Abstract
We present a comparative study (using PBE, PBE0, and HSE functionals) of electronic and atomic structure, magnetism, and phonon dispersion relations of -CuPO. Four possible magnetic configurations are considered, FM, AFM-1, AFM-2, and AFM-3. The calculations reveal that -CuPO is mechanically and thermodynamically stable. The elastic moduli indicate a weak resistance of the compound to volume and shear deformations. The electronic structure at the valence band maximum is dominated by O, with a small admixture of Cu- states. The conduction band results from the hybridization between Cu and O states which, in the case of AFM-2, produces the largest band gap of 3.966 eV and the smallest magnetic moment of ±0.785 on Cu. AFM-2 is found to be the lowest-energy structure that may be viewed as consisting of quasi-one-dimensional −CuCuCuCu chains along the b axis; the antiferromagnetism is due to two identical Cu−O−Cu paths with a bond angle of 100.301. The phonon spectra exhibit four distinct frequency ranges corresponding to different vibrational modes of ions and ionic groups. Thus, a quantitative description of the structural, electronic, and magnetic properties of -CuPO is possible using the HSE hybrid functional, which enables computational studies of transition metal pyro compounds.
1. Introduction
Transition metal pyro compounds MXO with M-cations (M = Cr, Mn, Fe, Ni, Co, Cu, Zn, Cd, Mg, Hg) and X = P, As, Se, Ge have been widely used in catalysts [1,2], supercapacitors [3,4,5,6], photoluminescence devices [7], and some other applications [8,9,10] due to their excellent chemical stability, facile preparation, magnetic properties, and non-toxicity [11,12,13]. The MXO compounds are observed in two types of structure, i.e., dichromate structure type and thortveitite structure type, depending on the ratio of the M to X ionic radii. Amongst these, copper pyrophosphate (CuPO) has been well characterized and received much attention as a prospective material for advanced energy storage devices, lightning, photovoltaic devices, sensors, catalysts, etc. [2,3,4,5,6,7,10].
The room-temperature phase of CuPO with space group undergoes a solid–solid phase transition to -phase with space group at a slightly elevated temperature of about 339–383 K, as has been established by X-ray diffraction (XRD) [14]. The structure is composed of pyrophosphate (diphosphate) [PO] ionic groups arranged into layers that are parallel to the (001) lattice plane, and Cu cations in irregular octahedral coordination by oxygen atoms. Cu octahedra form a pseudo-hexagonal network between the adjacent [PO] layers. Each pyrophosphate unit [PO] consists of two monophosphate tetrahedra [PO] which share one corner via the bridging O(1) atom. In the phase, the P−O(1)−P bond angle is 157, whereas it is 180 in the phase [15]. Robertson and Calvo reported that the phase could be described simply as a disordered phase of CuPO, which manifests interesting thermal and dielectric properties [15]. Additionally, the phase exhibits twice as small c lattice parameter and a disordered arrangement of the bridging O atoms shared by the two [PO] tetrahedra in a pyrophosphate group [16]. Since the phase is structurally similar to the phase of CuPO, in the present work we focus on the phase which is fully ordered and stable at room temperature.
It is well understood that the properties of materials strongly depend on the crystal structure, dimensionality, purity, morphology, and size of their constituent particles [17,18,19,20]. Thus, various methods to synthesize CuPO with controllable microstructure, shape, and particle size have been considered [5,6,13,17,21], such as solid-state routes, sol-gel synthesis, chemical bath deposition, co-precipitation, successive ionic layer and adsorption reaction strategy, carbothermal reduction method, and a simple hydrothermal method. Correspondingly, the physical and chemical properties of CuPO such as structural, morphological, optical, magnetic, and electrochemical properties, thermodynamic stability and phase transitions have been investigated experimentally and theoretically [6,12,13,16,17,22,23].
The magnetic behavior of -CuPO was studied in the early 1970s. Tentative models of the magnetic structure were derived from neutron diffraction and nuclear magnetic resonance data; an antiferromagnetic (AFM) ordering was reported at 27 K [24,25]. Theoretically, the AFM order in -CuPO was confirmed by Janson et al., in which the magnetism was found to originate from the half-filled Cu 3 orbital states [12]. The interlayer Cu–Cu coupling is relatively weak and short-ranged since it is mediated by a thick pyrophosphate [PO] layer composed of [PO] tetrahedral units. Moreover, based on a comparison of -CuPO and -CuAsO with -CuVO, Janson et al. found that p cations of P and As atoms donate electrons to the neighboring O anions, leading to more ionic P–O bonds, which confines the interlayer superexchange to the Cu–O–Cu path [12]. However, the electronic band structure calculations employing local density approximation (LDA) wrongly predict a metallic ground state [12], implying that strong electron correlations in Cu states, included in that work via Hubbard-U corrections (LDA+U), are critical for obtaining microscopic insight into the electronic and magnetic properties of complex compounds.
Previous studies concluded that generalized gradient approximation (GGA) combined with the Hubbard-U correction (GGA+U) gives correct results for the electronic and magnetic properties of CuVO[23,26]. Thus, the calculations yielded band gap values of 2.85 eV [26] and 3.17 eV [23] and magnetic moments on Cu atoms of 0.84 [26] and 0.8 [23] in the AFM order, which is compatible with experimental findings [12,23]. Moreover, a negative thermal expansion behavior of -CuPO was disclosed recently by the X-ray and neutron diffraction investigations, which is caused by the transverse vibrations of O atoms [13]. Although the Raman spectra of CuPO in and phases have been studied experimentally [16] and also theoretically including the full phonon dispersion relations [23], the whole set of elastic, electronic, and magnetic properties of CuPO have not been systematically studied.
Motivated by the studies mentioned above, here we systematically study the atomic, electronic, and magnetic structure, as well as the elastic properties and lattice dynamics, of -CuPO using hybrid-functional calculations based on density functional theory (DFT). Importantly, we find that PBE0 and HSE hybrid functionals give more accurate descriptions of the electronic structure and thermodynamic properties of -CuPO relative to the accuracy of semi-local PBE functional calculations, thus demonstrating the usefulness of the hybrid-functional approach in studies of transition metal pyro compounds. The remainder of this article is organized as follows. The first-principles calculation methods are briefly described in Section 2.1. Section 2.2 presents the details of the elastic constant calculations. In Section 3 we report and discuss the crystal structure, thermodynamic stability, and other properties derived from the calculated electronic and phonon spectra. Finally, the conclusions are drawn in Section 4.
2. Methodology
2.1. Computational Details
In this work, density functional theory (DFT) [27] based calculations for -CuPO have been performed using the projector-augmented-wave (PAW) type pseudopotentials with the Vienna Ab initio Simulation Package (VASP) code [28,29]. Four possible magnetic configurations are considered, FM, AFM-1, AFM-2, and AFM-3, see Figure 1. Three different approximations to treat the exchange and correlation contributions have been applied: (i) the PBE approach utilizing a standard generalized gradient approximation scheme [30], (ii) the hybrid PEB0 approach [31,32] and (iii) the hybrid HSE method [33,34] as implemented in VASP. In the PBE0 functional, 25% of PBE exchange has been replaced by the exact non-local exchange showing the normal kernel. When the degree of exact exchange is allowed to vary, ; such a case is referred to as the generalized class of functionals [31,32]. Both PBE0 and HSE functionals used in this work mix 25% of the exact Hartree-Fock (HF) exchange with 75% of the PBE exchange functional in their short-range part. However, in the HSE functional, the long-range part of the exact exchange is smoothly replaced with the local expression of the PBE exchange.
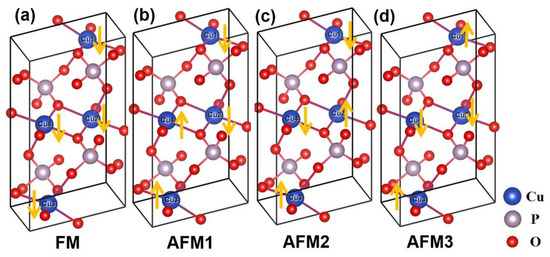
Figure 1.
Optimized crystal structures for four collinear configurations of magnetic moments on Cu atoms in -CuPO, one ferromagnetic (FM) and three antiferromagnetic (AFM). (a) FM; (b) AFM-1; (c) AFM-2; (d) AFM-3. Large blue, grey, and red balls denote copper, phosphorus, and oxygen atoms, respectively.
The valence basis set included 3d and 4s states on Cu, 3s and 3p states on P, and 2s and 2p states on O. A cutoff energy of 520 eV was chosen for the plane-wave basis set. For each considered functional and magnetic configuration, full structural optimization was performed including the shape and volume of the unit cell. Internal atomic positions were fully relaxed until the Hellmann-Feynman forces on them were less than 0.01 eV/Å. A 3 × 3 × 3 Monkhorst-Pack k-point mesh [35] was used to integrate over the Brillouin zone (BZ) for each configuration of -CuPO, whose unit cell contained 22 atoms. A dense 5 × 5 × 3 k-point mesh was selected to compute the density of states (DOS) and the charge density distributions. Spin polarization was turned on in all the calculations. To have a quantitative description of the charge distribution and transfer, the Bader charge analysis was employed [36]. Phonon frequencies were calculated based on the density functional perturbation theory (DFPT) as implemented in the VASP and PHONOPY [37] packages for the lowest-energy AFM-2 configuration using PBE functional at the PBE-optimized volume. In the interpolation of the force constants for the phonon dispersion curve calculations, 3 × 3 × 3 k-point mesh in the first BZ was used. Vibrational contributions to thermodynamic properties such as (free) energy, entropy, and heat capacity were evaluated, as functions of temperature, from the computed phonon dispersion relationships of -CuPO using the methodology detailed in Section 3.5.
The relative chemical stability of -CuPO was calculated according to the definition of formation energy (per formula unit),
where is the total ground-state energy of the primitive cell containing two CuPO formula units and , and are the chemical potentials associated with the pure elements in their standard states [38]. Thus a positive formation energy means that energy must be supplied to form the material, while a negative formation energy implies that the formation is energetically favorable and spontaneous.
For the purpose of comparison of DFT functionals, the formation energy is not a very informative indicator because Equation (1) mixes together the energies of substances with very different types of primary chemical bonding (metallic, ionic, and covalent) for which the DFT functionals perform very differently. For instance, semi-local functionals like PBE perform very well for metals, while hybrid functionals describe the bonding in insulators and molecular substances more accurately than in metals [39].
Another approach [40,41] is to compute the cohesive energy, that is the energy needed to form a solid crystal from the free atoms, which may be defined by replacing the chemical potentials in Equation (1) with the energies of the corresponding atomic species. Still, such a definition operates the energies of very different systems (namely, a strongly bonded solid versus free atoms), so it also suffers from the transferability errors of DFT functionals that are quite large for atomization energies [42].
Yet another possibility is to consider the energy of a chemical reaction in which CuPO compound is formed from a basic oxide, CuO, and an acidic oxide, PO [22],
calculated as follows:
Here CuO represents the monoclinic unit cell of CuO structure, space group , containing four formula units (Z = 4) with an antiferromagnetic alignment of magnetic moments on Cu ions. The calculated lattice parameters by PBE functional is a = 4.667 Å, b = 3.419 Å, c = 5.157 Å, = 99.4767, which is very well consistent with the experimental findings [43]. PO crystallizes in the orthorhombic structure, space group Pnma, whose unit cell contains four formula units. The calculated lattice parameters by PBE functional are a = 9.613 Å, b = 4.941 Å, c = 7.386 Å, and = = = 90. The optimized crystal structures of CuO and PO are shown in Figure S1.
2.2. Elastic Properties
Elastic constants are critical indicators to understand the response of a material to applied macroscopic stress. The tensor of elastic constants is defined by the generalized Hooke’s law expressing linear relationships between stress tensor components and strain tensor components where each of the indices i, j, k, and l runs over the Cartesian basis vectors x, y, and z. The elastic tensor is commonly represented as a 6 × 6 matrix using the following short-hand notations for pairs of indices and : , , , , , and . Owing to the structural symmetry, the number of independent non-zero elastic constants decrease from 21 to 13 for the monoclinic system, which is expressed as follows,
The strain energy, which is defined as the change in the total energy per unit cell relative to the ground state energy of the unstrained crystal, may be expanded as a Taylor series in terms the components of the strain tensor
where is the undistorted unit cell volume. For the monoclinic structure, elastic constants include 13 independent non-zero tensor components, namely , , , , , , , , , , , and [44]. They may be calculated using the following thirteen different strains,
The strain amplitude was varied in steps of 0.005 from −0.025 to 0.025. A detailed description of the calculation procedure used here can be found in Refs. [44,45].
3. Results and Discussion
3.1. Magnetic Structure of -CuPO
The crystal structure of monoclinic -CuPO with space group includes two layers of pyrophosphate [PO] groups; the layers are parallel to the (100) plane. The optimized crystal structures for the four magnetic configurations considered in this work are shown in Figure 1 with the local magnetic moments on the four Cu atoms indicated by arrows. The primitive cell parameters for the ferromagnetic (FM) state, as calculated by PBE functional, are = = 5.485 Å, c = 9.411 Å, = = 75.725, = 97.330, = 260.53 Å and Z = 2 (two CuPO formula units per primitive cell). The calculated (standardized, Z = 4) unit cell parameters are listed in Table 1 where they are compared with some of the available theoretical and experimental data [14,15,22,23].

Table 1.
Unit cell parameters of -CuPO in comparison with previous theoretical [23] and room-temperature experimental [14,15,22,23] results. Up and down arrows denote a possible order of magnetic moments on sites Cu1–Cu4, see Figure 1.
We note that a hydrated form of nanocrystalline copper pyrophosphate exists, whose unit cell (incorporating four CuPO2HO formula units) is bigger than that of the anhydrous form: a = 11.466 Å, b = 10.204 Å, c = 6.123 Å [46]. Naturally, the molecular volume for the hydrated form, = 196.054 Å per CuPO2HO formula unit, is bigger than that for the anhydrous form, = 130.265 Å per CuPO formula unit, by the volume of two HO molecules.
In copper pyrophosphate, only Cu ions are calculated to carry local magnetic moments corresponding to spin 1/2. Each Cu ion is surrounded by five O atoms forming a [CuO] polyhedron with the volume of 6.905 Å and Cu–O bond lengths in the range 1.913–2.316 Å. Besides, the shortest Cu–Cu distance (along the crystallographic direction b) is calculated to be ∼3.12 Å. Importantly, for this Cu–Cu pair there are two identical Cu–O–Cu paths with a bond angle ∠Cu–O–Cu of 101.735, as shown in Table 2. According to previous studies on the nearest-neighbor (NN) exchange coupling, the closeness of ∠Cu–O–Cu to 90 indicates a sizeable ferromagnetic contribution, based on the Goodenough-Kanamori rules [12,47,48]. Hence, it is natural to expect the superexchange interaction to favor an antiferromagnetic order for the Cu–Cu NN pair, due to the deviation of Cu–O–Cu bond angle from 90. The calculated Cu–Cu distance and Cu–O–Cu bond angle is in agreement with the results of previous studies, 3.05–3.33 Å and 100.4–100.8 [12,14,24].
Table 2.
Calculated Cu–O–Cu bond angles (), Cu–Cu distances (), and volumes of [CuO] and [PO] polyhedra in -CuPO.
For the obtained parameters of Cu–O–Cu paths, competing exchange interactions may be expected to exist along the shortest Cu–Cu distance, giving rise to four possible magnetic configurations FM, AFM-1, AFM-2, and AFM-3 shown in Figure 1. Experimentally, -CuPO is antiferromagnetic at cryogenic temperatures [12,23,24,25,26] and Curie-Weiss paramagnetic (PM) at room temperature, with an effective paramagnetic moment of 1.89 corresponding to spin-1/2 moment on each Cu ion [12].
Another structural degree of freedom is related to the [PO] polyhedron, consisting of two corner-sharing [PO] tetrahedra, as shown in Figure S2. The structure of [PO] polyhedron may be perturbed by changing the angle between these two [PO] tetrahedral sub-units and the values of P–O bond lengths. The P–O(1)–P bond angle is calculated to be about 154.736, which is consistent with the experimental value of 157[15]. The obtained inner and outer P–O bond lengths for [PO] polyhedron are 1.585 Å and 1.539 Å, respectively, in agreement with the corresponding values of 1.58 Å and 1.53 Å measured in Ref. [15]. The calculated volume of [PO] tetrahedron is about 1.9 Å. Intriguingly, the lattice parameters in Table 1 and Table 2 show very slight differences among the four studied magnetic configurations. It is worth mentioning that, compared with the PBE functional, all of these values shrink when a hybrid density functional, PBE0 or HSE, is used.
The calculated formation energies ( and ) of -CuPO in the four considered states of magnetic order are collected in Table 3. The obtained negative values indicate an exothermic formation process and the thermodynamic stability of the compound. By comparing the experimental and calculated formation energies listed in Table 3, one may get an impression that PBE works slightly better than the hybrid functionals for -CuPO. However, this is due to the fact that the energies of substances with very different types of chemical bonding are involved into Equation (1), so that the errors of DFT functionals do not cancel out. However, the experimental result for the enthalpy of formation of -CuPO from the binary oxides at 298 K, , is −279.0 ± 1.4 kJ/mol [22] while the calculated values (for the ground-state AFM-2 structure) using the PBE, PBE0, and HSE functional translate into −145.8, −252.2 and −252.8 kJ/mol, respectively. One can clearly see that hybrid functionals PBE0 and HSE give formation energy values that are consistent with the experimental result for -CuPO, while the value derived using a semi-local PBE functional is considerably off.
Table 3.
Calculated formation energy (eV/f.u.), formation energy from oxides (eV/f.u.), band gap (eV), and effective atomic charges , , and , in -CuPO. Elementary charge units (e) are used; negative charge values denote a lack of electrons. Previous experimental and theoretical values are quoted for comparison.
Notably, the calculated and are the lowest for the AFM-2 ordered state as compared with the other three states of magnetic order, when calculated with any of the PBE, PBE0, or HSE functionals. This demonstrates that AFM-2 is indeed the most stable ordered structure, which supports the findings of previous studies that -CuPO exhibits an antiferromagnetic ordering bellow 22–27 K [12,23,24,25,26]. In particular, the ground-state energy () of AFM-2 ordered state is smaller than that of FM state by 0.189, 0.158, or 0.116 eV (per primitive cell) when calculated using PBE, PBE0, or HSE functionals, respectively, see Table 4 for further details.
Table 4.
Calculated total energy (eV per primitive cell), total magnetic moment (Bohr magnetons per primitive cell), and local magnetic moments , , and () on the respective atoms in -CuPO. Previous theoretical data are also quoted for comparison [23].
3.2. Elastic Properties and Mechanical Stability of -CuPO
Before discussing the calculated elastic and other properties of -CuPO in detail, it is instructive to demonstrate its mechanical stability. For a monoclinic structure, the Born criteria of mechanical stability [49,50] require that the following equations should be satisfied [51],
Here , , , , , and .
The calculated elastic constants for -CuPO are collected in Table 5. According to Equation (6) and the obtained values, we can definitely conclude that -CuPO is mechanically stable since the calculated elastic constants meet the Born stability criteria. The calculated elastic constants , and are lower than 200 GPa, indicating that -CuPO has a relatively weak deformation resistance along the a-, b- and c-axes. Also, the elastic constants , and are calculated to be small. It can therefore be concluded that -CuPO has a weak shear deformation resistance.
Table 5.
Calculated elastic constants (GPa) of -CuPO.
Ab initio determination of mechanical properties of anisotropic crystalline materials is a formidable multi-scale problem. In this connection, it is common to make simplified qualitative judgments about the mechanical properties of materials using empirical relationships formulated in terms of directionally-averaged (polycrystalline) elastic moduli such as bulk modulus (B) and shear modulus (G). In our work, the elastic moduli of -CuPO have been calculated using the Voigt-Reuss-Hill theory [52,53,54]. More details are presented in the Supporting information. The average Young’s modulus (Y) and Poisson ratio () are calculated through and . The calculated averaged moduli and other indicators of mechanical properties are listed in Table 6. The averaged bulk modulus measures the resistance of a polycrystalline material to a volume change. The calculated Voigt-, Reuss-, and Hill-averaged bulk moduli B are 93.298 GPa, 80.624 GPa, and 86.961 GPa, respectively. Thus, -CuPO has a relatively weak volume deformation resistance, which is consistent with the conclusions from the single-crystal elastic constants. The averaged shear modulus G reflects the shear deformation resistance of a polycrystalline material. The obtained values of Voigt-, Reuss-, and Hill-averaged shear moduli are close to each other, around 35 GPa, which shows that -CuPO has relatively weak shear deformation resistance. The calculated averaged Young’s moduli are 120.468 GPa, 80.112 GPa, and 100.533 GPa, indicating low elastic stiffness. In addition, the ratio is a common parameter to judge the ductility of a material [55,56]. If the value of is larger than 1.75, the material is expected to exhibit ductile behavior; otherwise, the material is expected to exhibit brittle behavior. It can be seen that the calculated ratio for -CuPO is greater than 1.75, indicating that the material should be ductile. Poisson’s ratio is another indicator of the ductility of a material, predicting the material to exhibit ductile behavior if 0.26. The obtained values of indicate that -CuPO should be ductile, in agreement with the analysis based on ratio.
Table 6.
Direction-averaged bulk modulus B, shear modulus G, Young’s modulus Y, Poisson’s ratio , bulk-to-shear modulus ratio , and hardness H of -CuPO calculated according to Voigt, Reuss, and Hill approximations.
3.3. Electronic and Magnetic Structure of -CuPO
The spin-resolved band structure of -CuPO has been calculated to evaluate the exchange coupling using PBE, PBE0, and HSE functionals. As Figure S3a shows, the band structure calculated within PBE functional for -CuPO in FM state is metallic. Specifically, the states at the Fermi level (chosen to be the zero of energy) belong to both spin-up and spin-down channels, while the states from just above the Fermi level to the top of the valence band have pure spin-down character (exhibit 100% spin polarization). It is well known that PBE functional tends to underestimate band gaps [57]; therefore, additional calculations using hybrid functionals PBE0 and HSE have been performed to get better estimates of the band gap, as depicted in Figure S3 and Figure 2, respectively.
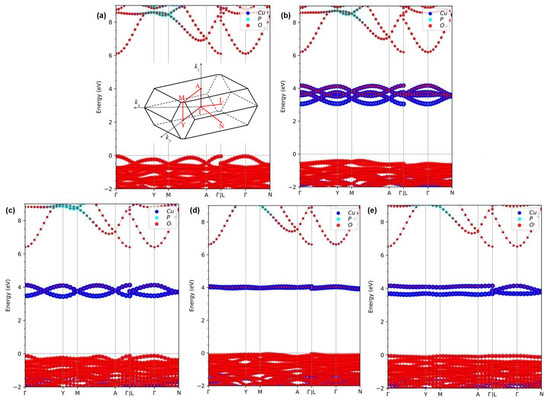
Figure 2.
Fat band structures for -CuPO in (a) FM spin-up channel, (b) FM spin-down channel, (c) AFM-1, (d) AFM-2, and (e) AFM-3 magnetic states calculated within HSE density functional. The zero energy is chosen at the highest occupied level at 0 K. The inset in panel (a) shows the BZ shape and paths used in the electronic and phonon spectrum calculations.
Surprisingly, a band gap as large as 3.784 eV and 3.084 eV opens up in the electronic structure of FM -CuPO upon going from PBE to, respectively, PBE0 and HSE functionals. In particular, we observe a sizeable enhancement of the band gap value in the spin-down channel. According to a previous study, the local Coulomb interaction U in GGA+U or LDA+U calculations appears to produce a similar band gap opening in the electronic structure of FM -CuPO, indicating that the insulating ground state of -CuPO is of the Mott type [12,23].
In the case of AFM-1 magnetic ordering, due to the shifts of the valence band maximum (VBM) and conduction band minimum (CBM) positions, the band gap values have been calculated to be 0.390 eV, 4.344 eV, and 3.566 eV within PBE, PBE0, and HSE functionals, respectively. The calculated band gap values for AFM-2 state of magnetic order are 0.730 eV, 4.670 eV, and 3.966 eV within PBE, PBE0, and HSE functional, respectively. Finally, the obtained band gap values for the AMF-3 state are 0.362 eV, 4.395 eV, and 3.683 eV within PBE, PBE0, and HSE functionals, respectively. All calculations that consider an antiferromagnetic order of Cu magnetic moments predict -CuPO to be a semiconductor. Additionally, the same variation trend of the band structures is observed under PBE0 and HSE hybrid functional calculations. All these results are collected in Table 3. The band gap values calculated in this work using hybrid functionals agree reasonably well with the band gap values of 3.17 eV, 2.85 eV, 3.36 eV, and 3.80 eV calculated previously using LDA+U and GGA+U approaches [12,16,17,26]. But as a whole, HSE results seem to be in better agreement with the experiment than PBE0 results. Intriguingly, the total energy calculations show that the most favorable magnetic structure of -CuPO is AFM-2, for which the largest band gap value is obtained no matter which functional (PBE, PBE0 or HSE) is used. Besides, the fat band structures calculated within HSE functional are depicted in Figure 2. They are resolved into the atoms to gain more physical insights into the electronic structure variations, in which blue-, cyan- and red-colored balls denote the contribution by Cu, P, and O atoms, respectively. Similar to the four magnetic states, one finds that the valence bands are mainly dominated by O atoms with little contribution from Cu atoms. In contrast, the conduction bands are dominated by hybridized states of Cu and O atoms. What makes FM ordering special is that the conduction band states, in that case, are completely in the spin-down channel and belong to both Cu and O atoms, as shown in Figure 2b. It is worth mentioning that the hybridization of Cu and O atoms at the bottom of the conduction band of AFM-2 configuration is so strong that it produces an almost flat level, which leads to the smallest magnetic moments on Cu atoms, as shown in Figure 2d. Relative to that case, the exchange coupling of Cu and O atoms is weak and split into wider band energy regions in the cases of FM, AFM-1, and AFM-3 ordering. Based on above discussion one can conclude that -CuPO is an antiferromagnetic semiconductor with a band gap of ∼3.96 eV.
To get a deeper insight into the band structure, the spin-polarized total density of states (TDOS) and partial density of states (PDOS), computed using HSE functional, have been plotted together in Figure 3. In all the four cases of magnetic ordering, the broad oxygen bands are located between −9 and 0 eV, with an admixture of Cu 3d band states above −5.5 eV corresponding to the [CuO] pyramid. Specifically, the top of the valence band (VBM) is primarily composed of O-2p electron states with a slight admixture of Cu-3d states, particularly Cu orbital states. Oppositely, the CBM is mainly composed of Cu-3d electron states hybridized with O-2p states, especially in the AFM-2 case, which is resolved as the magnetically active orbital [11]. This relevant hybridization was identified using Wannier functions and LDA for Cu states [11,58]. Figure S5 zooms into the magnetic layers of [CuO] structural dimers, containing two edge-sharing [CuO] plaquettes, which are separated from each other by nonmagnetic [PO] tetrahedra in -CuPO. More exactly, the spacer layers consist of [PO] pyrophosphate groups formed by joining two [PO] tetrahedra into a corner-sharing structural unit. Thus, the magnetic orbital lies in the plane of [CuO] plaquette which for two possible magnetic coupling pathways, i.e., Cu–O–Cu and Cu–O–Cu pathways.
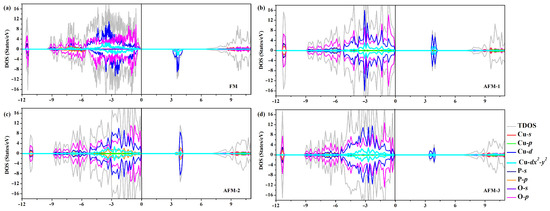
Figure 3.
(a–d) Electronic total density of states (TDOS) and atom-projected density of states for -CuPO in the four studied states of magnetic order, as calculated within HSE density functional. The zero of energy is chosen at the highest occupied level at 0 K.
The charge density cross-sections along the (−1.5, 1, −2.5) plane for [CuO] structural dimers in -CuPO at 0.3 e/Å are presented in Figure S6 for the four different magnetic states considered in this work. The covalent bonding component between Cu and O atoms can be clearly seen as streaks of the charge density, which may be attributed to the overlap and hybridization of Cu-3d and O-2p orbitals. The Bader analysis [59] shows the transfer of electrons from Cu and P cations to be about 1.35 e and 3.78 e, respectively. In comparison, the nearest O ion gains 1.42 e, indicating that the covalent and ionic contributions to Cu–O and P–O bonding are significant. More details can be found in Table 3.
3.4. Magnetic Properties of -CuPO
The alignment of magnetic moments arises from a competition between two physical mechanisms, the intraatomic exchange splitting and the interatomic interactions involving Cu 3d states. The magnitude of the magnetic moment increases with the strength of the exchange interaction. As shown in Table 2 and Figure 4, the shortest Cu–Cu separation distance of 3.056 Å, occurs between Cu and Cu ionic positions, as well as between Cu and Cu positions in the structure. For comparison, the bond length between Cu and Cu positions is 3.261 Å. The short interatomic distance is expected to result in a strong direct interaction between the open 3d shells of these Cu ions. The unoccupied Cu 3d states contribute to the narrow conduction band lying at about 4 eV in Figure 2d that forms a sharp DOS peak at the same energy in Figure 3c. All calculated information about the total and partial magnetic moments is collected in Table 4. Previously calculated values of magnetic moments are also given for comparison. With regards to FM ordering, the conduction bands come totally from spin-down states, with the dominant contribution due to Cu 3d states with a slight exchange splitting, as shown in Figure 2b. The splitting signifies a higher magnetic moment of the Cu atom. Within HSE functional the partial magnetic moments of Cu, P, and O in the FM case of orders are calculated to be 0.791 , 0.008 and 0.063 , respectively. The spin density in the interstitial region is rather small: it adds up to about 0.12 electrons per primitive cell in the calculations employing HSE functional. The total magnetic moment of the FM state is 3.944 per primitive cell, which is close to the integer value corresponding to one d-orbital hole per each of the four Cu ions in the cell. For comparison, the local atomic moments are 0.785 for Cu, 0.002 for P, and 0.028 for O in AFM-2 ordered state, in which the net magnetic moment is 0 since the total DOS is symmetrical in the spin up and spin down channels. As Table 4 shows, the magnetic moments obtained in this work are in good agreement with the results of previous GGA+U calculations [23]. The local magnetic moments computed in this work are characteristic of Cu oxidation state 2+, which is in line with the paramagnetic moment of 0.15 emu/g [60] or 1.89 [12] experimentally reported for CuPO in the rage from −123 to +127 C. Some nanocrystalline specimens at room temperature have been found to exhibit a weak ferromagnetic or superparamagnetic response to an applied strong magnetic field [17,60]. Unfortunately, no experimental data on local moments are available for the ordered antiferromagnetic state.
Theoretically, -CuVO has been proposed to be a quasi-one-dimensional spin system composed of magnetic Cu ions and a local magnetic moment of 0.73 per Cu atom was obtained from GGA+U calculations [61]. It is consistent with the values calculated in this work for -CuPO, due to structural similarities between the two phases where the Cu ions are arranged into the [CuO] plaquettes linked via two corner-sharing [PO] tetrahedra. For comparison, the shortest Cu–Cu distance is of 2.9974 Å in -CuVO, which is slightly shorter than the Cu–Cu (or Cu–Cu) bond length of 3.056 Å calculated here for -CuPO in AFM-2 ordered state, see Figure 4. The length of the next-nearest neighbor bond, Cu–Cu, is 3.266 Å as obtained here from HSE functional calculations for -CuPO in AFM-2 state. From Figure S6, one can see that the covalent component of bonding between Cu and O ions is very pronounced; the effect facilitates itself in the form of electron density streaks connecting the neighboring atoms and may be mainly attributed to the hybridization between Cu-3d and O-2p atomic orbitals upon the formation of the chemical bond. Also, Figure 4 shows that electron density accumulates near the short Cu–Cu bonds; somewhat weaker density accumulation may be seen near the longer Cu–Cu bonds. Thus, chemical bonding produces zig-zag −CuCuCuCu chains along the b-axis in the structure of -CuPO with AFM-2 ordering. The occurrence of these chains provides valuable theoretical support and a qualitative explanation to the hypothesis that transition metal pyro compounds (MXO) are quasi-one-dimensional spin systems composed of magnetic Cu ions, as proposed by Janson et al. [12] for -CuPO and by Arongo et al. [11] for -CuAsO.

Figure 4.
(a) Atomic structure of -CuPO for AFM-2 state in the (0, 1, 29.5) cross-section plane chosen for charge density calculation. (b) Contour plot of the charge density calculated in the (0, 1, 29.5) cross-section plane within HSE density functional. The isosurface level of the charge density is set at 0.3 e/Å; the image is produced using VESTA [62]. The white solid and dotted lines indicate, respectively, the distances of 3.056 Å for bond and 3.266 Å for bond.
Figure 4.
(a) Atomic structure of -CuPO for AFM-2 state in the (0, 1, 29.5) cross-section plane chosen for charge density calculation. (b) Contour plot of the charge density calculated in the (0, 1, 29.5) cross-section plane within HSE density functional. The isosurface level of the charge density is set at 0.3 e/Å; the image is produced using VESTA [62]. The white solid and dotted lines indicate, respectively, the distances of 3.056 Å for bond and 3.266 Å for bond.

Contour plots (isosurfaces) of the spin density are depicted in Figure 5 for the studied magnetic configurations of -CuPO. The Figure clearly shows that the magnetic moments of -CuPO arise from Cu atoms; the O and P atoms are essentially nonmagnetic and the spin density in the interstitial region is negligible. This is in accordance with the above-discussed results on the calculated magnetic moments. Spin densities for these four cases look similar: the colored clouds, indicating loci of accumulated spin-up and spin-down density, are almost identical for the different Cu positions. Particularly, the spin density along the Cu chains is localized in one orbital, i.e., . Besides, one can see small clouds of spin density accumulated on O atoms, which is of importance for opening the superexchange channels like CuOCu and CuOCu. Based on the above discussion, we can generally conclude that all four studied cases of magnetic ordering are reasonable with the consideration of electron interactions of each Cu atom with its first, second, and third nearest-neighbor atoms. After a more comprehensive analysis, we can conclude that the energetically preferable magnetic state in -CuPO is AFM-2 ordering with quasi-one-dimensional antiferromagnetic −CuCuCuCu chains along the b axis, in which two identical Cu−O−Cu paths make an angle of 100.301 to each other.
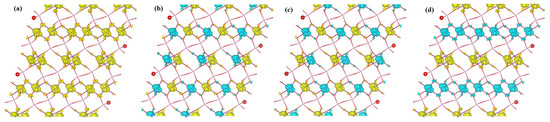
Figure 5.
Spin density of -CuPO in (a) FM, (b) AFM-1, (c) AFM-2 and (d) AFM-3 states of magnetic order, as calculated within HSE density functional. The isosurface level of the charge density is set at 0.2 e/Å; the image is produced using VESTA [62].
3.5. Phonon Dispersion Curves and Lattice Thermodynamic Properties
In addition to thermodynamic stability and mechanical stability, dynamical stability can also decide the structural stability of a material. Thus the phonon dispersion curves along the YA−ML−N directions in the BZ have been calculated for the lowest-energy AFM-2 configuration using PBE functional at the PBE-optimized volume and are displayed in Figure 6a; the corresponding phonon density of state (PhDOS) is plotted in Figure 6b, see also Figure 2a for BZ path notations. It can be seen that there are no imaginary phonon frequencies, implying the dynamical stability of -CuPO which depends on the vibration frequencies of all atoms in the material. The entire phonon spectrum consists of five separate frequency bands. The low-frequency band below 12 THz includes the acoustic and optical branches of vibrations involving all ions, Cu, P, and O. The broad optical band between 13 and 18 THz may be associated with bending modes of pyrophosphate [PO] groups consisting in changing the P–O–P bridge angle. Then there occurs a distinct gap in the phonon dispersion relations with a narrow vibrational band at about 20 THz associated with stretching modes of the P–O–P bridge. The two high-frequency bands in the range from 25 to 34 THz above the gap are mainly due to O–P bond stretching modes.
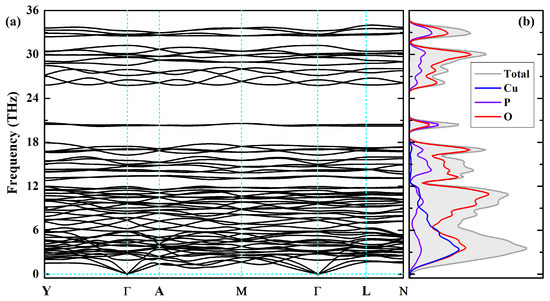
Figure 6.
Phonon dispersions (a) and phonon density of states (b) of -CuPO, as calculated within PBE density functional. For BZ path notations, see Figure 2.
Temperature dependencies of the total energy, Helmholtz free energy, entropy, and specific heat contributions from the lattice vibrations have been evaluated by integrating the phonon density of states obtained from the interaction force constants calculated in the quasi-harmonic approximation (QHA) [56,63]. In particular, the Helmholtz free energy (F) as a function of temperature (T) and volume (V) is expressed as following equation,
where is the electronic ground-state total energy of the system, denotes the phonon-free energy, and is the electronic excitation energy at a given unit cell volume (V) and temperature (T). For wide-band-gap semiconductors at moderate temperatures, the electronic free energy contribution may safely be neglected. Under the QHA, the phonon contributions to Helmholtz free energy , the internal energy (E), and the vibrational entropy (S) can be obtained from PhDOS g() using the following formulas [64],
where stands for the frequency of the phonon mode calculated at a primitive cell volume V, is the phonon density of state with the norm . and at zero temperature are both equal to the zero-point energy, which is given by , where n is the number of atoms per primitive cell, N is the number of primitive cells. Here ℏ and are the Plank constant and the Boltzmann constant, respectively.
The internal energy E, Helmholtz free energy F and entropy S of -CuPO, calculated in the temperature range of 0…1000 K using a 2 × 2 × 1 supercell (Z = 8) and expressed per mole of CuPO molecules at the molecular volume of 130.268 Å, are shown in Figure 7a–c, respectively. Due to the vibrational contributions, E and S increase gradually with increasing the temperature, while F exhibits a non-linear decrease. Besides, the calculated zero-point energy of -CuPO phase is = 89.04 kJ/mol. (Note that the values of thermodynamic properties plotted in Figure 7 are expressed per mole of CuPO formula units).
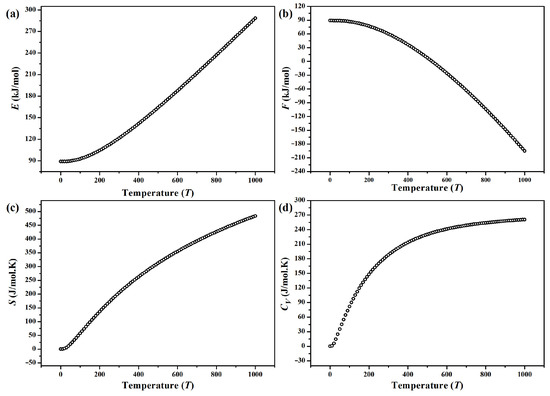
Figure 7.
Temperature-dependent (a) total (internal) energy E, (b) Helmholtz free energy F, (c) entropy S and (d) heat capacity of -CuPO calculated per mole (of formula units) at a molecular volume of 130.268 Å.
A critical parameter to understand the thermal behavior of -CuPO is the heat capacity at a constant volume, which can be expressed as
The calculated temperature-dependent heat capacity of -CuPO is shown in Figure 7d. One can see that increases sharply at low temperatures (0–300 K). Then increases smoothly up to 270 J/K per mole of CuPO formula units.
4. Conclusions
To summarize, we have carried out a comparative hybrid-density functional study (using PBE, PBE0, and HSE functionals) of electronic structure, magnetic ordering, phonon dispersion relations, and thermodynamic properties of -CuPO. First of all, the calculated formation energy, cohesive energy, elastic constants, and phonon dispersion relations indicate that -CuPO is mechanically and thermodynamically stable. The mechanical property indicators based on elastic moduli reveal that -CuPO should exhibit a ductile behavior with a relatively weak volume deformation resistance and shear deformation resistance. Secondly, the spin-polarized calculations indicate that the FM, AFM-1, AFM-2, and AFM-3 magnetic configurations have rather similar electronic structures, except for some slight differences at the bottom of the conduction band where the hybridization between the electron states of Cu and O atoms in the AFM-2 structure is so strong that the conduction band is shaped almost like a flat level. Thus AFM-2 magnetic ordering produces the largest band gap of 3.966 eV and the smallest local magnetic moments on Cu atoms within HSE functional calculations. Correspondingly, the states at the valence band maximum are mainly dominated by O states with a small contribution due to Cu electron states, particularly from Cu- orbital. Thirdly, by comparing the energies for the different types of magnetic ordering in -CuPO, we find that AFM-2 ordering is the most stable structure having the strongest antiferromagnetic interactions. Specifically, the proposed one-dimensional spin system in AFM-2 ordering originates from −CuCuCuCu chains along the b axis, in which there are two identical Cu−O−Cu paths, each forming a bond angle of 100.301. Within HSE functional, the partial magnetic moments on Cu atoms in FM, AFM-1, AFM-2 and AFM-3 ordered states are 0.791 , ±0.790 , ±785 and ±0.788 , respectively. Fourthly, phonon dispersion relations indicate that there are four ranges of vibrational frequencies: (1) 0–12 THz contributed by acoustic and optical modes involving Cu, P, and O atoms, (2) 13–18 THz mainly dominated by vibrations corresponding to bending of pyrophosphate groups; (3) a narrow band at 20 THz due to the stretching modes of a P–O–P bridge; (4) two bands in the range 25–34 THz due to O–P stretching modes. Our systematic study on structural stability, electronic and magnetic properties, and phonon dispersion relations of -CuPO opens a way for further computational studies of related materials such as transition metal pyro compounds, MXO.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app13010498/s1, Formalism based on Voigt-Reuss-Hill (VRH) approximation for the evaluation of direction-averaged bulk modulus (B) and shear modulus (G) [52,53,54]; Figure S1: Optimized crystal structures of (a) CuO and (b) PO; Figure S2: The local structure of a pyrophosphate group [PO] in -CuPO; Figure S3: Electronic energy band structures calculated using PBE functional for -CuPO in (a) FM, (b) AFM-1, (c) AFM-2, and (d) AFM-3 magnetic states; Figure S4: Electronic energy band structures calculated using PBE0 functional for -CuPO in (a) FM, (b) AFM-1, (c) AFM-2, and (d) AFM-3 magnetic states; Figure S5: Different representations of the Cu local environment: (a) the [CuO] pyramid and (b) the [CuO] distorted plaquette; Figure S6: Charge density in the (−1.5, 1, −2.5) plane with [CuO] layer in -CuPO in four different magnetic states calculated using HSE functional. The isosurface level of the charge density is set at 0.3 e/Å.
Author Contributions
Conceptualization, P.K.; methodology, X.Y.; software, P.K.; validation, X.Y. and P.K.; formal analysis, X.Y. and P.Z.; investigation, X.Y. and P.K.; resources, P.K.; data curation, X.Y.; writing—original draft preparation, X.Y.; writing—review and editing, P.K.; visualization, P.K. and P.Z.; supervision, P.K.; project administration, P.K.; funding acquisition, X.Y. and P.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was performed at the Vinnova Competence Centre “Hero-m 2i”, funded jointly by the Swedish Governmental Agency for Innovation Systems (Vinnova, grant number 2016-00668), Swedish industry, the Swedish steel producers’ association Jernkontoret, and KTH Royal Institute of Technology. P.K. acknowledges support by Svensk Kärnbränslehantering AB (SKB). X.Y. sincerely appreciates the support of National Natural Science Foundation of China (Grant No. 11705152 and 11974055) and by Stiftelsen för Tillämpad Termodynamik, Sweden.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and supplementary material.
Acknowledgments
The computational resources are provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC) in Linköping, Sweden, and at the Center for High-Performance Computing (PDC) in Stockholm, Sweden, partially funded by the Swedish Research Council through grant agreement No. 2018-05973.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AFM | Antiferromagnetic |
| BZ | Brillouin zone |
| DFT | Density functional theory |
| DOS | Density of states |
| FM | Ferromagnetic |
| GGA | Generalized gradient approximation |
| HSE | Heyd-Scuseria-Ernzerhof hybrid functional |
| LDA | Local density approximation |
| PAW | Projector-augmented-wave |
| PBE | Perdew-Burke-Ernzerhof semi-local functional |
| PBE0 | Perdew-Burke-Ernzerhof hybrid functional |
| PDOS | Partial density of states |
| PhDOS | Phonon density of states |
| PM | Paramagnetic |
| QHA | Quasi-harmonic approximation |
| TDOS | Total density of states |
| VASP | Vienna Ab initio Simulation Package |
| XRD | X-ray diffraction |
References
- Dosen, A.; Marinkovic, B.A. Negative thermal expansion and cationic migration in zeolite Y used in FCC catalysts. Bull. Mater. Sci. 2019, 42, 86. [Google Scholar] [CrossRef]
- In-noi, O.; Daorattanachai, P.; Rungnim, C.; Prasitnok, K.; Rungtaweevoranit, B.; Faungnawakij, K.; Khemthong, P. Insight into Fructose Dehydration over Lewis Acid α-Cu2P2O7 Catalyst. Chem. Nano Mat. 2021, 7, 292–298. [Google Scholar]
- Li, S.Y.; Liu, Q.L.; Zhou, J.J.; Pan, T.; Gao, L.; Zhang, W.D.; Fan, L.; Lu, Y.Y. Hierarchical Co3O4 Nanofiber-Carbon Sheet Skeleton with Superior Na/Li-Philic Property Enabling Highly Stable Alkali Metal Batteries. Adv. Funct. Mater. 2019, 29, 1808847. [Google Scholar] [CrossRef]
- Agarwal, A.; Majumder, S.; Sankapal, B.R. Multi-walled carbon nanotubes supported copper phosphate microflowers for flexible solid-state supercapacitor. Int. J. Energy Res. 2022, 46, 6177–6196. [Google Scholar] [CrossRef]
- Meganathan, K.L.; BoopathiRaja, R.; Parthibavarman1, M.; Sharmila, V.; Shkir, M.; Gaikwad, S.A.; Praveenkumar, M. Design and fabrication of Cu2P2O7@Ppy electrode for extraordinary capacitance and long-term stability for ideal asymmetric supercapacitor application. J. Mater. Sci. Mater. Electron. 2021, 32, 24736–24747. [Google Scholar] [CrossRef]
- Agarwal, A.; Sankapal, B.R. Ultrathin Cu2P2O7 nanoflakes on stainless steel substrate for flexible symmetric all-solid-state supercapacitors. Chem. Eng. J. 2021, 422, 130131. [Google Scholar] [CrossRef]
- Zou, H.; Yang, X.Q.; Chen, B.; Du, Y.Y.; Ren, B.Y.; Sun, X.W.; Qiao, X.S.; Zhang, Q.W.; Wang, F. Thermal enhancement of upconversion by negative lattice expansion in orthorhombic Yb2W3O12. Angew. Chem. Int. Ed. 2019, 58, 17255–17259. [Google Scholar] [CrossRef]
- Magnusson, H.; Frisk, K. Thermodynamic evaluation of the copper-rich part of the Cu–H–O–S–P system at low temperatures. CALPHAD 2014, 47, 148–160. [Google Scholar] [CrossRef]
- Li, Y.G.; Korzhavyi, P.A. Interactions of point defects with stacking faults in oxygen-free phosphorus-containing copper. J. Nucl. Mater. 2015, 462, 160–164. [Google Scholar] [CrossRef]
- Sang, J.; Wei, P.; Liu, T.; Lv, H.; Ni, X.; Gao, D.; Zhang, J.; Li, H.; Zang, Y.; Yang, F.; et al. A Reconstructed Cu2P2O7 Catalyst for Selective CO2 Electroreduction to Multicarbon Products. Angew. Chem. 2022, 134, 202114238. [Google Scholar] [CrossRef]
- Arango, Y.C.; Vavilova, E.; Hafiez, M.A.; Janson, O.; Tsirlin, A.A.; Rosner, H.; Drechsler, S.L.; Weil, M.; Nenert, G.; Klingeler, R.; et al. Magnetic properties of the low-dimensional spin-1/2 magnet α-Cu2As2O7. Phys. Rev. B 2011, 84, 134430. [Google Scholar] [CrossRef]
- Janson, O.; Tsirlin, A.A.; Sichelschmidt, J.; Skourski, Y.; Weickert, F.; Rosner, H. Long-range superexchange in Cu2A2O7 (A = P, As, V) as a key element of the microscopic magnetic model. Phys. Rev. B 2011, 83, 094435. [Google Scholar] [CrossRef]
- Shi, N.; Sanson, A.; Gao, Q.L.; Sun, Q.; Ren, Y.; Huang, Q.Z.; de Souza, D.O.; Xing, X.R.; Chen, J. Strong Negative Thermal Expansion in a Low-Cost and Facile Oxide of Cu2P2O7. J. Am. Chem. Soc. 2020, 142, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, H. Structural refinement of low-temperature copper(II) pyrophosphate. Acta Cryst. C 1990, 46, 691–692. [Google Scholar] [CrossRef]
- Robertson, B.E.; Calvo, C. The crystal structure and phase transformation of α-Cu2P2O7. Acta Cryst. 1967, 22, 665–672. [Google Scholar] [CrossRef]
- Pogorzelec-Glaser, K.; Pietraszko, A.; Hilczer, B.; Poomska, M. Structure and phase transitions in Cu2P2O7. Phase. Transit. 2006, 79, 535–544. [Google Scholar] [CrossRef]
- Karaphun, A.; Chirawatkul, P.; Maensiri, S.; Swatsitang, E. Influence of calcination temperature on the structural, morphological, optical, magnetic and electrochemical properties of Cu2P2O7 nanocrystals. J. Sol.-Gel. Sci. Technol. 2018, 88, 407–421. [Google Scholar] [CrossRef]
- Yang, X.Y.; Lu, Y.; Zhang, P. First-principles study of native point defects and diffusion behaviors of helium in zirconium carbide. J. Nucl. Mater. 2015, 465, 161–166. [Google Scholar] [CrossRef]
- Ren, K.; Wang, K.; Zhang, G. Atomic Adsorption-Controlled Magnetic Properties of a Two-Dimensional (2D) Janus Monolayer. ACS Appl. Electron. Mater. 2022, 4, 4507–4513. [Google Scholar] [CrossRef]
- Ren, K.; Ma, X.; Liu, X.; Xu, Y.; Huo, W.; Li, W.; Zhang, G. Prediction of 2D IV–VI semiconductors: Auxetic materials with direct bandgap and strong optical absorption. Nanoscale 2022, 14, 8463–8473. [Google Scholar] [CrossRef]
- Bamberger, C.E.; Specht, E.D.; Anovitz, L.M. Crystalline Copper Phosphates: Synthesis and Thermal Stability. J. Am. Ceram. Soc. 1997, 80, 3133–3138. [Google Scholar] [CrossRef]
- Le, S.N.; Navrotsky, A.; Pralong, V. Energetics of copper diphosphates-Cu2P2O7 and Cu3(P2O6OH)2. Solid State Sci. 2008, 10, 761–767. [Google Scholar] [CrossRef]
- Pastukh, S.; Laskowska, M.; Dulski, M.; Krzykawski, T.; Parlinski, K.; Piekarz, P. Ab initio studies for characterization and identification of nanocrystalline copper pyrophosphate confined in mesoporous silica. Nanotechnology 2021, 32, 415701. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.A.R.; Stager, C.V. Magnetic Structure of Manganese Pyrophosphate and Copper Pyrophosphate. Can. J. Phys. 1972, 50, 3079. [Google Scholar] [CrossRef]
- Stiles, J.A.R.; Stager, C.V. Nuclear Magnetic Resonance in Antiferromagnetic Cu2P2O7. Can. J. Phys. 1973, 51, 87. [Google Scholar] [CrossRef]
- Pastukh, S.; Piekarz, P. Ab initio calculations of the structural and dynamical properties of copper pyrophosphate. Proceedings 2020, 4, 32. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof funtionals. Phys. Rev. B 1999, 59, 7413. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Bruke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Assessment and validation of a screened Coulomb hybrid density functional. J. Chem. Phys. 2004, 120, 7274. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Ewing, M.B.; Lilley, T.H.; Olofsson, G.M.; Ratzsch, M.T.; Somsen, G. Standard quantities in chemical thermodynamics. Fugacities, activities and equilibrium constants for pure and mixed phases (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 533–552. [Google Scholar] [CrossRef]
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, I.C.; Ángyán, J.G. Screened hybrid density functionals applied to solids. J. Chem. Phys. 2006, 124, 154709. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Movlarooy, T.; Kompany, A. First-principle calculations of the cohesive energy and electronic properties of PbTiO3. Phys. B 2007, 391, 316e21. [Google Scholar] [CrossRef]
- Yang, X.Y.; Yang, Y.; Lu, Y.; Sun, Z.Y.; Hussain, S.; Zhang, P. First-principles GGA+U calculation investigating the hydriding and diffusion properties of hydrogen in PuH2+x, 0≤x≤ 1. Int. J. Hydrogen Energy 2018, 43, 13632–13638. [Google Scholar] [CrossRef]
- Pedroza, L.; da Silva, A.J.R.; Capelle, K. Gradient-dependent density functionals of the Perdew-Burke-Ernzerhof type for atoms, molecules, and solids. Phys. Rev. B 2009, 79, 201106. [Google Scholar] [CrossRef]
- Forsyth, J.B.; Brown, P.J.; Wanklyn, B.M. Magnetism in cupric oxide. J. Phys. C Solid State Phys. 1988, 21, 2917–2929. [Google Scholar] [CrossRef]
- Watt, J.P. Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with monoclinic symmetry. J. Appl. Phys. 1980, 51, 1520–1524. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, B.T.; Zhao, X.G. Ground-state properties and high-pressure behavior of plutonium dioxide: Density functional theory calculations. Phys. Rev. B 2010, 82, 144110. [Google Scholar] [CrossRef]
- Majtyka-Pilat, A.; Wojtyniak, M.; Laskowski, L.; Chrobak, D. Structure and Properties of Copper Pyrophosphate by First-Principle Calculations. Materials 2022, 15, 842. [Google Scholar] [CrossRef]
- Hase, M.; Kohno, M.; Kitazawa, H.; Tsujii, N.; Suzuki, O.; Ozawa, K.; Kido, G.; Imai, M.; Hu, X. 1/3 magnetization plateau observed in the spin-1/2 trimer chain compound Cu3(P2O6OH)2. Phys. Rev. B 2006, 73, 104419. [Google Scholar] [CrossRef]
- Goodenough, J.B. Theory of the role of covalence in the Perovskite-Type Manganites [La, M(II)]MnO3. Phys. Rev. 1995, 100, 564. [Google Scholar] [CrossRef]
- Born, M. On the stability of crystal lattices I. Proc. Camb. Philos. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Grimvall, G.; Magyari-Köpe, B.; Ozoliņš, V.; Persson, K.A. Lattice instabilities in metallic elements. Rev. Mod. Phys. 2012, 84, 945–986. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Voigt, W. Lehrburch der Kristallphysik; Teubner: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Lond. A 1952, 65, 350. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Phil. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Yang, X.Y.; Lu, Y.; Wei, Z.F.; Ping, Z. Mechanical, electronic, and thermodynamic properties of zirconium carbide from first-principles calculations. Chin. Phys. B 2015, 24, 116301. [Google Scholar] [CrossRef]
- Li, Y.G.; Korzhavyi, P.A. Physical and chemical properties of Cu (I) compounds with O and/or H. Dalton Trans. 2017, 46, 529–538. [Google Scholar] [CrossRef]
- Eschrig, H.; Koepernik, K. Tight-binding models for the iron-based superconductors. Phys. Rev. B 2009, 80, 104503. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990; p. 155. [Google Scholar]
- Xiang, Y.; Hao, X.; Liu, X.; Wang, M.; Tian, J.; Kang, C.; Liang, E.; Zhang, W.; Jia, Y. Tailoring Thermal Expansion of (LiFe)0.5xCu2-xxP2O7 via Codoping LiFe Diatoms in Cu2P2O7 Oxide. Inorg. Chem. 2022, 61, 1504–1511. [Google Scholar] [CrossRef]
- Yashima, M.; Suzuki, R.O. Electronic structure and magnetic properties of monoclinic β-Cu2V2O7: A GGA+U study. Phys. Rev. B 2009, 79, 125201. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Yang, X.Y.; Lu, Y.; Zhang, P. The temperature-dependent diffusion coefficient of helium in zirconium carbide studied with first-principles calculations. J. Appl. Phys. 2015, 117, 164903. [Google Scholar] [CrossRef]
- Lee, C.; Gonze, X. Ab initio calculation of the thermodynamic properties and atomic temperature factors of SiO2α-quartz and stishovite. Phys. Rev. B 1995, 51, 8610–8613. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).