
Monitoring the Microbiomes of Agricultural and Food Waste Treating Biogas Plants over a One-Year Period

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Analytical Methods
2.3. Sequencing Data Analysis and Statistical Correlations
3. Results
3.1. Chemical Characterization and Composition of the Digestates of the Selected Full-Scale Anaerobic Digesters
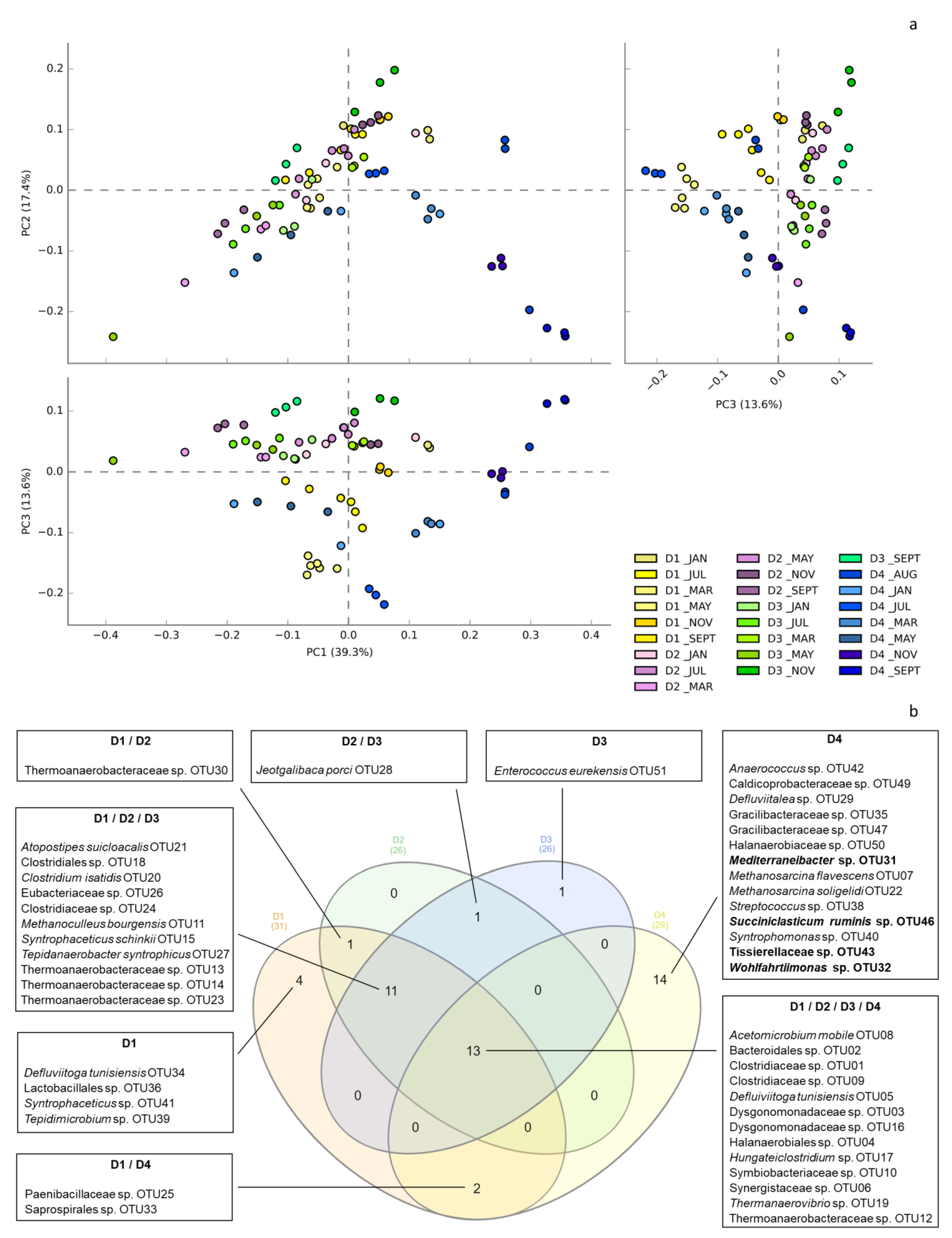
3.2. Global Overview of the Microbial Ecology in the Monitored AD Plants
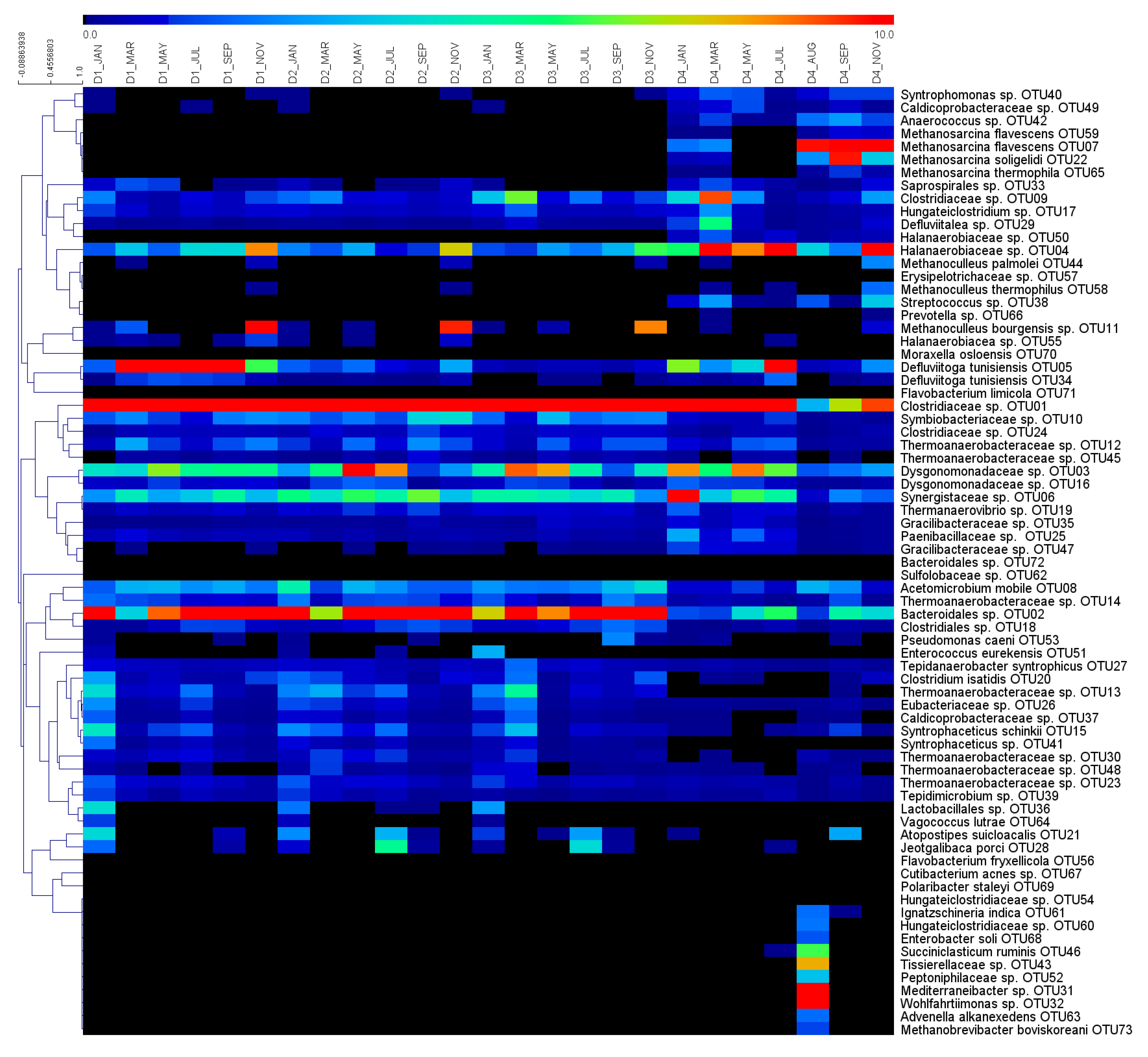
3.3. Dynamics of Microbial Populations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atelge, M.R.; Atabani, A.E.; Banu, J.R.; Krisa, D.; Kaya, M.; Eskicioglu, C.; Kumar, G.; Lee, C.; Yildiz, Y.Ş.; Unalan, S.; et al. A Critical Review of Pretreatment Technologies to Enhance Anaerobic Digestion and Energy Recovery. Fuel 2020, 270, 117494. [Google Scholar] [CrossRef]
- Brojanigo, S.; Alvarado-Morales, M.; Basaglia, M.; Casella, S.; Favaro, L.; Angelidaki, I. Innovative Co-Production of Polyhydroxyalkanoates and Methane from Broken Rice. Sci. Total Environ. 2022, 825, 153931. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Wu, J. Enhancement of Methane Production in Anaerobic Digestion Process: A Review. Appl. Energy 2019, 240, 120–137. [Google Scholar] [CrossRef]
- Basile, A.; Campanaro, S.; Kovalovszki, A.; Zampieri, G.; Rossi, A.; Angelidaki, I.; Valle, G.; Treu, L. Revealing Metabolic Mechanisms of Interaction in the Anaerobic Digestion Microbiome by Flux Balance Analysis. Metab. Eng. 2020, 62, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhao, L.; Li, C.; Angelidaki, I.; Lv, N.; Ning, J.; Cai, G.; Zhu, G. Deep Insights into the Network of Acetate Metabolism in Anaerobic Digestion: Focusing on Syntrophic Acetate Oxidation and Homoacetogenesis. Water Res. 2021, 190, 116774. [Google Scholar] [CrossRef] [PubMed]
- Theuerl, S.; Klang, J.; Heiermann, M.; De Vrieze, J. Marker Microbiome Clusters Are Determined by Operational Parameters and Specific Key Taxa Combinations in Anaerobic Digestion. Bioresour. Technol. 2018, 263, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Maus, I.; Klocke, M.; Derenkó, J.; Stolze, Y.; Beckstette, M.; Jost, C.; Wibberg, D.; Blom, J.; Henke, C.; Willenbücher, K.; et al. Impact of Process Temperature and Organic Loading Rate on Cellulolytic/Hydrolytic Biofilm Microbiomes during Biomethanation of Ryegrass Silage Revealed by Genome-Centered Metagenomics and Metatranscriptomics. Environ. Microbiome 2020, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Kougias, P.G.; Campanaro, S.; Treu, L.; Angelidaki, I. Evolution of the Microbial Community Structure in Biogas Reactors Inoculated with Seeds from Different Origin. Sci. Total Environ. 2021, 773, 144981. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New Insights from the Biogas Microbiome by Comprehensive Genome-Resolved Metagenomics of Nearly 1600 Species Originating from Multiple Anaerobic Digesters. Biotechnol. Biofuels 2020, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Abu Hanifa Jannat, M.; Hyeok Park, S.; Chairattanawat, C.; Yulisa, A.; Hwang, S. Effect of Different Microbial Seeds on Batch Anaerobic Digestion of Fish Waste. Bioresour. Technol. 2022, 349, 126834. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Luo, G.; Angelidaki, I. Metagenomic Binning Reveals the Functional Roles of Core Abundant Microorganisms in Twelve Full-Scale Biogas Plants. Water Res. 2018, 140, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Kougias, P.G.; Treu, L.; Kovalovszki, A.; Valle, G.; Cappa, F.; Morelli, L.; Angelidaki, I.; Campanaro, S. Microbial Activity Response to Hydrogen Injection in Thermophilic Anaerobic Digesters Revealed by Genome-Centric Metatranscriptomics. Microbiome 2018, 6, 194. [Google Scholar] [CrossRef]
- Cardona, L.; Cao, K.A.L.; Puig-Castellví, F.; Bureau, C.; Madigou, C.; Mazéas, L.; Chapleur, O. Integrative Analyses to Investigate the Link between Microbial Activity and Metabolite Degradation during Anaerobic Digestion. J. Proteome Res. 2020, 19, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, S.; Hashemi, S.E.; Lien, K.M.; Lamb, J.J. Molecular microbial community analysis as an analysis tool for optimal biogas production. Microorganisms 2021, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Joshi, C.; Paritosh, K.; Thakur, J.; Pareek, N.; Masakapalli, S.K.; Vivekanand, V. Reprint of Organic waste conversion through anaerobic digestion: A critical insight into the metabolic pathways and microbial interactions. Metab. Eng. 2022, 71, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Mistretta, M.; Gulotta, T.M.; Caputo, P.; Cellura, M. Bioenergy from anaerobic digestion plants: Energy and environmental assessment of a wide sample of Italian plants. Sci. Total Environ. 2022, 843, 157012. [Google Scholar] [CrossRef] [PubMed]
- EBA. EBA Statistical Report 2022; EBA: Brussels, Belgium, 2022. [Google Scholar]
- Costa, P.; Basaglia, M.; Casella, S.; Favaro, L. Polyhydroxyalkanoate Production from Fruit and Vegetable Waste Processing. Polymers 2022, 14, 5529. [Google Scholar] [CrossRef]
- Cucchiella, F.; D’Adamo, I.; Gastaldi, M. Sustainable Italian cities: The added value of biomethane from organic waste. Appl. Sci. 2019, 9, 2221. [Google Scholar] [CrossRef]
- Czubaszek, R.; Wysocka-Czubaszek, A.; Tyborowski, R. Methane Production Potential from Apple Pomace, Cabbage Leaves, Pumpkin Residue and Walnut Husks. Appl. Sci. 2022, 12, 6128. [Google Scholar] [CrossRef]
- Favaro, L.; Basaglia, M.; Rodriguez, J.E.G.; Morelli, A.; Ibraheem, O.; Pizzocchero, V.; Casella, S. Bacterial production of PHAs from lipid-rich by-products. Appl. Food Biotechnol. 2019, 6, 45–52. [Google Scholar] [CrossRef]
- Gupte, A.P.; Basaglia, M.; Casella, S.; Favaro, L. Rice waste streams as a promising source of biofuels: Feedstocks, biotechnologies and future perspectives. Ren. Sustain. Energy Rev. 2022, 167, 112673. [Google Scholar] [CrossRef]
- APHA. Standard Methods for the Examination of Water and Wastewater; APHA: Washington, DC, USA, 2005. [Google Scholar]
- Brojanigo, S.; Parro, E.; Cazzorla, T.; Favaro, L.; Basaglia, M.; Casella, S. Conversion of starchy waste streams into polyhydroxyalkanoates using Cupriavidus necator DSM 545. Polymers 2020, 12, 1496. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. [9] TM4 Microarray Software Suite. Methods Enzymol. 2006, 411, 134–193. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Beiko, R.G. Identifying Biologically Relevant Differences Between Metagenomic Communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef]
- Calusinska, M.; Goux, X.; Fossépré, M.; Muller, E.E.; Wilmes, P.; Delfosse, P. A year of monitoring 20 mesophilic full-scale bioreactors reveals the existence of stable but different core microbiomes in bio-waste and wastewater anaerobic digestion systems Biotechnol. Biofuels 2018, 11, 196. [Google Scholar] [CrossRef]
- Liu, T.; Goux, X.; Calusinska, M.; Westerholm, M. Analyzing Microbial Core Communities, Rare Species, and Interspecies Interactions Can Help Identify Core Microbial Functions in Anaerobic Degradation. In Assessing the Microbiological Health of Ecosystems; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2022; pp. 127–157. [Google Scholar] [CrossRef]
- Zampieri, G.; Campanaro, S.; Angione, C.; Treu, L. Metatranscriptomics-Guided Genome-Scale Metabolic Modeling of Microbial Communities. Cell Rep. Methods 2023, 3, 100383. [Google Scholar] [CrossRef]
- Cotta, M.A.; Whitehead, T.R.; Falsen, E.; Moore, E.; Lawson, P.A. Erratum to: Two Novel Species Enterococcus lemanii Sp. Nov. and Enterococcus eurekensis Sp. Nov., Isolated from a Swine-Manure Storage Pit. Antonie Leeuwenhoek 2013, 103, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.W.; Park, T.; Tong, Y.W.; Yu, Z. The microbiome driving anaerobic digestion and microbial analysis. In Advances in Bioenergy; Elsevier: Amsterdam, The Netherlands, 2020; Volume 5, pp. 1–61. [Google Scholar]
- Sposob, M.; Moon, H.S.; Lee, D.; Yun, Y.M. Microbiome of seven full-scale anaerobic digestion plants in South Korea: Effect of feedstock and operational parameters. Energies 2021, 14, 665. [Google Scholar] [CrossRef]
- Maus, I.; Cibis, K.G.; Bremges, A.; Stolze, Y.; Wibberg, D.; Tomazetto, G.; Blom, J.; Sczyrba, A.; König, H.; Pühler, A.; et al. Genomic Characterization of Defluviitoga tunisiensis L3, a Key Hydrolytic Bacterium in a Thermophilic Biogas Plant and Its Abundance as Determined by Metagenome Fragment Recruitment. J. Biotechnol. 2016, 232, 50–60. [Google Scholar] [CrossRef]
- Owusu-Agyeman, I.; Plaza, E.; Cetecioglu, Z. Long-Term Alkaline Volatile Fatty Acids Production from Waste Streams: Impact of PH and Dominance of Dysgonomonadaceae. Bioresour. Technol. 2022, 346, 126621. [Google Scholar] [CrossRef]
- Hahnke, S.; Langer, T.; Koeck, D.E.; Klocke, M. Description of Proteiniphilum saccharofermentans Sp. Nov., Petrimonas mucosa Sp. Nov. and Fermentimonas caenicola Gen. Nov., Sp. Nov., Isolated from Mesophilic Laboratory-Scale Biogas Reactors, and Emended Description of the Genus Proteiniphilum. Int. J. Syst. Evol. Microbiol. 2016, 66, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, L.; Li, F.; Zheng, D.; Yang, H. Effect of Air Mixing on High-Solids Anaerobic Digestion of Cow Manure: Performance and Mechanism. Bioresour. Technol. 2023, 370, 128545. [Google Scholar] [CrossRef]
- Pradel, N.; Fardeau, M.-L.; Bunk, B.; Spröer, C.; Boedeker, C.; Wolf, J.; Neumann-Schaal, M.; Pester, M.; Spring, S. Aminithiophilus ramosus Gen. Nov., Sp. Nov., a Sulphur-Reducing Bacterium Isolated from a Pyrite-Forming Enrichment Culture, and Taxonomic Revision of the Family Synergistaceae. Int. J. Syst. Evol. Microbiol. 2023, 73, 005691. [Google Scholar] [CrossRef] [PubMed]
- Suksong, W.; Kongjan, P.; Prasertsan, P.; O-Thong, S. Thermotolerant Cellulolytic Clostridiaceae and Lachnospiraceae Rich Consortium Enhanced Biogas Production from Oil Palm Empty Fruit Bunches by Solid-State Anaerobic Digestion. Bioresour. Technol. 2019, 291, 121851. [Google Scholar] [CrossRef] [PubMed]
- Basak, B.; Ahn, Y.; Kumar, R.; Hwang, J.-H.; Kim, K.-H.; Jeon, B.-H. Lignocellulolytic Microbiomes for Augmenting Lignocellulose Degradation in Anaerobic Digestion. Trends Microbiol. 2022, 30, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Brulc, J.M.; Antonopoulos, D.A.; Berg Miller, M.E.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-Centric Metagenomics of the Fiber-Adherent Bovine Rumen Microbiome Reveals Forage Specific Glycoside Hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Kougias, P.G.; Campanaro, S.; Treu, L.; Tsapekos, P.; Armani, A.; Angelidaki, I. Spatial Distribution and Diverse Metabolic Functions of Lignocellulose-Degrading Uncultured Bacteria as Revealed by Genome-Centric Metagenomics. Appl. Environ. Microbiol. 2018, 84, e01244-18. [Google Scholar] [CrossRef]
- Ben Hania, W.; Godbane, R.; Postec, A.; Hamdi, M.; Ollivier, B.; Fardeau, M.-L. Defluviitoga tunisiensis Gen. Nov., Sp. Nov., a Thermophilic Bacterium Isolated from a Mesothermic and Anaerobic Whey Digester. Int. J. Syst. Evol. Microbiol. 2012, 62, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Simankova, M.V.; Chernych, N.A.; Osipov, G.A.; Zavarzin, G.A. Halocella cellulolytica Gen. Nov., Sp. Nov., a New Obligately Anaerobic, Halophilic, Cellulolytic Bacterium. Syst. Appl. Microbiol. 1993, 16, 385–389. [Google Scholar] [CrossRef]
- Li, Y.; Fang, W.; Xue, H.; Yang, X.; Xie, S.; Wang, L. Wohlfahrtiimonas Populi Sp. Nov., Isolated from Symptomatic Bark of a Populus × Euramericana Canker. Int. J. Syst. Evol. Microbiol. 2017, 67, 4424–4428. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kumar, S.; Lee, G.-H.; Chang, D.-H.; Rhee, M.-S.; Yoon, M.-H.; Kim, B.-C. Methanobrevibacter boviskoreani Sp. Nov., Isolated from the Rumen of Korean Native Cattle. Int. J. Syst. Evol. Microbiol. 2013, 63, 4196–4201. [Google Scholar] [CrossRef]
- Kern, T.; Fischer, M.A.; Deppenmeier, U.; Schmitz, R.A.; Rother, M. Methanosarcina flavescens Sp. Nov., a Methanogenic Archaeon Isolated from a Full-Scale Anaerobic Digester. Int. J. Syst. Evol. Microbiol. 2016, 66, 1533–1538. [Google Scholar] [CrossRef]
- Wagner, D.; Schirmack, J.; Ganzert, L.; Morozova, D.; Mangelsdorf, K. Methanosarcina soligelidi Sp. Nov., a Desiccation- and Freeze-Thaw-Resistant Methanogenic Archaeon from a Siberian Permafrost-Affected Soil. Int. J. Syst. Evol. Microbiol. 2013, 63, 2986–2991. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.H.; Diop, A.; Bittar, F.; Maraninchi, M.; Valero, R.; Armstrong, N.; Dubourg, G.; Labas, N.; Richez, M.; Delerce, J.; et al. Description of Mediterraneibacter massiliensis, Gen. Nov., Sp. Nov., A New Genus Isolated from the Gut Microbiota of an Obese Patient and Reclassification of Ruminococcus faecis, Ruminococcus lactaris, Ruminococcus torques, Ruminococcus gnavus and Clostridium glycyrrhizinilyticum as Mediterraneibacter faecis Comb. Nov., Mediterraneibacter lactaris Comb. Nov., Mediterraneibacter Torques Comb. Nov., Mediterraneibacter gnavus Comb. Nov. and Mediterraneibacter glycyrrhizinilyticus Comb. Nov. Antonie Leeuwenhoek 2018, 111, 2107–2128. [Google Scholar] [CrossRef]
- Van GYLSWYK, N.O. Succiniclasticum ruminis Gen. Nov., Sp. Nov., a Ruminal Bacterium Converting Succinate to Propionate as the Sole Energy-Yielding Mechanism. Int. J. Syst. Evol. Microbiol. 1995, 45, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Kurade, M.B.; Saha, S.; Salama, E.-S.; Patil, S.M.; Govindwar, S.P.; Jeon, B.-H. Acetoclastic Methanogenesis Led by Methanosarcina in Anaerobic Co-Digestion of Fats, Oil and Grease for Enhanced Production of Methane. Bioresour. Technol. 2019, 272, 351–359. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature Range (°C) | HRT (Days) | Operation Volume (m3) | Location | Main Feeding Substrates | |
|---|---|---|---|---|---|
| D1 | 50–55 | 28–32 | 3000 | Mira, Venezia, Italy | Agricultural by-products, manure and bovine slurry |
| D2 | 50–55 | 30–31 | 3200 | Noventa Vicentina, Vicenza, Italy | Residues of agro-food activities; effluent of poultry manure/pig manure; by-products of terrestrial and aquatic animals |
| D3 | 50–55 | 30–31 | 3200 | Ospedaletto Euganeo, Padova, Italy | Agri-food products and sub-products; poultry manure and similar; pig slurry |
| D4 | 50–55 | 36–37 | 2500 | Este, Padova, Italy | OFMSW |
| D1 | D2 | D3 | D4 | |
|---|---|---|---|---|
| Raw reads | 241,360 | 227,000 | 235,940 | 225,250 |
| Assigned reads | 100,600 | 109,500 | 113,700 | 90,510 |
| OTUs | 387 | 389 | 395 | 393 |
| Alpha diversity | 86.6 | 83.5 | 83.2 | 84.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostini, S.; Moriconi, F.; Zampirolli, M.; Padoan, D.; Treu, L.; Campanaro, S.; Favaro, L. Monitoring the Microbiomes of Agricultural and Food Waste Treating Biogas Plants over a One-Year Period. Appl. Sci. 2023, 13, 9959. https://doi.org/10.3390/app13179959
Agostini S, Moriconi F, Zampirolli M, Padoan D, Treu L, Campanaro S, Favaro L. Monitoring the Microbiomes of Agricultural and Food Waste Treating Biogas Plants over a One-Year Period. Applied Sciences. 2023; 13(17):9959. https://doi.org/10.3390/app13179959
Chicago/Turabian StyleAgostini, Sara, Francesco Moriconi, Mauro Zampirolli, Diego Padoan, Laura Treu, Stefano Campanaro, and Lorenzo Favaro. 2023. "Monitoring the Microbiomes of Agricultural and Food Waste Treating Biogas Plants over a One-Year Period" Applied Sciences 13, no. 17: 9959. https://doi.org/10.3390/app13179959
APA StyleAgostini, S., Moriconi, F., Zampirolli, M., Padoan, D., Treu, L., Campanaro, S., & Favaro, L. (2023). Monitoring the Microbiomes of Agricultural and Food Waste Treating Biogas Plants over a One-Year Period. Applied Sciences, 13(17), 9959. https://doi.org/10.3390/app13179959