium | 6.07 | 22.46 | 14.86 |
| Ascomycota | Davidiellaceae | Davidiella | 5.49 | 12.45 | 11.72 |
| Ascomycota | Didymellaceae | Phoma | 3.17 | 4.68 | 4.76 |
| Ascomycota | Saccharomycetaceae | Issatchenkia | 2.72 | 0.00 | 0.00 |
| Ascomycota | Metschnikowiaceae | Metschnikowia | 2.53 | 0.00 | 0.00 |
| Ascomycota | Didymellaceae | Epicoccum | 1.34 | 2.84 | 0.78 |
| Ascomycota | Botryosphaeriaceae | Botryosphaeria | 0.66 | 1.59 | 1.09 |
| Ascomycota | Pleosporaceae | Lewia | 0.51 | 1.78 | 1.47 |
| Ascomycota | Glomerellaceae | Glomerella | 0.09 | 0.77 | 2.81 |
| Basidiomycota | Ustilaginaceae | Rhodosporidium | 1.74 | 3.81 | 0.76 |
| The rest of genera | 2.63 | 3.7 | 3.39 | ||
| B | |||||
| Phyllum | Family | Genus | Harvest | Berry Set | Bark and Bud |
| Proteobacteria | Acetobacteraceae | Gluconacetobacter | 41.95 | 0.12 | 0.16 |
| Proteobacteria | Enterobacteriaceae | Erwinia | 15.50 | 4.81 | 0.10 |
| Proteobacteria | Acetobacteraceae | Gluconobacter | 14.21 | 0.03 | 0.03 |
| Proteobacteria | Halomonadaceae | Zymobacter | 7.71 | 0.01 | 0.00 |
| Proteobacteria | Oxalobacteraceae | Massilia | 1.81 | 0.02 | 9.15 |
| Proteobacteria | Halomonadaceae | Halomonas | 1.50 | 0.00 | 0.00 |
| Proteobacteria | Enterobacteriaceae | Tatumella | 1.25 | 0.02 | 0.00 |
| Proteobacteria | Enterobacteriaceae | Pantoea | 1.23 | 25.80 | 1.62 |
| Proteobacteria | Pseudomonadaceae | Pseudomonas | 1.16 | 12.37 | 4.40 |
| Proteobacteria | Acetobacteraceae | Acetobacter | 0.79 | 0.01 | 0.00 |
| Proteobacteria | Sphingomonadaceae | Sphingomonas | 0.72 | 0.19 | 11.12 |
| Proteobacteria | Enterobacteriaceae | Buchnera | 0.07 | 39.41 | 0.05 |
| Proteobacteria | Enterobacteriaceae | Raoultella | 0.03 | 6.20 | 0.00 |
| Proteobacteria | Enterobacteriaceae | Cronobacter | 0.01 | 0.82 | 0.00 |
| Proteobacteria | Enterobacteriaceae | Arsenophonus | 0.00 | 0.60 | 0.00 |
| Bacteroidetes | Cytophagaceae | Hymenobacter | 0.41 | 0.33 | 43.75 |
| Bacteroidetes | Sphingobacteriaceae | Pedobacter | 0.05 | 0.15 | 11.44 |
| Bacteroidetes | Cytophagaceae | Flexibacter | 0.03 | 0.00 | 0.83 |
| Actinobacteria | Microbacteriaceae | Curtobacterium | 0.21 | 0.01 | 1.72 |
| Actinobacteria | Microbacteriaceae | Frigoribacterium | 0.18 | 0.04 | 7.87 |
| Actinobacteria | Kineosporiaceae | Kineococcus | 0.07 | 0.06 | 1.69 |
| Firmicutes | Lactobacillaceae | Lactobacillus | 0.32 | 1.45 | 0.02 |
| Firmicutes | XI. Incertae Sedis | Tissierella | 0.03 | 0.00 | 0.70 |
| The rest of genera | 10.78 | 7.55 | 5.36 | ||
| Factor | PCoA Axes | α-Diversity (Shannon) | β-Diversity (Bray-Curtis) | |||||
|---|---|---|---|---|---|---|---|---|
| ANOVA | PERMANOVA | ANOSIM | ||||||
| F | P | F | P | R | P | |||
| 16S Data | Phenological stages | 1= 22.98%, 2= 10.8% | 4.58 | 0.0124 | 24.9 | 0.0001 | 0.64 | 0.0001 |
| Bark and bud | 1= 34.5%, 2= 14.2% | NA | NA | 12.5 | 0.0001 | 0.66 | 0.0001 | |
| Berry Set | 1= 36%, 2= 17.1% | NA | NA | 13.1 | 0.0001 | 0.76 | 0.0001 | |
| Harvest | 1= 18.1%, 2= 9.1% | NA | NA | 04.6 | 0.0001 | 0.42 | 0.0001 | |
| ITS2 Data | Phenological stages | 1= 21.05%, 2= 19.9% | 1.25 | 0.2903 | 12.1 | 0.0001 | 0.21 | 0.0001 |
| Bark and bud | 1= 34.1%, 2= 26% | NA | NA | 17.5 | 0.0001 | 0.82 | 0.0001 | |
| Berry Set | 1= 41.4%, 2= 19.2% | NA | NA | 23.6 | 0.0001 | 0.81 | 0.0001 | |
| Harvest | 1= 50.5%, 2= 10.5% | NA | NA | 15.5 | 0.0001 | 0.71 | 0.0001 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awad, M.; Giannopoulos, G.; Mylona, P.V.; Polidoros, A.N. Comparative Analysis of Grapevine Epiphytic Microbiomes among Different Varieties, Tissues, and Developmental Stages in the Same Terroir. Appl. Sci. 2023, 13, 102. https://doi.org/10.3390/app13010102
Awad M, Giannopoulos G, Mylona PV, Polidoros AN. Comparative Analysis of Grapevine Epiphytic Microbiomes among Different Varieties, Tissues, and Developmental Stages in the Same Terroir. Applied Sciences. 2023; 13(1):102. https://doi.org/10.3390/app13010102
Chicago/Turabian StyleAwad, Murad, Georgios Giannopoulos, Photini V. Mylona, and Alexios N. Polidoros. 2023. "Comparative Analysis of Grapevine Epiphytic Microbiomes among Different Varieties, Tissues, and Developmental Stages in the Same Terroir" Applied Sciences 13, no. 1: 102. https://doi.org/10.3390/app13010102