
High-Temperature Thermodynamics of Uranium from Ab Initio Modeling

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Ab Initio Computational Methodology
3. CALPHAD Methodology
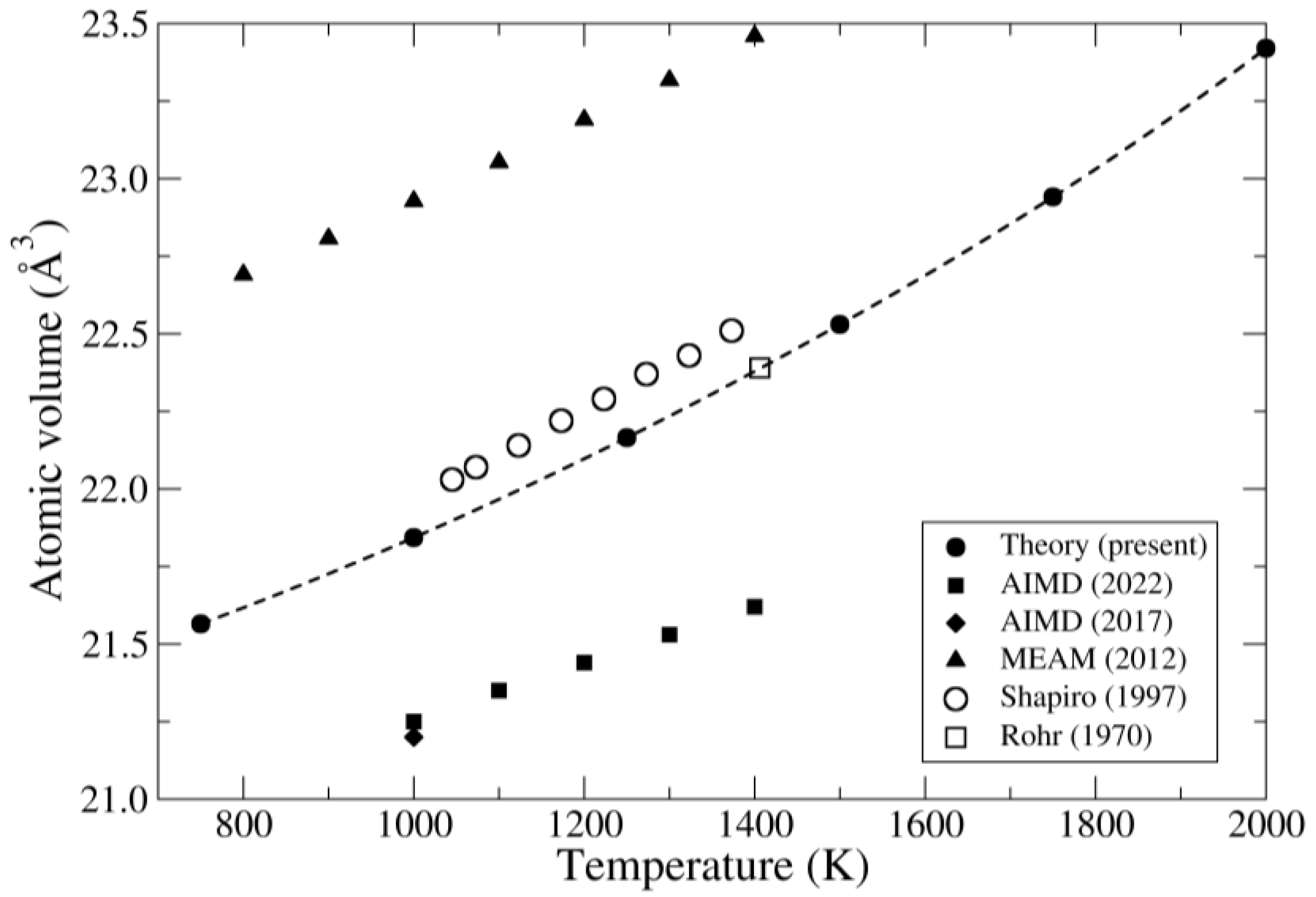
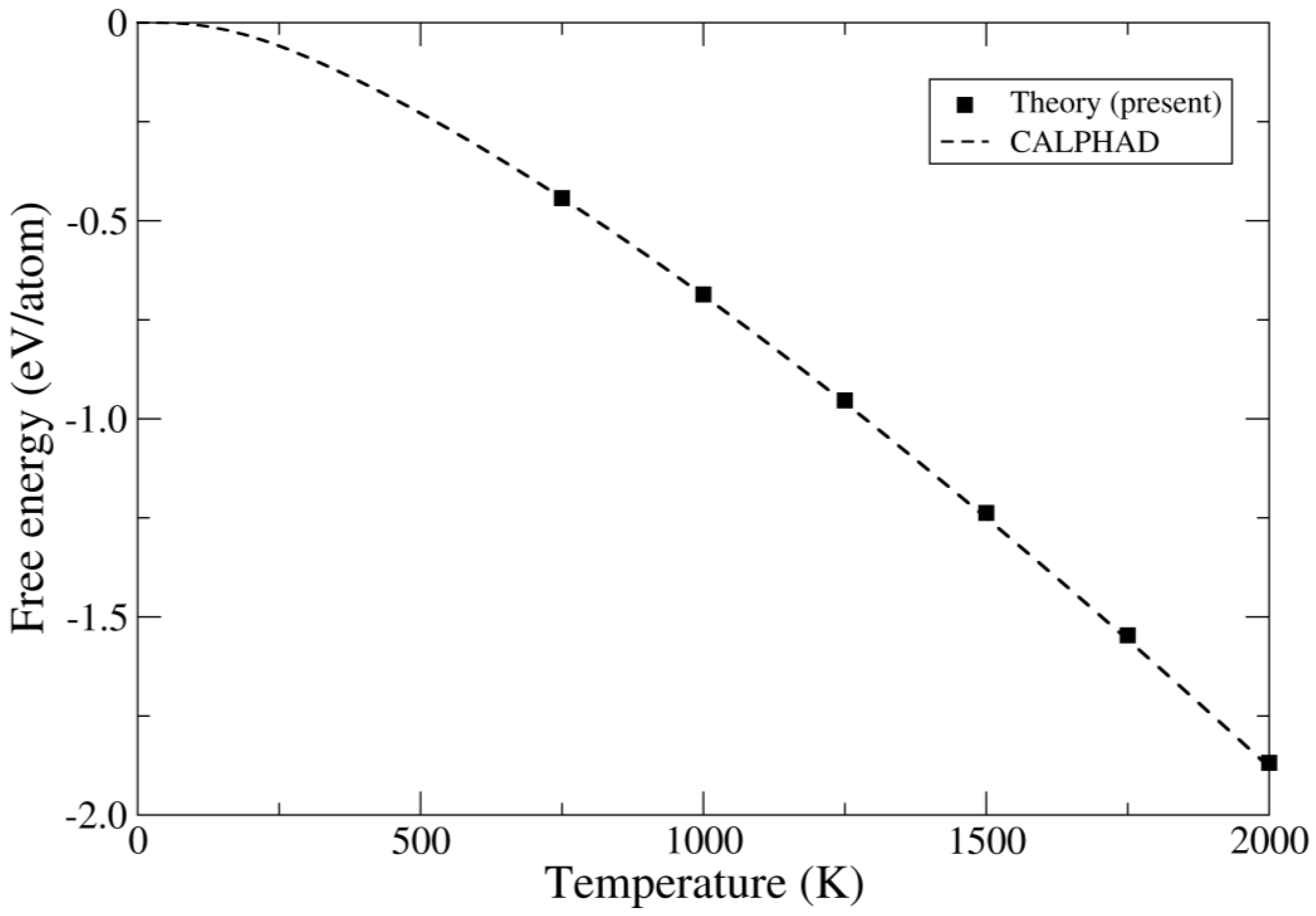
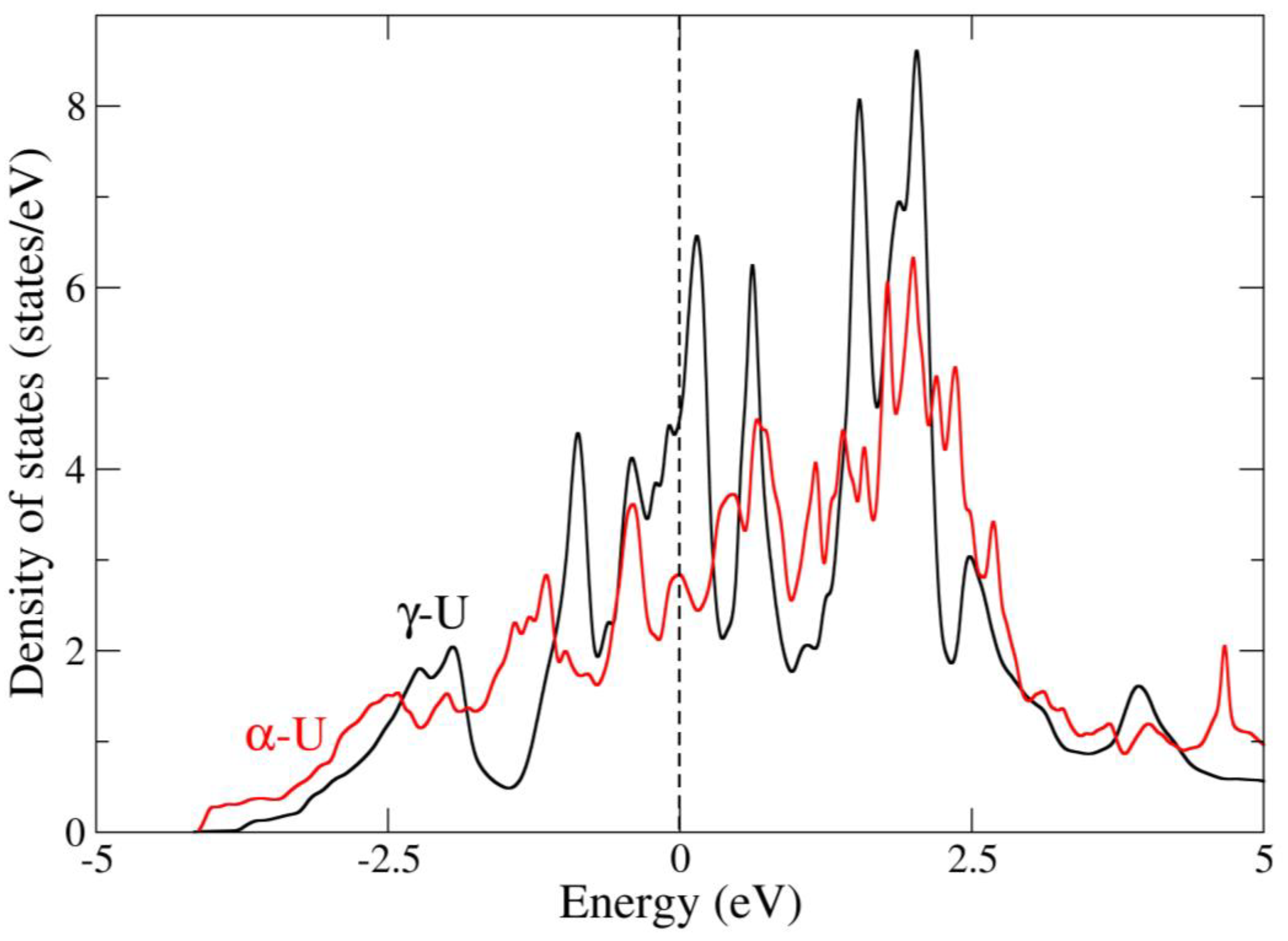
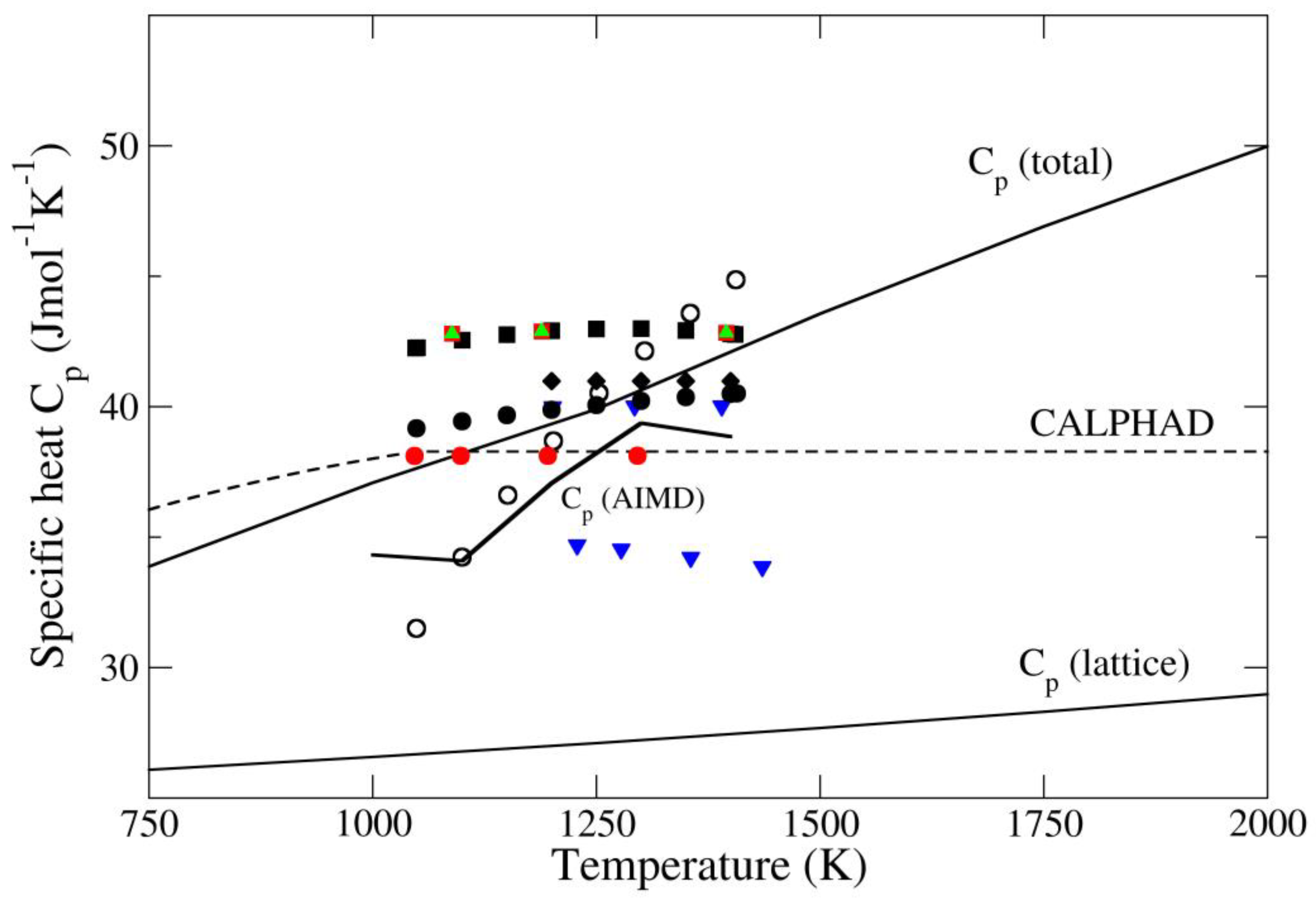
4. Results
5. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hofman, G.; Walters, L.; Bauer, T. Metallic fast reactor fuels. Prog. Nucl. Energy 1997, 31, 83–110. [Google Scholar] [CrossRef]
- Meyer, M.K.; Hofman, G.L.; Hayes, S.L.; Clark, C.R.; Wiencek, T.C.; Snelgrove, J.L.; Strain, R.V.; Kim, K.-H. Low-temperature irradiation behavior of uranium–molybdenum alloy dispersion fuel. J. Nucl. Mater. 2002, 304, 221–236. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hofman, G.; Yacout, A. Migration of minor actinides and lanthanides in fast reactor metallic fuel. J. Nucl. Mater. 2009, 392, 164–170. [Google Scholar] [CrossRef]
- Carmack, W.J.; Porter, D.L.; Chang, I.; Hayes, S.L.; Meyer, M.K.; Burkes, D.E.; Lee, C.B.; Mizuno, T.; Delage, F.; Somers, J.; et al. Metallic fuels for advanced reactors. J. Nucl. Mater. 2009, 392, 139–150. [Google Scholar] [CrossRef]
- Todreas, N.E. Thermal-hydraulics challenges in fast reactor design. Nucl. Tech. 2009, 167, 127–144. [Google Scholar] [CrossRef]
- Janney, D.E. Metallic Fuels Handbook, Part 1: Alloys Based on U-Zr, Pu-Zr, U-Pu, or U-Pu-Zr, Including Those with Minor Actinides (Np, Am, Cm) Rare-Earth Elements (La, Ce, Pr, Nd, Gd), and Y; Idaho National Laboratory: Idaho Falls, ID, USA, 2017. [Google Scholar]
- Capriotti, L.; Bremier, S.; Inagaki, K.; Poml, P.; Papaioannou, D.; Ohta, H.; Ogata, T.; Rondinella, V.V. Characterization of metallic fuel for minor actinides trans mutation in fast reactor. Prog. Nucl. Energy 2017, 94, 194–201. [Google Scholar] [CrossRef]
- Imhoff, S.D. Uranium Density, Thermal Conductivity, Specific Heat, and Thermal Diffusivity; LA-UR-21-21810; OSTI.GOV: Idaho National Lab: Idaho Falls, ID, USA, 24 February 2021. [Google Scholar]
- Söderlind, P. Theory of the crystal structures of cerium and the light actinides. Adv. Phys. 1998, 47, 959–998. [Google Scholar] [CrossRef]
- Söderlind, P.; Johansson, B.; Yongming, L.; Nordström, L. Calculated thermal expansion of the actinide elements. Int. J. Thermophys. 1991, 12, 611–615. [Google Scholar] [CrossRef]
- Söderlind, P.; Eriksson, O.; Wills, J.M.; Boring, A.M. Elastic constants of cubic f-electron elements: Theory. Phys. Rev. B 1993, 48, 9306–9312. [Google Scholar] [CrossRef]
- Söderlind, P.; Eriksson, O.; Wills, J.; Boring, A. A unified picture of the crystal structures of metals. Nature 1995, 374, 524–525. [Google Scholar] [CrossRef]
- Söderlind, P.; Grabowski, B.; Yang, L.; Landa, A.; Björkman, T.; Souvatzis, P.; Eriksson, O. High-temperature phonon stabilization of γ-uranium from relativistic first-principles theory. Phys. Rev. B 2012, 85, 60301. [Google Scholar] [CrossRef]
- Yoo, C.-S.; Cynn, H.; Söderlind, P. Phase diagram of uranium at high pressures and temperatures. Phys. Rev. B 1998, 57, 10359. [Google Scholar] [CrossRef]
- Söderlind, P. First-principles elastic and structural properties of uranium metal. Phys. Rev. B 2002, 66, 85113. [Google Scholar] [CrossRef]
- Wills, J.M.; Eriksson, O. Crystal-structure stabilities and electronic structure for the light actinides Th, Pa, and U. Phys. Rev. B 1992, 45, 13879. [Google Scholar] [CrossRef]
- Richard, N.; Bernard, S.; Jollet, F.; Torrent, M. Plane-wave pseudopotential study of the light actinides. Phys. Rev. B 2002, 66, 235112. [Google Scholar] [CrossRef]
- Hood, R.; Yang, L.H.; Moriarty, J. Quantum molecular dynamics simulations of uranium at high pressure and temperature. Phys. Rev. B 2008, 78, 24116. [Google Scholar] [CrossRef]
- Taylor, C.D. Evaluation of first-principles techniques for obtaining materials parameters of alpha uranium and the (001) alpha uranium surface. Phys. Rev. B 2008, 77, 94119. [Google Scholar] [CrossRef]
- Xiang, S.; Huang, H.; Hsiung, L.M. Quantum mechanical calculations of uranium phases and niobium defects in γ-uranium. J. Nucl. Mater. 2008, 375, 113–119. [Google Scholar] [CrossRef]
- Bouchet, J. Lattice dynamics of α uranium. Phys. Rev. B 2008, 77, 24113. [Google Scholar] [CrossRef]
- Beeler, B.; Good, B.; Rashkeev, S.; Deo, C.; Baskes, M.; Okuniewski, M. First principles calculations for defects in U. J. Phys. Condens. Matter. 2010, 22, 505703. [Google Scholar] [CrossRef]
- Raymond, S.; Bouchet, J.; Lander, G.H.; Le Tacon, M.; Garbarino, G.; Hoesch, M.; Rueff, J.-P.; Krisch, M.; Lashley, J.C.; Schulze, R.K.; et al. Understanding the complex phase diagram of uranium: The role of electron-phonon coupling. Phys. Rev. Lett. 2011, 107, 136401. [Google Scholar] [CrossRef]
- Bouchet, J.; Albers, R. Elastic properties of the light actinides at high pressure. J. Phys. Condens. Matter. 2011, 23, 215402. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-Y.; Wirth, B.D. First-principles study of diffusion of interstitial and vacancy in α U-Zr. J. Phys. Condens. Matter 2011, 23, 205402. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-Y.; Wirth, B.D. First-principles study of bubble nucleation and growth behaviors in α U-Zr. J. Phys. Condens. Matter 2012, 24, 415404. [Google Scholar] [CrossRef]
- Akella, J.; Weir, S.; Wills, J.M.; Söderlind, P. Structural stability in uranium. J. Phys. Condens. Matter 1997, 9, L549–L555. [Google Scholar] [CrossRef]
- Smirnova, D.E.; Starikov, S.V.; Stegailov, V.V. Interatomic potential for uranium in a wide range of pressures and temperatures. J. Phys. Condens. Matter. 2012, 24, 15702. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, D.E.; Starikov, S.V.; Stegailov, V.V. New interatomic potential for computation of mechanical and thermodynamic properties of uranium. Phys. Met. Metallogr. 2012, 113, 107–116. [Google Scholar] [CrossRef]
- Beeler, B.; Deo, C.; Baskes, M.; Okuniewski, M. Atomistic properties of γ uranium. J. Phys. Condens. Matter 2012, 24, 75401. [Google Scholar] [CrossRef]
- Pascuet, M.I.; Bonny, G.; Fernández, J.R. Many-body interatomic U and Al–U potentials. J. Nucl. Mater. 2012, 424, 158–163. [Google Scholar] [CrossRef]
- Beeler, B.; Deo, C.; Baskes, M.; Okuniewski, M. First principles calculations of the structure and elastic constants of α, β and γ uranium. J. Nucl. Mater. 2013, 433, 143–151. [Google Scholar] [CrossRef]
- Smirnova, D.E.; Kuksin, A.Y.; Starikov, S.V.; Stegailov, V.V.; Insepov, Z.; Rest, J.; Yacout, A.M. A ternary EAM interatomic potential for U–Mo alloys with xenon. Modell. Simul. Mater. Sci. Eng. 2013, 21, 35011. [Google Scholar] [CrossRef]
- Smirnova, D.E.; Kuksin, A.Y.; Starikov, S.V. Investigation of point defects diffusion in bcc uranium and U–Mo alloys. J. Nucl. Mater. 2015, 458, 304–311. [Google Scholar] [CrossRef]
- Smirnova, D.E.; Kuksin, A.Y.; Starikov, S.V.; Stegailov, V.V. Atomistic modeling of the self-diffusion in γ-U and γ-U–Mo. Phys. Met. Metallogr. 2015, 116, 445–455. [Google Scholar] [CrossRef]
- Moore, A.P.; Beeler, B.; Deo, C.; Baskes, M.I.; Okuniewski, M.A. Atomistic modeling of high temperature uranium-zirconium alloy structure and thermodynamics. J. Nucl. Mater. 2015, 467, 802–819. [Google Scholar] [CrossRef]
- Bouchet, J.; Bottin, F. Thermal evolution of vibrational properties of α-U. Phys. Rev. B 2015, 92, 174108. [Google Scholar] [CrossRef]
- Tseplyaev, V.I.; Starikov, S.V. The atomistic simulation of pressure-induced phase transition in uranium mononitride. J. Phys. Conf. Ser. 2015, 653, 012092. [Google Scholar] [CrossRef]
- Ren, Z.; Wu, J.; Ma, R.; Hu, G.; Luo, C. Thermodynamic properties of α-uranium. J. Nucl. Mater. 2016, 480, 80–87. [Google Scholar] [CrossRef]
- Kuksin, A.Y.; Starikov, S.V.; Smirnova, D.E.; Tseplyaev, V.I. The diffusion of point defects in uranium mononitride: Combination of DFT and atomistic simulation with novel potential. J. Alloys Comp. 2016, 658, 385–394. [Google Scholar] [CrossRef]
- Kolotova, L.N.; Kuksin, A.Y.; Smirnov, D.E.; Starikov, S.V.; Tseplyaev, V.I. Features of structure and phase transitions in pure uranium and U–Mo alloys: Atomistic simulation. J. Phys. Conf. Ser. 2016, 774, 12036. [Google Scholar] [CrossRef]
- Kolotova, L.N.; Starikov, S.V. Anisotropy of the U–Mo alloy: Molecular-dynamics study. Phys. Met. Metallogr. 2016, 117, 487–493. [Google Scholar] [CrossRef]
- Tseplyaev, V.I.; Starikov, S.V. The atomistic simulation of pressure-induced phase transition in uranium mononitride. J. Nucl. Mater. 2016, 480, 7–14. [Google Scholar] [CrossRef]
- Starikov, S.V.; Kolotova, L.N. Features of cubic and tetragonal structures of U–Mo alloys: Atomistic simulation. Script. Mater. 2016, 113, 27–30. [Google Scholar] [CrossRef]
- Huang, S.-Q.; Ju, X.-H. First-principles study of properties of alpha uranium crystal and seven alpha-uranium surfaces. J. Chem. 2017, 2017, 8618340. [Google Scholar] [CrossRef]
- Bouchet, J.; Bottin, F. High-temperature and high-pressure phase transitions in uranium. Phys. Rev. B 2017, 95, 54113. [Google Scholar] [CrossRef]
- Kolotova, L.N.; Starikov, S.V. Atomistic simulation of defect formation and structure transitions in U-Mo alloys in swift heavy ion irradiation. J. Nucl. Mater. 2017, 495, 111–117. [Google Scholar] [CrossRef]
- Starikov, S.; Kuksin, A.; Smirnova, D.; Dolgodvorov, A.; Ozrin, V. Multiscale modeling of uranium mononitride: Point defects diffusion, self-diffusion, phase composition. Defect Diffus. Forum 2017, 375, 101–113. [Google Scholar] [CrossRef]
- Starikov, S.V.; Kolotova, L.N.; Kuksin, A.Y.; Smirnova, D.E.; Tseplyaev, V.I. Atomistic simulation of cubic and tetragonal phases of U-Mo alloy: Structure and thermodynamic properties. J. Nucl. Mater. 2018, 499, 451–463. [Google Scholar] [CrossRef]
- Starikov, S.; Korneva, M. Description of phase transitions through accumulation of point defects: UN, UO2 and UC. J. Nucl. Mater. 2018, 510, 373–381. [Google Scholar] [CrossRef]
- Lunev, A.V.; Starikov, S.V.; Aliev, T.N.; Tseplyaev, V.I. Understanding thermally-activated glide of 1/2 110 {110} screw dislocations in UO2—A molecular dynamics analysis. Intern. J. Plast. 2018, 110, 294–305. [Google Scholar] [CrossRef]
- Torres, E.; Kaloni, T.P. Projector augmented-wave pseudopotentials for uranium-based compounds. Comp. Mater. Sci. 2020, 171, 109237. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Perron, A.; Sadigh, B.; Heo, T.W. Ground-state and thermodynamical properties of uranium mononitride from anharmonic first-principles theory. Appl. Sci. 2019, 9, 3914. [Google Scholar] [CrossRef]
- Kolotova, L.N.; Starikov, S.V.; Ozrin, V.D. Atomistic simulation of the fission-fragment-induced formation of defects in a uranium–molybdenum alloy. J. Exp. Theor. Phys. 2019, 129, 59–65. [Google Scholar] [CrossRef]
- Castellano, A.; Bottin, F.; Dorado, B.; Bouchet, J. Thermodynamic stabilization of γ-U-Mo alloys: Effect of Mo content and temperature. Phys. Rev. B 2020, 101, 184111. [Google Scholar] [CrossRef]
- Ladygin, V.V.; Korotaev, P.Y.; Yanilkin, A.V.; Shapeev, A.V. Lattice dynamics simulation using machine learning interatomic potentials. Comput. Mater. Sci. 2020, 172, 109333. [Google Scholar] [CrossRef]
- Beeler, B.; Casagranda, A.; Aagesen, L.; Zhang, Y.; Novascone, S. Atomistic calculations of the surface energy as a function of composition and temperature in γ U-Zr to inform fuel performance modeling. J. Nucl. Mater. 2020, 540, 152271. [Google Scholar] [CrossRef]
- Beeler, B.; Andersson, D.; Jiang, C.; Zhang, Y. Ab initio molecular dynamics investigation of point defects in γ-U. J. Nucl. Mater. 2020, 545, 152714. [Google Scholar] [CrossRef]
- Kolotova, L.; Gordeev, I. Structure and phase transition features of monoclinic and tetragonal phases in U–Mo alloys. Crystals 2020, 10, 515. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Perron, A.; Moore, E.E.; Wu, C. Thermodynamics of plutonium monocarbide from anharmonic and relativistic theory. Appl. Sci. 2020, 10, 6524. [Google Scholar] [CrossRef]
- Beeler, B.; Mahbuba, K.; Wang, Y.; Jokisaari, A. Determination of thermal expansion, defect formation energy, and defect-induced strain of α-U via ab initio molecular dynamics. Front. Mater. 2021, 8, 661387. [Google Scholar] [CrossRef]
- Migdal, K.; Yanilkin, A. Cold and hot uranium in DFT calculations: Investigation by the GTH pseudopotential, PAW, and APW + lo methods. Comput. Mater. Sci. 2021, 199, 110665. [Google Scholar] [CrossRef]
- Ouyang, W.; Lai, W.; Li, J.; Liu, J.; Liu, B. Atomic simulations of U-Mo under irradiation: A new angular dependent potential. Metals 2021, 11, 1018. [Google Scholar] [CrossRef]
- Söderlind, P.; Yang, L.H.; Landa, A.; Wu, A. Mechanical and thermal properties for uranium and U–6Nb alloy from first-principles theory. Appl. Sci. 2021, 11, 5643. [Google Scholar] [CrossRef]
- Söderlind, P.; Moore, E.E.; Wu, C.J. Thermodynamics modeling for actinide monocarbides and mononitrides from first principles. Appl. Sci. 2022, 12, 728. [Google Scholar] [CrossRef]
- Aly, A.; Beeler, B.; Avramova, M. Ab initio molecular dynamics investigation of γ-(U,Zr) structural and thermal properties as a function of temperature and composition. J. Nucl. Mater. 2022, 561, 153523. [Google Scholar] [CrossRef]
- Flotow, H.E.; Lohr, H.R. The heat capacity and thermodynamic functions of uranium from 5 to 350 K. J. Phys. Chem. 1960, 64, 904–906. [Google Scholar] [CrossRef]
- Jones, W.; Gordon, J.; Long, E. The heat capacities of uranium, uranium trioxide, and uranium dioxide from 15 K to 300 K. J. Chem. Phys. 1952, 20, 695–699. [Google Scholar] [CrossRef]
- Nakamura, J.-I.; Takashi, Y.; Izumi, S.-I.; Kanno, M. Heat capacity of metallic uranium and thorium from 80 to 1000 K. J. Nucl. Mater. 1980, 88, 64–72. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials; Elsevier BV: Amsterdam, The Netherlands, 1999; pp. 157–172. [Google Scholar]
- Konings, R.J.M.; Beneš, O. The thermodynamic properties of the f-elements and their compounds. I. The lanthanide and actinide metals. J. Phys. Chem. Ref. Data 2010, 39, 43102. [Google Scholar] [CrossRef]
- Marchidan, D.I.; Ciopec, M. Enthalpy of uranium to 1500 K by drop calorimetry. J. Chem. Therm. 1976, 8, 691–701. [Google Scholar] [CrossRef]
- Gathers, G. Dynamic methods for investigating thermophysical properties of matter at very high temperatures and pressures. Rep. Prog. Phys. 1986, 86, 341–396. [Google Scholar] [CrossRef]
- Belashchenko, D.K.; Smirnova, D.E.; Ostrovsky, O.I. Molecular-dynamic simulation of the thermophysical properties of liquid uranium. High Temp. 2010, 48, 363–375. [Google Scholar] [CrossRef]
- Souvatzis, P.; Eriksson, O.; Katsnelson, M.I.; Rudin, S.P. Entropy driven stabilization of energetically unstable crystal structures explained from first principles theory. Phys. Rev. Lett. 2008, 100, 95901. [Google Scholar] [CrossRef] [PubMed]
- Söderlind, P.; Landa, A.; Hood, R.Q.; Moore, E.E.; Perron, A.; McKeown, J.T. High-temperature thermodynamics modeling of graphite. Appl. Sci. 2022, 12, 7556. [Google Scholar] [CrossRef]
- Grimvall, G. Spin disorder in paramagnetic fcc iron. Phys. Rev. B 1989, 39, 12300–12301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Classical mean-field approach for thermodynamics: Ab initio thermophysical properties of cerium. Phys. Rev. B 2000, 61, R11863–R11866. [Google Scholar] [CrossRef]
- Grimvall, G.; Häglund, J.; Fernández Guillermet, A. Spin fluctuations in paramagnetic chromium determined from entropy considerations. Phys. Rev. B 1993, 47, 15338–15341. [Google Scholar] [CrossRef]
- Wills, J.M.; Eriksson, O.; Andersson, P.; Delin, A.; Grechnyev, O.; Alouani, M. Full-Potential Electronic Structure Method; Springer Series in Solid-State Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 167. [Google Scholar]
- Sadigh, B.; Kutepov, A.; Landa, A.; Söderlind, P. Assessing relativistic effects and electron correlation in the actinide metals Th-Pu. Appl. Sci. 2019, 9, 5020. [Google Scholar] [CrossRef]
- Söderlind, P. First-principles phase stability, bonding, and electronic structure of actinide metals. J. Electron. Spectr. Rel. Phenom. 2014, 194, 2–7. [Google Scholar] [CrossRef]
- Söderlind, P.; Moore, K.T. When magnetism can stabilize the crystal structure of metals. Scripta Mat. 2008, 59, 1259–1262. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Sadigh, B. Density-functional theory for plutonium. Adv. Phys. 2019, 68, 1–47. [Google Scholar] [CrossRef]
- Eriksson, O.; Brooks, M.S.S.; Johansson, B. Orbital polarization in narrow-band systems: Application to volume collapses in light lanthanides. Phys. Rev. B 1990, 41, 7311(R). [Google Scholar] [CrossRef]
- Eschrig, H.; Sargolzaei, M.; Koepernik, K.; Richter, M. Orbital polarization in the Kohn-Sham-Dirac theory. EPL 2005, 72, 611–617. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Saunders, N.; Miodownik, A. CALPHAD Calculation of Phase Diagrams: A Comprehensive Guide; Elsevier Science: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Lukas, H.; Fries, S.; Sundman, B. Computational Thermodynamics: The CALPHAD Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Andersson, J.O.; Helander, T.; Höglund, L.; Shi, P.F.; Sundman, B. Thermo-Calc & DICTRA, computational tools for material science. Calphad 2002, 26, 273–312. [Google Scholar]
- Thermo-Calc Software PURE5/Pure Substances Database Version 5. Available online: https://thermocalc.com/products/databases/general-alloys-and-pure-substances/ (accessed on 3 February 2023).
- Dinsdale, A.T. SGTE data for pure elements. Calphad 1991, 15, 317–425. [Google Scholar] [CrossRef]
- Wilson, A.S.; Rundle, R.E. The structures of uranium metal. Acta Cryst. 1949, 2, 126–127. [Google Scholar] [CrossRef]
- Crocombette, J.; Jollet, F.; Nga, L.; Petit, T. Plane-wave pseudopotential study of point defects in uranium dioxide. Phys. Rev. B 2001, 64, 104107. [Google Scholar] [CrossRef]
- Shang, S.L.; Saengdeejing, A.; Mei, Z.G.; Kim, D.E.; Zhang, H.; Ganeshan, S.; Wang, Y.; Liu, Z.K. First-principles calculations of pure elements: Equations of state and elastic stiffness constants. Comp. Mater. Sci. 2010, 48, 813–826. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Summers, L.T.; Eckels, D.J.; Sahai, V. Modeling of Casting Microstructures and Defects; UCRL-ID-128519; LLNL Internal Report: Livermore, CA, USA, 1997. [Google Scholar]
- Rohr, W.G.; Wittenberg, L.J. Density of liquid uranium. J. Phys. Chem. 1970, 74, 1151–1152. [Google Scholar] [CrossRef]
- Moore, G.E.; Kelley, K.K. High-temperature heat contents of uranium, uranium dioxide, and uranium trioxide. J. Am. Chem. Soc. 1947, 69, 2105–2107. [Google Scholar] [CrossRef] [PubMed]
- Ginnings, D.C.; Corruccini, R.J. Heat capacities at high temperatures of uranium, uranium trichloride, and uranium tetrachloride. J. Res. N. B. S. 1947, 39, 309–316. [Google Scholar] [CrossRef]
- Levinson, L.S. Heat content of molten uranium. J. Chem. Phys. 1964, 40, 3584–3585. [Google Scholar] [CrossRef]
- Rand, H.; Kubaschewski, O. Thermochemical Properties of Uranium Compounds; Oliver and Boyd; Ltd.: Edinburgh, Scotland; London, UK, 1963; p. 36. [Google Scholar]
- Fei, Y.; Saxena, S.K. An equation for the heat capacity of solids. Geochim. Cosmochim. Acta 1987, 51, 251–254. [Google Scholar] [CrossRef]
- Holley, C.E., Jr.; Storms, E.K. Thermodynamics of Nuclear Materials; Proc. IAEA.: Vienna, Austria, 1968; p. 411. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Atomic Volume (Å3) | Bulk Modulus (GPa) |
|---|---|---|
| MEAM [30] | 21.49 | 115 |
| Pseudopotential [95] | 19.06 | 170 |
| Pseudopotential [19] | 20.18 | 176 |
| Pseudopotential [96] | 20.32 | 133 |
| Pseudopotential [32] | 20.12 | 132 |
| All-electron [15] | 20.76 | 120 |
| All-electron DFT + OP + SCAILD | 21.00 | 114 |
| Experiment [14,94] | 20.89 | 113 |
| Temperature | µspin | µorbital | Fmag | Fel | Flat |
|---|---|---|---|---|---|
| 750 | 0.1150 | −0.0870 | −0.0035 | −0.0053 | −0.4342 |
| 1000 | 0.1383 | −0.1088 | −0.0049 | −0.0239 | −0.6572 |
| 1250 | 0.1580 | −0.1290 | −0.0061 | −0.0470 | −0.9005 |
| 1500 | 0.1740 | −0.1480 | −0.0066 | −0.0738 | −1.1571 |
| 1750 | 0.1860 | −0.1650 | −0.0062 | −0.1041 | −1.4362 |
| 2000 | 0.1950 | −0.1810 | −0.0048 | −0.1372 | −1.7264 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Söderlind, P.; Landa, A.; Moore, E.E.; Perron, A.; Roehling, J.; McKeown, J.T. High-Temperature Thermodynamics of Uranium from Ab Initio Modeling. Appl. Sci. 2023, 13, 2123. https://doi.org/10.3390/app13042123
Söderlind P, Landa A, Moore EE, Perron A, Roehling J, McKeown JT. High-Temperature Thermodynamics of Uranium from Ab Initio Modeling. Applied Sciences. 2023; 13(4):2123. https://doi.org/10.3390/app13042123
Chicago/Turabian StyleSöderlind, Per, Alexander Landa, Emily E. Moore, Aurélien Perron, John Roehling, and Joseph T. McKeown. 2023. "High-Temperature Thermodynamics of Uranium from Ab Initio Modeling" Applied Sciences 13, no. 4: 2123. https://doi.org/10.3390/app13042123
APA StyleSöderlind, P., Landa, A., Moore, E. E., Perron, A., Roehling, J., & McKeown, J. T. (2023). High-Temperature Thermodynamics of Uranium from Ab Initio Modeling. Applied Sciences, 13(4), 2123. https://doi.org/10.3390/app13042123