Abstract
Natural organic matter, including humic substances (HS), comprises complex secondary structures with no defined covalent chemical bonds and stabilized by inter- and intra-molecular interactions, such as hydrogen bonding, Van der Waal’s forces, and pi-pi interactions. The latest view describes HS aggregates as a hydrogel-like structure comprised by a hydrophobic core of aromatic residues surrounded by polar and amphiphilic molecules akin a self-assembled soft material. A different view is based on the classification of this material as either mass or surface fractals. The former is intended as made by the clustering of macromolecules generating dendritic networks, while the latter have been modelled in terms of a solvent-impenetrable core surrounded by a layer of lyophilic material. This study reviews the evolution of the increasingly refined models that appeared in the literature, all capable to describing the physicochemical properties of HS. All the models are critically examined and revisited in terms of their ability to provide key information on the structural organization of HS. Understanding how the molecular association pathway influences aggregation of HS also provides a key acknowledgment of their role in the environment.
1. Introduction
The organic matter in terrestrial and aquatic environments is being classified as particulate organic matter (POM) and dissolved organic matter (DOM). POM includes particles of natural organic matter (NOM) characterized by average sizes ranging from 50 μm to 2 mm. [1]. DOM is the corresponding fraction of dissolved POM that can pass through a 0.45 mm mesh filter [2]. In the marine environment this rather artificial distinction between POM and DOM is sometimes questioned and replaced by a broader definition where organic matter is considered as a continuum of gel-like polymers, replete with colloids cross-linked by polymer strings, sheets, and bundles while the size can reach hundreds of micrometers [3]. An important part of DOM comes from the carbon mobility processes occurring in the environment. However, much of it is lost through various mechanisms such as substrate to soil microorganisms or adsorbed on soil sediments and eventually diffuses into the ocean. Much of the carbon DOM present in terrestrial and aquatic compartments is oxidized to CO2 through microbial metabolism and then accumulates in the atmosphere [4]. DOM is a parameter that responds to changes in ecological processes and is capable to promote the dissolution and transport of metals such as micronutrients and organic compounds, including pollutants [5,6,7,8]. Organic compounds in DOM have been shown to affect the chemical reactivity of soil minerals [9] and the complex process of podzolization of soil [10]. The production of greenhouse gases such as methane, and soil denitrification is connected to the bacterial degradation of DOM [11]. Furthermore, the localization of the DOM in the soil, for example, located in pores of various sizes, influences its mobility, ecological fate and availability [2].
The major pools of DOM are molecules solubilized and leached from soil organic matter (SOM). Those can be biomolecules originating from both fresh materials, such as leaf and grass litter, root exudates, decaying fine roots, soil microorganisms etc. and microbiologically processed, humified, fractions such as humic substances (HS) [12]. Due to the environmental conditions, the dissolved organic carbon cycle is relatively fast. The controlling factors for DOM biodegradability, and thus carbon dynamics, are influenced by several factors. Higher content of non-humified hydrophilic compounds such as carbohydrates, organic acids and proteins increase DOM degradability. On the contrary, the presence of aromatic and hydrophobic residues leads to a reduction of its biodegradability, due to the high resistance shown by the aromatic compounds towards degradation phenomena and to the inhibitory action of the enzymatic activity. Effects of heavy metal concentrations on DOM biodegradability is not clear yet since various studies reported conflicting results. As degradation is dependent on microbial activity, the type and concentration of microbial population must be taken into account as well [13].
HS are the most abundant part of DOM and their chemical structure and even their concept is still a matter of discussion [14]. For decades, it had been accepted that HS are macromolecules formed through a two-step process. In the first step, molecular building blocks are produced from the degradation of plant-, microbial-, and animal-derived organic systems. In the second step, secondary reactions are believed to occur, including condensation of resulting fragments into larger products, each hardly referable to the parent feedstock [15]. This view has been revised and new models have been developed to explain the chemical-physical properties of HS related with their long-term persistence and their molecular architecture [16]. From a chemical-physical point of view, HS are described as dark-coloured, chemically, and presumably biologically refractory, heterogeneous organic compounds playing the main role in soil fertility [17].
This description does not coincide with a specific path of production of a set of organic compounds nor a characteristic molecular mass identified by a chemical reactivity (recalcitrant carbon fraction). However, it excludes insoluble organic matter present in sedimentary rocks (kerogen) [18], and other complex mixtures of organic compounds produced by combustion processes (carbon black) [19]. Strong debate still exists about the chemical nature of HS in various forms, i.e., as aqueous dispersions, within soils and at mineral interfaces, as well as on their role in carbon stabilization mechanisms [20,21]. Recently the carbon stabilization via interaction with soil mineral surfaces is referred to as mineral-associated organic matter (MAOM) [22]. In a small aggregate (40–50 µm), MAOM consists of low-molecular weight compounds potentially sorbed to mineral surfaces, via either incorporation into microbial biomass or extracellular enzymes [23]. However, the new view of stabilisation mechanisms and observed longevity of MAOM [24], is still unclear, because the organo-mineral interactions are easy alterable by root exudates [25].
This is fundamentally a paradigm shift in how the interactions act between SOM components and soil minerals [26], i.e., MAOM aggregates, since they are not permanent. Instead, the persistence of SOM assisted by both the microbiome and the physical structure of the soil is considered the main carbon stabilization mechanism [27]. For example, according to Baveye et al. [28], no significant advances have been made in the last century about the basic (bio)chemistry of HS and carbon stabilization. In fact, it appears that all the modern views about a better definition of HS or on a widely accepted operational extraction procedures do not provide all answer able to overcome the Waksman model as hypothesized in the early years of the Twentieth century [28,29,30,31]. How much is this true? Certainly, the investigation on structure and aggregation of HS can provide useful information about the relevant environmental processes and models in which these natural substances are involved. In this study, we intend to review the existing literature about HS to understand how much is known and where humic research is addressed in order to aid solution of the environmental problems such as the global climatic changes whose effects are there for all to see. Thus, after an overview of the current models available in literature on the structure of the HS (Section 2), in Section 3 the reader will find details on the recent scientific results achieved with the most modern experimental techniques used to unravel the structure of HS both in the solid and liquid state. Finally, a comprehensive exposition of future challenges and concluding remarks will be outlined in Section 4.
2. Models of the Structure of Humic Substances
Along all the research activities carried out to elucidate the nature of aggregation state of HS, various conceptual models have been proposed, among which we can begin to mention the oldest one indicated as the rigid sphero-colloid model elaborated by Ghosh et al. [32]. In particular, those authors linked the structure of HS dissolved in solution to their concentration, being able to form either spherical or flexible linear colloids at high and low HS concentrations, respectively. Vapor-pressure osmometry (VPO) was carried out to investigate the aggregation of standard fulvic acid (FA) [33]. Swift and coworkers argued that HS dissolved in aqueous solutions could consist of high molecular weight macromolecules capable of stabilizing a random coil configuration [34,35]. The polymer gel theory was also invoked to interpret the supramolecular dynamics of HS in marine environment [36]. Beginning in 1990, Wershaw launched the first hypothesis of molecular aggregation, in which HS was described as a self-assembled material consisting of a hydrophobic interior and a hydrophilic exterior [37,38,39]. That hypothesis was also supported by Guetzloff et al. [40,41], who demonstrated through surface tension measurements, that purified HA was able to form pseudo-micelles in alkaline aqueous solutions at concentrations greater than 7.2 g∙L−1. Evidences for the formation of hydrophobic micro-environments following the dissolution of HS in water were formerly documented through fluorescence quenching studies. In particular, the HA-promoted reduction of fluorescence quenching of both pyrene [42] and positively charged synthetic organic fluorophores [43], provided the evidence that HS in aqueous solutions were capable of configuring domains with a micellar cage structure.
2.1. The Micellar Model
Differently from orthodox micellar solutions, humic pseudo-micelles were initially considered as a type of aggregates characterized by a broad distribution of both size and functional groups. According to that vision, pseudo-micelles still retain the ability of traditional micelles to sequester hydrophobic species that partition into the nonpolar microdomains [44,45,46]. Using ultrasonic velocimetry Kučerík et al. [47] demonstrated that the aggregation can be observed at concentrations as low as 1 mg∙L−1 under various pH and ionic strength conditions. The micelle-like behavior of HS was also investigated by Piccolo et al. [48] who verified through size exclusion chromatography (SEC), the effect of the concentration of organic acids on the HS dissociation into small-sized micellar aggregates. HS aggregation was also studied in aqueous solution by HPSEC [49].
The formation of aggregates in solutions of HS was studied through capillary zone electrophoresis (CZE) for the first time by Fetsch et al. [50]. In the late 1990s, Jones et al. [51] published a review in which they surveyed the major findings reported up to that time on the colloidal properties of HS, by emphasizing the aggregation behavior in both artificial and natural environments. By varying the composition of the mobile phase at fixed ionic strength of a SEC system, Conte et al. [52] were able to monitor the conformational changes of different types of dissolved HA and FA. In particular, through high- and low-pressure chromatography, the authors verified that HS complied with the model describing the association of small heterogeneous molecules into larger dimensional structures. The work of Piccolo et al. [53] on HA and FA offered further direct evidence more in favor of the model based on the reversible self-association of small molecules rather than the polymer random coil picture. The reductions in HS dimensions that occurred as a result of variation of composition of the mobile phase were attributed by the authors to a drastic attenuation of weak intermolecular associations giving rise to separate smaller molecules rather than to the compaction of macromolecular chains. Other studies have rejected the idea that the micellar structure of HS could only be inferred by changing the retention volumes following the addition of organic acids.
According to Varga et al. [54], secondary (ionic, hydrophobic) interactions should always be considered for the interpretation of SEC developed for HS. Young et al. [55] interpreted the SEC and fluorescence experiments on several type of HA aqueous solutions in terms of the formation of ‘pseudo-micelles’ assisted under the influence of cations in solution.
The former concept of ‘pseudo-macromolecularity’ was proposed by Piccolo [56] to suggest that the macromolecular properties of HS could be compatible with those produced by pseudo-micellar structures or by other molecular assemblies involving long-chain hydrocarbons, fatty acids, esters and suberin-like components. Subsequently, Sutton et al. [57] published a new model on the molecular structure of HS based on supramolecular associations of light molecular mass components, stabilized by hydrophobic interactions and H-bonds, which self-assemble into micellar aggregates in suitable aqueous milieu.
This model was based on the use of several techniques, such as gel permeation chromatography (GPC), size exclusion chromatography at high pressure (HPSEC), and ultraviolet visible spectroscopy (UV-VIS). Indirect evidence in favor of the micellar model includes, (i) the extent of the reduction in surface tension of aqueous HS solutions observed with increasing HS concentration [58], (ii) the improved solubility of hydrophobic organic compounds at following dispersion in aqueous solutions of HS [37,38,39,40,41] and (iii) the preferred distribution of fluorescent probes such as pyrene, within the hydrophobic microdomain of HA aggregates in aqueous solutions [42,45].
Criticisms against the micellar model can be summarized with the following questions: (i) how can HS lacking well-defined amphiphilic substances self-assemble in aqueous solutions in the form of colloidal aggregates? (ii) What are the typical sizes, shapes and internal structures of HS aggregates in aqueous media? (iii) and how do these parameters depend on the pH and Concentration of HS, dissolved salts and metal ions?
2.2. The Supramolecular Aggregate Model
Alongside the development of polymeric and micellar-like models, a flourishing line of research was developed on the use of the fractal theory in the study of the aggregation of HS [59]. The most salient scientific results concerning the application of the fractal model to HS will be illustrated in Section 3.3.1 dedicated to the scattering studies.
After the advent of polymeric and micellar models, more credits are given to the hypothesis that HS are supramolecular aggregates of small organic compounds, held together by weak dispersive forces other than covalent bonding [60,61,62,63,64]. Hence, according to the concept of supramolecular structures, HS can be considered as relatively small and heterogeneous molecules of various origin, which self-organize in supramolecular conformations. Humic superstructures of relatively small molecules are now imagined as complex aggregates stabilized only by weak forces such as dispersive hydrophobic interactions (van der Waals, π–π, and CH–π bonding) and H-bonds, the latter being progressively more important at low pH values. The representation of HS according to the model based on the spontaneous association of molecular components through weak bonds, provides several important implications: (1) accumulation of organic matter in soils is driven by hydrophobic interactions; (2) any HS supramolecular structure can undergo interchange processes with hydrophilic molecules generated through the degradation of biological tissues; (3) the hydrophobic bonds also lead to the consequent stabilization of the organo-mineral complexes and the general structure of soil [60].
Experimental evidences that HS have a hierarchical or “structure within a structure” architecture with at least two sequential levels of organization were provided by Chilom et al. [65,66,67]. In details, they developed a method in which a given HA0 sample was firstly subjected to Soxhlet extraction using a C6H6:CH3OH azeotrope for 72 h to collect two main fractions, respectively, a lipid-like L0 and a non-amphiphilic HA1, corresponding to about 67% of the total organic carbon (TOC). Then, the L0 fraction was separated into further two sub-fractions: HA2 accounting for almost one-third of TOC (possessing amphiphilic properties and soluble in an alkaline aqueous solution) and L1 (lipid-like and soluble in nonpolar solvents).
Differences in heat capacities between the pristine HA and the reconstituted physical mixtures (HA1 + L0 and HA1 + HA2 + L1) were compared. Thus, it was deduced a first level of organization correspondent to self-assembled nanoparticles formed by the amphiphilic HA2 and the lipid L1 fractions, which further interact with the components of HA1 yielding the overall HA0 composite (second level of organization). However, the interactions that trigger and stabilize the structure of these assembled molecular complexes are still unknown.
The diffusion coefficients of carbohydrates, carboxyl-rich alicyclic molecules and aliphatic compounds in DOM strongly depend on their concentrations indicating their gradual association [68]. Based on this observation, Drastik et al. [69] hypothesized that free hydrophilic molecules at lower concentration can play a role as hydrotropic compounds and stabilize and solubilize the aromatic aggregates in DOM while at higher concentration slowly aggregate forming physically stabilized gel-like structures.
Earlier results showing the different stability of HS aggregates at increasing temperatures [70], led to a new model in which HS are described as a hydrophobic inner core stabilized by outer layers of amphiphilic and polar molecules that impart a hydrogel-like structure [71]. Recent research developed by Wells et al. [72], emphasized the role played by “strong” H-bonds among polar domains, in the formation of stable hydrogels-like structure. In such hypothesis, HS aggregates with size > 1 μm have been proposed to consist of “metachemical” hydrogels < 1 mm embedded within water dispersible porous physical hydrogel scaffolds [73], which can form, dissipate, and reassemble along a sequence of alternating cycles of mechanical agitation and quiescence [74].
In the following section we will see how the methodologies and instrumental techniques employed in the study of the microstructure of HS both in solid and liquid state, are very useful to highlight morphological and dynamic aspects complementary to each other.
3. Selected Methods and Experimental Approaches for Analysis of HS Structure
3.1. Extraction and Fractionation of Humic Substances
The chemical properties of HS are basically ascertained after extraction and fractionation of DOM or SOM according to the different solubilities. The soil sample, initially dispersed in concentrated alkaline solution, is first separated from the insoluble material (called humin) and then acidified with the addition of concentrated hydrochloric acid. The obtained precipitate is called humic acid (HA), while the remaining soluble fraction is called fulvic acid (FA) (see Figure 1).
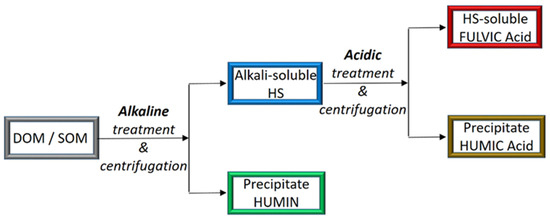
Figure 1.
Generalized flowchart for the traditional extraction and fractionation procedure of HS [75].
Further purification processes of the HA and FA fractions involve centrifugation operations and treatments with ion exchange resins to remove inorganic impurities and weakly associated organic compounds. HA, which precipitate out of solution below pH 2, have pKa ≈ 4.5 and apparent molecular weight ranging from 103 to greater than 106 Da. The chemical structure is characterized by randomly condensed aromatic rings and aliphatic residues containing different functional groups such as carboxylic acid, amine, carbonyl and alcohol [76]. On average, 30–50% of DOM in rivers is HS [51,77]. Recent studies criticize the traditional definition of HS and the related extraction protocol by using a strongly alkaline solution [29]. In fact, according to these objections, the treatment with alkali, in addition to representing an extraction method, could induce processes of chemical alteration of the HS. [78].
While other studies support the traditional approach [79], great attention has been devoted to the fraction of HS that can be easily released into water without recurring to the drastic use of alkaline extraction [80].
3.2. Solid State Investigations
The structure of solid-state HS is less detailed than that present in the liquid state. However, various powerful methodologies such as solid-state NMR, electron microscopy and scattering of electromagnetic radiation, are available for researchers. Most of those investigations were analyzed in the framework of fractal theory applied to HS in solid state.
3.2.1. Microscopy Techniques: Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), Atomic Force Microscopy AFM
Microscopy and imaging techniques have been extensively used to assess particle shape and aggregate structure of HS [81,82]. The electron micrographs are able to highlight the fractal structure of microscopic irregular objects [83] whose fractal dimension df (defined in the section Light/X-ray/Neutron Scattering Studies and Fractal Architecture of HS Aggregates), can be estimated by means of counting methods that make use of appropriate software for analyzing the digitized images. For example, a transition from collapsed structures with relatively smooth surfaces (at acidic pH) to open and more irregular structures with asymmetrical shapes (near neutral pH) was revealed from SEM microphotographs of soil HA particles, previously separated by aqueous suspensions at various pH values (3–7) after an equilibration time of 4 h [84,85]. TEM image processing analysis performed on HA aggregates at different pH and equilibration times, confirmed the existence of fractal structures described by a power law between the mass and the characteristic length of the aggregates [86]. In particular, HA clusters exhibited large and compact structures at pH 3, which transformed into open and elongated structures at pH 6 at short equilibration times. However, with an increase in the equilibration time, larger and more compact structures were observed, as a result of the occurrence of aggregation process followed by internal restructuring of HA aggregates.
Observing the structure of HS is challenging owing to their architectural complexity and dynamic nature. Additionally, these molecules can adopt multiple interchanging conformational states depending on the pH and metal complexation [57,87].
Atomic force microscopy (AFM) and scanning electron microscopy (SEM) have been employed for a long time to study the micro-morphologies of HA and FA [88,89,90]. Compared to electron microscopy, AFM offers more flexibility in sample preparation, three-dimensional imaging and, in many cases, better image resolution [91].
The advantage of AFM techniques is the ability to image microstructures not only on dried samples, but also in aqueous solutions, with a spatial resolution in the nanometer range [92]. AFM images can be obtained at ambient temperature and pressure, and the force can change during image acquisition. Thus, AFM is a valuable tool allowing to visualize small colloids, as well as colloid agglomeration, metals adsorption onto surfaces, or morphological changes affected by interaction with anions and pH [93].
However, imaging techniques require sophisticated sample preparation (e.g., mounting on slides or sample dewatering/drying), which is highly perturbing to the structure of the sample under study. Imaging also refers to the morphological analysis of the single particle which can be very laborious and can only visualize a sample that is not representative from a statistical point of view, even with the aid of automated image recognition systems. HA or FA were dispersed on mica as the better substrate not only because of its easily available atomically smooth surface but also its similarity in surface properties with the mineral materials in natural aquatic and terrestrial environments. Thus, AFM study results are very important for understanding the structure and function of HS in water and soil environments [94].
Indeed, HA, FA, and their respective metal complexes can form many different structures on mica surfaces as revealed by recent AFM studies [95]. For investigations performed on soft materials, the AFM non-contact mode (or AFM intercept mode) is preferred, in which an oscillating cantilever is used to maintain a constant distance between the tip of the AFM and the sample surface. Characterization of HS aggregates by AFM suggested that FA and HA could form aggregates and clusters at nanometer scale with globular shape according the pH of the aqueous solutions [96].
An image of HA particles (extracted from Leonardite) adsorbed on mica surface is shown in Figure 2A. AFM images in tapping mode of HA aqueous dispersion absorbed on mica, showed separated small globular particles with sub-micron size [97]. In the isolated particle cluster (Figure 2B) continuous HA layers, in ring format, were observed.
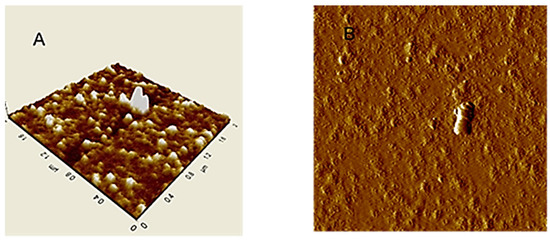
Figure 2.
AFM images in tapping mode of Leonardite dispersed in water and then adsorbed and dried on mica surface: (A) Typical rounded particles; (B) Isolated particle cluster.
These homogeneous ring structures (see Figure 3 and Figure 4), formed in saturated particle dispersed in water at low pH values, indicate evidences of supramolecular structures of HA in the natural environment, which in turn are strongly dependent on the dispersion method and pH. The hydrophilic surface of the HA aggregates interacts with the negatively charged mica surface, and promotes a large, swollen shape. If the structure is composed of isolated molecules, the dispersion procedure makes either slowly removing of water on the HA from mica surface and keep the initial ring structure, with small changes.
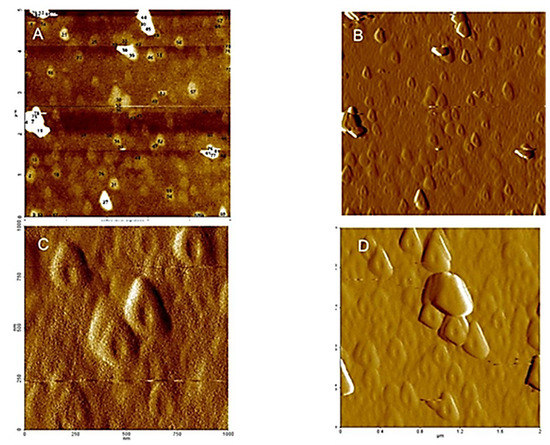
Figure 3.
AFM−NC (non-contact tapping mode): (A,C) microtopography amplitude and (B,D) 3D microtopography images of HA dispersed in pure water at pH 5, adsorbed and then dried on a mica surface.
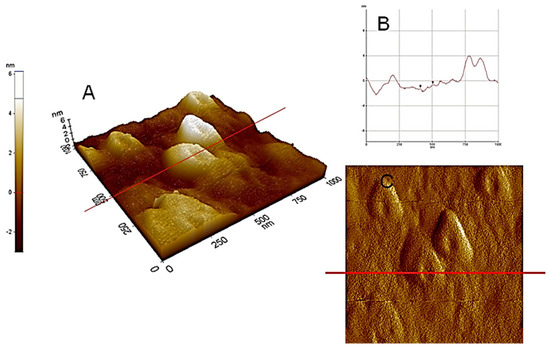
Figure 4.
AFM−C (contact mode): (A) microtopography with 3D images of HA dispersed in pure water at pH 5, adsorbed and dried on a mica surface with section analysis (B) and amplitude (C).
Figure 3 and Figure 4, illustrating high nanoscale resolution AFM-C images, show several spherical ring-like structures of HA bearing a shallow depression in the middle, with apparent colloidal diameters < 500 nm and vertical size of 3–4 nm. The image of a mica surface is very smooth and the adsorption of HA on the mica is quite clear. The size and shape of the pseudo-spherical colloids are in agreement with those observed by Colombo et al. [97] and also reported by other research groups for similar HS [98,99].
It should be noted that those ring-like structures were better observed at nanoscale with few drops of water suspension directly dried on the mica heated at 80–90 °C. The pseudo-spherical structure may be due to the rapid air-drying process occurring on the mica surface. It is generally accepted that the hydrophobic character of HA, due to the hydrophobic compounds (long alkyl-chain alkanes, alkenes, fatty acids, sterols, terpenoids, and phenyl-alkyl residues of lignin degradation), allows their self-assembly into supramolecular structures separated from the water milieu [100].
From the sectional analysis results, shown in Figure 4B, it is observed that the average dimensions of the ring-like particles ranged from about 350 to 550 nm in lateral dimension and 3–4 nm in height. In Figure 4B, the images illustrate a cut along the solid line shown in the vertical profile. Moreover, particle sizes and shapes appear quite uniform inside the field of view. These ring structures, formed at subacid pH values, again indicate evidences of supramolecular arrangements [101]. Consequently, it can be deduced that the analysis of the AFM images supports the HA aggregation model, based on the cooperative association of small molecules. It is important to highlight that aggregation process of HA depends strictly on the solvent used for the dispersions and the dehydration conditions. One example is showed in Figure 5 where HA from Leonardite was observed with SEM after a dispersion in alcohol and adsorption/dehydration on mica surface, compared with the same sample of HA dispersed in alcohol observed with TEM. In both cases, the alcohol dehydration favours the collapse of HA molecules into fibrous structures. Comparing AFM images with SEM and TEM, the solution conditions of Figure 2, Figure 3 and Figure 4 for AFM are very similar and the results are comparable. The hydrophobic effect has been invoked to justify the formation of similar ring-shaped structures observed with AFM at low salt concentration and low pH [102,103].
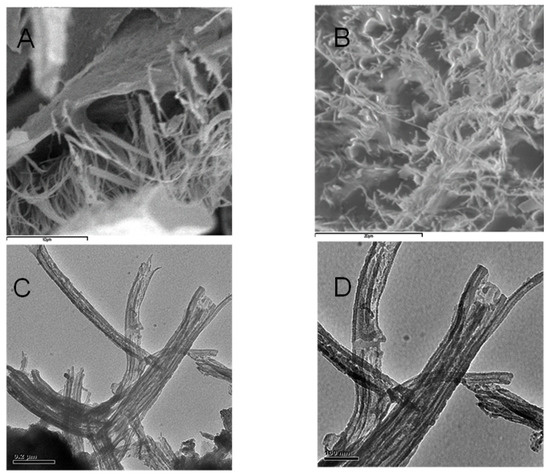
Figure 5.
(A,B) SEM microtopographies of HA dispersed in alcohol and then adsorbed and dried on mica surface. (C,D) TEM microtopographies of HA dispersed in alcohol in forward film.
By contrast, it is well known that the sample for SEM and TEM analysis must be treated under high vacuum conditions and on dehydrated conditions. Thereby, the first step with alcohol treatment followed by spraying carbon in the vacuum tank, changes the morphology of HA. However, it is worth noting that the sizes of the fibrous aggregates in Figure 5 are similar in both SEM and TEM images. This may be attributed to the strong alcohol dehydration conditions. The influence of Na+ on the macromolecular structure of HA at pH 4.0–6.0 favours pseudo-spherical colloids, while the alcohol dehydration stabilizes elongated fibrous structures. Figure 3 and Figure 4 show that dimensional parameters such as particle diameter, length and height of the irregular aggregates are much larger than those observed with AFM. The dehydration treatment with alcohol could be responsible for this effect, which may favour the formation of bridges between cation ions and carboxyl groups or other polar groups. This behavior confirms that the dispersion in water used for AFM observation is very useful for obtaining an in-depth analysis of the HS structure analogous to that occurring in natural aqueous environments. The amphiphilic properties of different chemical groups present within the structure of HA such as, e.g., Leonardite, can give rise to the formation of micellar-like aggregates in solution and also to assemblies of various shapes onto mineral surfaces.
3.2.2. Nuclear Magnetic Resonance Techniques
Nuclear magnetic resonance (NMR) is a locution referring to an ensemble of different techniques all based on the same principles and used to unveil chemical-physical characteristics in solid, liquid, and semi-solid phases [104,105,106,107,108]. The general theory about NMR technique is reported elsewhere [109]. Here we intend to give only a brief overview about the NMR principles. In details, nuclei with an odd number of protons and/or neutrons have a nuclear spin associated with a magnetic moment that orients when immersed in an external magnetic field (B0). At equilibrium, the orientation of the nuclear magnetic moments is distributed over different energy levels, characterized by having a slightly accessing population on the lowest energy level. This distribution of population can be perturbed by irradiating the sample with pulses of radiofrequency waves with an oscillating magnetic field (B1). After this excitation, the distribution of the orientations of the nuclear magnetic moments returns to the equilibrium configuration through two relaxation mechanisms. The first, defined as longitudinal relaxation, is linked to the recovery of the magnetization vector towards its equilibrium value, oriented along the direction of B0 with consequent loss of energy. The second, called transversal relaxation, is responsible for the loss of phase coherence and the decay of the magnetization vector in the plane perpendicular to B0. Both mechanisms act simultaneously, although they are characterized by different relaxation times defined as longitudinal or spin-lattice relaxation time, (T1) and transverse or spin-spin relaxation time (T2).
The signals acquired by NMR technique can be analyzed in many different ways. As an example, in the NMR spectroscopy, the application of the Fourier transform provides plots (also referred to as spectra) where resonance signals are reported versus frequency. In order to make signal position independent of the strength of the applied magnetic field, the resonant frequency is expressed as chemical shift measured in part-per-millions (ppm). According to the number of the frequency axes, it is possible to differentiate among mono- (1D), bi- (2D) and three-dimensional (3D) NMR spectroscopy [110,111]. While signal intensity is directly related to the number of nuclei which are excited by the applied pulse, signal position is dependent on the nuclear chemical environment. This is why NMR spectroscopy can be used both to investigate the structure and 3D conformation of complex organic molecules, and to determine the number of nuclei affected by the same chemical environment.
However, the quantitative analysis performed on complex mixtures can be limited by the type of sample phase, the pulse sequence used for the NMR investigation and by hardware constraints [104,112,113]. A number of parameters affect the time scale of the relaxation processes such as, e.g., the molecular size, temperature, presence of paramagnetic metals and other factors linked to physical state of the system under study. In particular, when experiments are designed to measure relaxation times T1 or T2, the time decays of the NMR signal can be analyzed by using the inverse Laplace transform which allows to obtain the distribution of relaxation times [114,115,116,117,118].
This method, called time-domain (TD) NMR, is used to acquire information on molecular dynamics for both simple systems and complex mixtures [119]. In the last few decades, the new technique called Fast Field Cycling (FFC) NMR relaxometry is appearing on the scene of different scientific fields [119]. It is based on the rapid changes of the proton Larmor frequencies (νL) by modulating the current intensity passing through a coil printed on a silver support [120]. Due to the low sensitivity, FFC NMR prevents any spectroscopic investigation, while it is applied to monitor molecular dynamics at frequency intervals lower than those span by the traditional NMR spectroscopy [119].
The Limits of the NMR Techniques in Humic Chemistry
All the aforementioned NMR techniques are widely applied to unveil the chemical-physical characteristics of HS. However, some issues must be accounted for when dealing with NMR data. From the one hand, solid state NMR spectroscopy suffers for the limits due to the cross-polarization (CP). CP is based on the energy transfer from hydrogens to the closest carbon nuclei, thereby “permitting” the observation of the less abundant 13C nuclei via the population of the most abundant 1H systems. However, when the carbon backbone is made mainly by aromatic systems or quaternary, and tertiary alkyl C, lack of hydrogen nuclei is obtained, thereby enabling low CP efficiency. As a consequence, NMR sensitivity lowers, and signal disappearance occurs. In addition, if humic samples are not well packed in the zirconia containers used to perform solid state NMR investigations, a vortex effect occurs thus providing NMR artifacts [104].
On the other hand, due to the colloidal nature of HS, liquid state NMR spectroscopy suffers for the lack of information contained in the solid parts of the colloidal systems. In fact, liquid state NMR spectroscopy performed on HS permits analyses only of the surface groups in colloids [121]. The best solution to try to overcome the problems of solid and liquid state NMR spectroscopy is the application of the high-resolution magic angle spinning (HRMAS) technique. By means of HRMAS, the colloidal dispersions made by HS are placed in the zirconia rotors used in solid state NMR and oriented at the magic angle (54.7°). The efficiency of the solid-state NMR spectroscopy is coupled with that of the liquid state NMR one, thus permitting the analysis of both surface and inner solid part of the colloidal humic particles. Sensitivity and lack of mathematical models for data elaboration are the main problems affecting application of FFC NMR relaxometry on HS [119].
NMR Results of Humic Substances
Over the years, modern NMR technology, such as the cryo-probe and hyphenated-NMR, along with the latest generation of NMR experiments, has been found very useful to explain the primary makeup and associations of HS. DOSY results, an NMR technique that directly correlates the diffusion coefficient of different molecular species to the proton chemical shift in a 2-D plot, allowed Simpson et al. [122] to identify a complex mixture of relatively simple compounds, such as fatty acids/esters, sugars, amino acids, and lignin-derived aromatic systems as the major components present in alkaline extractable soil HS. Those findings corroborated the concept of molecular aggregation of relatively low molecular weight components (<2000 Da), where metal ions play a crucial role in aggregate formation and stability [123].
Results similar to those revealed by Simpson et al. [122] have been obtained by Mao et al. [124] by applying 2-D solid state NMR pulse sequences. In particular, those authors could distinguish different humic hydrophobic/hydrophilic domains via the evaluation of 2-D maps where the x-axis reports the 1H-NMR spectrum, while the y-axis reports the 13C NMR spectrum. The interpretation of CPMAS-NMR spectra made by Conte et al. [125] allowed to speculate that humic aggregates can be considered as “highly condensed or strongly associated rigid aromatic systems interacting with carboxyl groups, involved in hydrogen-bond formation, and mobile C–O/C–N systems that, in turn, should be mainly displaced, as well as the mobile alkyl moieties, on the surface of the solid state HAs”.
Finally, recent applications of FFC 1H-NMR relaxometry revealed that a “hydrogel-like” structure is recognizable in HS as concentration increases. In fact, the proposed structure consists of a hydrophobic core made up of aromatic substances all stabilized together by van der Waals forces and pi-pi interactions, which in turn are coated by a hydrogel made up of both polar and amphiphilic molecules [71].
Notwithstanding the huge amount of solid and liquid state NMR data collected on HS (see, as an example, [126]), it has never been possible to go beyond a simple qualitative description of the conformational aspect of HS. In other words, no detailed information on the 3-D structure of HS have been ever obtained such as in the case of proteins [127] or polysaccharides [128]. This limit is due to the intrinsic complexity of HS, which prevents any evaluation of the way how the different humic subunits are faced to each other. In addition, it must be noticed that humic conformation after extraction from soil is conceivably different [129] from that present in nature due to the contemporary presence of clay minerals, soil aggregates and water system.
3.3. Liquid State Investigations
The progress in research of supramolecular arrangement of HS in solution has always been connected with progress in analytical instrumentation [4]. Current knowledge is mostly based on application of spectroscopy techniques and regards mainly to the unravelling of the DOM primary structure. On the contrary, the methods for determination of physical structure are still used less frequently. Nevertheless, increasing number of studies stress out the importance of supramolecular structures for their functioning [73].
The models based on the “average” model structure or superposition of individual molecules are gradually extended by models taking into account the interaction between individual molecules and assemblies, i.e., as a system of dynamically responding to the disruption of their equilibrium state.
3.3.1. Scattering Techniques
Studies of DOM and humic colloid structures in both natural aquatic and artificial aqueous environments have, until recently, largely relied upon scattering techniques [130]. Scattering techniques measure dimensional quantities on submicrometer length scales and on short time scales, for samples large enough to be considered statistically significant. For light scattering, an incident radiation having wavelengths much greater than the average size of the colloidal particles is selected. This implies that the light scattered by a particle is in phase. Consequently, the scattered intensities are greater and more efficient than X-ray scattering. Ultrasounds can be used to investigate even turbid or highly concentrated systems but use longer wavelengths than light. X-ray and light scattering methods can be applied to measure a similar range of colloidal properties such as the radius of gyration (Rg), the cross-sectional radius of gyration, colloid particle volume, the cross-sectional area of the particle, molecular weight per unit length, and particle shape.
Dynamic Light Scattering
Dynamic light scattering (DLS) also known as quasi-elastic or photon correlation spectroscopy (PCS), can measure particle sizes down to a few nanometers, but only by making assumptions about the shape and diffusive behaviour of the object being studied. It is an inelastic scattering technique, not a type of diffraction measurement. It uses a laser beam to measure the fluctuation in the intensity of the scattered light originating from Brownian motion, defined by the translational diffusion coefficient, which in turn is converted into the hydrodynamic particle diameter Dh (or radius Rh) using the Stokes-Einstein relationship [131]. Rh corresponds to the apparent particle size, which includes also the hydration shell that surrounds it. In particular, the polydispersity index (PdI) and the z-average mean diameter of particles are determined from intensity-based DLS spectra, and represent a single average even if spectra are multimodal. DLS is a technique that measures the dynamics of fluctuations of scattered light to obtain the mobility of colloidal particles subject to Brownian motion [132].
Details on DLS data treatment such as, e.g., converting autocorrelation functions to particle size distributions, can be found elsewhere [133,134]. Experimental data provided through DLS-based studies or in combination with other techniques, consolidated the hypothesis that HS consist of low molecular weight compounds able to aggregate step by step to give supramolecular structures.
DLS measurements of a peat HA in water and also in NaCl solutions of varying concentrations yielded low values for Dh = (2–9)·10−11 m2·s−1, leading to the conclusion that such particles can be interpreted as consisting of large macromolecules or aggregates of smaller molecular species [135]. The effect of pH and ionic strength on the aggregation of both peat and commercial HA was studied by Pinheiro et al. [136] through DLS and voltammetry, who reported large aggregates in the range 30–185 nm. Ren et al. [137] studied the fractal dynamics of HA colloids and data were interpreted according to the dynamic scaling theory developed by Martin and co-workers [138], that described power law-polydisperse colloids. For example, the assembly kinetics of DOM polymers in 0.22 μm-filtered seawater was monitored by measuring the particle size as a function of time using DLS and flow cytometry [36].
It is worth noting that both of these methods are complementary: while flow cytometry provides accurate measurements of large particles (>1 μm), DLS yields reliable measurements of particle size in the sub-micrometre range. DLS showed that fresh filtrate contained a polydisperse set of polymers with sizes ranging from 2 to 200 nm. About 30 min after filtration, DLS revealed the presence of a polydisperse collection of assembled polymeric aggregates with sizes in the range 0.2–1 μm. A nonlinear growth process was observed and the increase in particle size followed a sigmoidal trend recalling second-order kinetics. Both methods showed that microgels continued growing, reaching an average equilibrium size of about 5 μm in 50–83 h.
The aggregation process of FA and HA certified by the International Humic Substances Society (IHSS) [139] as a function of solution properties such as temperature, pH and metal ions of different charge, was explored through DLS technique by Palmer et al. [140]. The authors reported average particle sizes of 210–280 nm and showed that the evolution of particle sizes under different solution conditions progressed from unfolded anionic polymers at high pH, low cation concentrations, and low temperature, through micelle-like structures and finally to colloidal precipitates as these conditions were changed.
The observation of a limited ionic strength effect on the Rh derived from diffusion coefficients measured by Fluorescence Correlation Spectroscopy (FCS) was reported by Hosse et al. [141], who supported the hypothesis of HS as rigid spheres. The observed slight compression of the tested HS standards at high ionic strength was found in agreement with the viscosity measurements made by Avena et al. [142] on several HA.
Although viscometry was not sensitive to aggregation, the determinations of 35–65% molecular volume reduction for an increasing ionic strength (1–100 mM) followed the same trend of slightly decreasing Rh obtained from FCS. DLS technique was also used to study the effect of pH on the aggregate size of a gray HA extracted from a commercial HS derived from brown coal [143]. The decrease of zeta potential, determined from laser Doppler electrophoresis (LDE) technique, from –27 mV (pH 1.5) to –63 mV (pH 8.8) was accompanied by a concomitant drop in particle size from 310 to 70 nm within the same pH interval. The authors justified that behavior on the basis of dissociation of acidic groups, which accordingly could lead to depletion of protons able to form H-bonds and hindering HA aggregation. Baalousha et al. [144] determined the Rh of Suwannee River HA (SRHA) in NaCl and CaCl2 solutions at different concentrations in the range 10−3–0.5 M and pH 4.5–9.3. The small size of SRHA (<10 nm) measured at low electrolyte concentrations was attributed to the presence of small base units, which in turn were able to interconnect and produce supramolecular structures at higher salt concentrations.
DLS measurements, in combination with ultrafiltration (UF) and SEC techniques, were performed on alkaline aqueous dispersions of Aldrich humic acid (AHA) and HS derived from peat and compost [62]. The combined DLS-UF results indicated the presence of the following three main molecular fractions: Rh = 0.5–2 nm (corresponding to the UF molecular fraction lower than 10 kDa), Rh =2–5 nm (corresponding to the 10–100 kDa UF fraction) and Rh =11–20 nm (corresponding to the 100 kDa and 0.45 μm UF fraction). Those three main molecular fractions were also observed in the SEC study performed by the same authors. Using time–resolved DLS measurements of HS in seawater conducted over a time–scale of days, within the first 2 h following filtration Verdugo et al. [145] identified free dissolved organic carbon (DOC) polymers with average sizes in the range 5–50 nm. However, 100–200 nm sized nanogels appeared after 5–10 h, which in turn began to restructure to form larger particles until reaching final dimensions of 3–6 μm (microgels) at equilibrium after 60 h. Overall, the self–assembly process followed a nonlinear sigmoidal relationship, leading to the conclusion that low–energy physical interactions could be responsible for DOC self–assembly [146].
The aggregation process of HS isolated from Rendzic Leptosol (RLHA) and IHSS HA standard from Elliot soil (EHA) was examined in the 2–11 pH range at constant ionic strength through DLS to monitor particle sizes and LDE technique for the determination of zeta potential [147]. At pH values > 6, particle sizes smaller than 10 nm were interpreted as small individual HA molecules or basic units. With the decrease in pH, the evidence of a sharp increase in diameters in the 6–4 pH range was attributed to the occurrence of the aggregation process of the HA polymers, indicating carboxylic hydrogen as responsible for the intermolecular H-bond and, consequently of the formation of aggregates. Aggregates formed at pH 2 were found of different sizes (from 600 nm to 1.5 μm) and dependent on the HA origin and concentration in the aqueous dispersions. The zeta potential values were more negative with increasing pH in the range 2–6 as a result of the functional-group dissociation. Interestingly, the authors documented the existence of a zeta-potential minimum, reached in the 5–7 pH range, which was not yet reported in literature. The explanation of the increase in the surface charge of the particles observed at higher pH values has been articulated on the basis of the existence of two related mechanisms: dissociation of acidic functional groups and consequent dissociation of HA aggregates, caused by the breaking of H-bonds, with opposite effects on the zeta potential values. In the first part of the zeta-potential vs. pH curve (from pH 2 to pH where the zeta potential minimum was reached), dissociation of acidic groups could be considered as the prevailing process leading to a negative-charge increase. At higher pH values (from the zeta potential minimum upward), disaggregation process likely predominated over dissociation, resulting in the z-average particle size decrease at higher pH values. As a consequence of smaller particles formed, negative charge per HA particle decreased causing zeta potential to become less negative.
Experimental evidences of dynamic processes related to the HS aggregation kinetics were also reported by Angelico et al. [148], although for shorter time scales, in aqueous dispersions of HA extracted from Leonardite. Indeed, time-resolved DLS measurements revealed a particle size growth scaling linearly with time for HA aqueous suspension at pH 2 and NaCl 0.015 M. The zeta-average Dh of HA aggregates increased monotonically from ~750 nm up to ~1.3 μm in 70 min for a sample left mechanically unperturbed in the measuring cell for the duration of the DLS experiment. However, the selected time window did not allow to observe a threshold limit value for the growth of particle sizes. After an hour of monitoring, a bimodal particle size distribution (PSD) on a numerical basis was found showing maxima centered, respectively, at 300 nm and 1.5 μm. On the contrary, the time-dependent increase in the aggregates’ size was not detected at pH > 2, where instead a monomodal PSD on a numerical basis centered at about 80 nm was obtained (see Figure 5 in ref. [148]).
Both DLS measurements and zeta potential determined from LDE technique, have been proved valuable for increasing comprehension of the aggregation behavior of HS in simulated low electrolytic conductivity aqueous solutions (16–192 μS/cm) [74]. The humic materials selected by the authors were standard HS such as SRHA, SRFA and AHA sodium salt, and two types of actual environmental aqueous samples. The HS aggregate sizes measured within the range 0.01–1 μm were postulated to be composed of subunits as precursor molecules for the formation of the 5-μm-sized particles, which in turn were observed to occur on a time-scale of minutes not hours/days as observed, e.g., by Verdugo et al. [145,146], in both simulated and actual environmental water samples. Interestingly, the authors explored the influence of degree of turbulence on the extent of disaggregation and self-assembly process. Indeed, it was found that the self-assembly of the 5-μm-sized particle formed, dissipated, and spontaneously re-formed over turbulent/quiescent cycles, indicating reversible self-assembly in all HS tested samples.
In a systematic determination of both Dh and zeta potential of HA sodium salt performed in aqueous dispersions in the concentration range of 3.2·10−4–20 g/dm3 at pH 6.0 and in absence of background electrolytes, it was found that the particle size varied from 30 nm to 5.6 μm, with PSD depending on the HA concentration [149]. The results were interpreted in terms of the existence of HA sodium salt in the associated state.
The dependence on concentration, pH and time of the physicochemical properties of aqueous dispersions of HA, such as particle sizes, morphology, zeta potential, electrical conductance and surface tension, have been interpreted by considering aqueous solution of HS as self-organizing non-equilibrium systems [150].
DLS and zeta potential measurements have been extensively performed on aqueous solutions of HA and FA by Klučáková et al. [151,152,153,154,155]. Two main processes were found responsible for the dynamics of HA and FA in solutions: the dissociation of acidic functional groups and the dissociation of humic particles into smaller molecular associations and/or single molecules. In particular, fulvic particles in water were bigger than in alkaline solution as a result of suppressed dissociation. The decrease in particle sizes with increasing concentration was interpreted in terms of enhanced strength of intermolecular interactions favoring HA and FA particles to adopt a more collapsed or coiled configuration. From the measurement of zeta potential, it was found that the colloidal stability was higher for HA than FA, probably due to the lower dissociation degree of the functional groups in FA compared to HA. In another study, DLS technique was used on soil extracted HA to evaluate the effect of H+ on the aggregation kinetics in Na+, K+, Mg2+, and Ca2+ nitrate electrolyte solutions with different ionic concentrations [156,157].
The results showed that the specific ion effects in HA aggregation process could be related to the strong particle surface electric field and the polarization of adsorbed cations. In addition, the origin of the specific ion effect for H+ was its chemical adsorption onto surfaces, while the effects for alkali cations were interpreted on the basis of non-classic polarization concept [158].
Light/X-ray/Neutron Scattering Studies and Fractal Architecture of HS Aggregates
The concept of fractals, which are self-similar scale invariant geometric figures, can be applied to describe both the geometric properties of complex systems such as, e.g., the HS, and the aggregation mechanisms to which various natural compounds are subject. [74,75,76,159,160]. As described, for example, in the model developed by Chen et al. [161], the fractal properties of an aggregate or a particle are described through: (i) its fractal dimension df, which gives information of a morphological type (i.e., whether the material is compact or loose), (ii) the particle radius (Rp) and (iii) the particle-particle correlation length (ξ).
Structural investigations of colloidal particles of submicron dimensions, dispersed in aqueous solutions, can be carried out with small angle scattering methods using light, neutrons or X-rays. Within the diffraction limit, accessible length scales are much less than the wavelength of the radiation being used. Thus, light scattering can only directly detect the larger end of the colloid size range (>400 nm) and is also limited to systems that are not too turbid whereas shorter length scales can only be probed by radiation of shorter wavelength than light, for example, by X-rays and neutrons.
In many respects, small-angle X-ray (SAXS) and neutron (SANS) scattering can provide complimentary information over similar length-scales, a consequence of the difference in the way that each type of radiation interacts with matter [162]. Though X-rays are often ideal for studying samples with high-Z elements and therefore might seem an obvious choice for investigating mineral-based dispersions, the very presence of those high-Z elements masks the scattering from the low-Z elements in the organic component of natural aquatic colloids. Nevertheless, SAXS technique has been found to be useful for determining the fractal dimension, average molecular weight and particle size in aqueous dispersions of HS [163,164].
SANS, on the other hand, does not suffer from any of these drawbacks, is a very penetrating radiation (and so is well suited to the study of optically opaque samples) and, importantly, can capitalize on the large differences in scattering behavior between hydrogen and deuterium. By dispersing colloidal material in aqueous matrices composed of varying ratios of H2O and D2O, it is possible to probe the different components of the system without substantively altering the physical chemistry of the system. This is of enormous potential importance for the study of natural aquatic colloids because of their complex mixed composition.
SANS technique allows for the substitution of the water medium with its deuterated analogue (D2O) to increase the contrast between the organic compound of interest and the solvent, thanks to the substantial difference between the scattering lengths of hydrogen (H) and deuteron (D). The normalized SANS data are expressed as scattering cross section per unit volume of sample where is the scattering vector, λ is the wavelength of the incident radiation and θ is the scattering angle. The asymptotic expression of for is described by the Guinier’s Gaussian law:
valid in the approximation of non-interacting particles, where is the extrapolated intensity at origin and Rg is the particle radius of gyration or the mass-weighted root-mean-square deviation from the center of mass. Alternatively, assuming the shape of the particles, of polydisperse anisotropic particles can be analyzed by modelling the experimental curves in the whole range. The latter approach implies the use of various fitting models in the general expression , where the scattering law describes how the scattering is modulated by size, shape and local order, k is a scale factor and b the incoherent scattered intensity. There are many expressions for available in the literature that refer to a wide variety of 1D, 2D and 3D forms, different interaction potentials and different density correlation functions. The skill in interpreting small-angle scattering data is in finding a physically realistic that fits the data better than any other. The difficulty with natural colloidal systems is that the colloidal objects responsible for the scattering are unlikely to be the simple regular geometric shapes (spheres, rods, ellipsoids of revolution, circular discs, etc…) for which there are a plethora of scattering functions. They often also exhibit size polydispersity. Polydispersity is actually relatively straightforward to handle in the data modelling, but broadening the size distribution has an effect comparable to degrading the data resolution. In most cases therefore, derived shape information is ‘representative’, rather than definitive, and size information is averaged, albeit over a large statistical sample size.
The application of fractal geometry allows for the determination of the mass distribution of the material within (mass fractals) or on the surface of the particles (surface fractals) [165]. Mass fractals are defined as those molecular agglomerates having a reticular structure, likewise trees where the branches are dendritic networks made up of smaller networks. In contrast, surface fractals have dense internal structure in which the fractal aspect is evident only at the particle surface. Depending on the type of investigation technique employed, the network would assume a collapsed spherical shape while on closer inspection it could result that it is irregular and not space filling. Between mass and surface fractals, partially penetrable hybrid structures may be found, likewise a porous sponge (see Figure 6). For an aggregate or cluster of N particles with radius (Rp), the fractal dimension df is defined as the exponent of the power law [166] , which roughly reflects the shape of the aggregate itself (Figure 6).

Figure 6.
Pictorial representation of mass and surface fractal objects. A mass fractal is depicted by a dendritic structure while a surface fractal is an object similar to a filled tennis ball. Partially penetrable structures (hybrid fractals) can be modelled as a porous sponge. On the right side it is graphically represented how the fractal dimension df increases from approximately 1 for rod-like aggregates to 3 for ideally spherical structures, passing through intermediate shapes (e.g., df = 2).
For large , a power-law dependence of the scattering intensity , which in double logarithmic scale looks like a linear dependence within some range in called the fractal region, is a main indicator of the fractal structure. The power law exponent describes how the mass or surface in the system scales with the linear dimensions. In particular, if n < 3 the measured sample is a mass fractal, which often represents open aggregates of smaller subunits with a dendritic morphology, and n coincides with its respective fractal dimension df. However, if 3 < n < 4 then the particles are surface fractals, in which only the surface of the material exhibits fractal properties, and df satisfies n = 2D – df, where D = 3 for three-dimensional space. Both Rg and df values are used to calculate surface-to-volume ratios to identify a general shape for the particles under investigation. More details on the fractal properties obtained from small angle scattering techniques can be found, e.g., in refs. [162,167].
The fractal nature of HS has been extensively reported from various studies based on the application of small angle scattering techniques and turbidimetry (see, e.g., [168]). The major weakness of the fractal approach is in providing results and interpretations that can be valid only in an average or statistical sense for finite natural systems such as HS.
HS particles in solution that have been characterized to date have demonstrated mass fractal behaviour in solution [84,85] and surface fractal in the solid state [169].
The fractal properties of aquatic HA and FA were recognized by analyzing the power law exponents of SAXS intensities acquired on the humus samples, which were previously extracted, converted to their acid form and lyophilized [169]. The finding of the exponents falling within the range 3–4 identified the humic materials in solid state as surface fractals with df = 2.2–2.8. However, when both the aquatic HS were measured in aqueous solution they behaved as mass fractals with dimensions in the range df = 1.6–2.5.
Using scattering methods, the fractal dimensions of both fractal types have been generally found within the range 2 ≤ df ≤ 3. Particles with mass fractal dimensions that fall within this range can be visualized as open 3D aggregates, which get denser as df → 3.
Former SANS studies reported by Österberg et al. [170] indicated that SRHA could form fractal aggregates in D2O at pH 5 and solute concentration of 3.6 mg/mL, characterized by df = 2.3 ± 0.1. In absence of a clear Guinier region, the primary particles were difficult to determine, and instead, a lower limit of Rg was obtained. In subsequent investigations, the same authors reported an increase in the fractal dimensions of aggregates (from df = 1.85 to 2.35) for two types of HA suspended in D2O (concentration = 3 mg/mL, pH = 5.0 and 0.1 M NaCl) after 48 h of equilibration at 4 °C and 11 °C and after 60 h at 22 °C, demonstrating that the increase in temperature somehow allowed a compaction of the humic material [171,172]. One possible explanation is that the increase in kinetic energy was sufficient to allow functional group interactions to overcome some activation energy barrier.
In their investigation, Senesi et al. [84] measured the wavelength dependence of the turbidity of humic particles in dilute suspensions in absence of background electrolytes, to obtain the respective fractal dimensions at various pH values and for different equilibration times at 25 °C. From fractal analysis, HA particles in aqueous suspension were observed to evolve as the pH and/or the equilibration time increased, from an almost compact, dense structure with either smooth surfaces (non-fractal particles) or corrugated surfaces (surface fractals), to less compact and irregular structures with increasingly rough surfaces (mass fractal). In a subsequent turbidimetric investigation carried out on aqueous suspensions of three HA standards, a general reduction of df was observed upon increasing HA concentration, pH, or ionic strength due to added NaCI [85]. However, in the presence of CaCl2, the HA particles existed as condensed structures with rippled and rough surfaces.
Wagoner et al. [173] performed high-performance size exclusion chromatography (HPSEC) to preliminary fractionate HS from Suwannee River natural organic matter and then used the multi-angle laser light scattering (MALLS) technique to measure the time-averaged angular dependence of the scattering of light by humic material in two phosphate buffer solutions. In 2.5 mM phosphate buffer, the weighted average molecular mass (Mw) and z-average particle radius (Rz) were, respectively, Mw = 25.7 ± 7.5 kDa and Rz = 68.1 ± 20.4 nm, the latter value being almost halved in the higher ionic strength solutions as a consequence of an increased macromolecular coiling. Those data supported the random coil hypothesis for macromolecular NOM species in aqueous solutions. A combination of DLS and SAXS techniques performed on dilute solutions of HA extracted from peat, yielded particle sizes Rh of the order of 50–100 nm at pH ~ 9 and low ionic strength, characterized by a loose, sponge-like inner structure with mass fractal dimension df = 2.1–2.2 [174].
Manning et al. [175] analyzed the non-perturbed AHA structure in aqueous solutions by using the noninvasive MALLS technique, giving an accurate in situ value for size and molecular weight of aggregates. All measurements were made immediately after the solutions were prepared to avoid long-term aggregation effects. The authors stated that from SEC fractionation experiments, three peaks were obtained in the 1–30 kDa range attributable to as many building blocks of AHA, although they could not be representative of the aggregation found in naturally occurring samples. By measuring the intensity of scattered light at various set angles, the values of Mw = 116.4 MDa and Rg = 436 nm were determined. For the authors, those values represented an aggregation of numerous smaller molecular fragments joined together by weak forces such as H-bonds, ion-dipole interactions, and the attraction and aggregation of non-polar (i.e., aromatic and aliphatic) components of the AHA structure.
Myneni et al. [176], by carrying out in-situ X-ray imaging experiments, found globular (200–450 nm) and ring-like (0.3–1.2 μm) microstructures of SRFA in dilute acidic and high ionic strength solutions. As the concentration of SRFA in solution increased, the inclusion of the globular and ring-like aggregates into large sheet-like microstructures (2–8 μm) was also observed. From the data treatment of SAXS and SANS curves performed on several types of HS in D2O, Pranzas et al. [177] reported that HS solutions could be described as dynamic systems with concentration-dependent and reversible aggregation processes. In particular, spherical particles with maximal diameters of about 3.5 nm, single “huge” agglomerates with sizes up to 2 μm and (for some HS) regular chainlike structures at higher HS concentrations (up to 10 g/L) were found. In the presence of copper ions large disordered agglomerates and network structures were formed, depending on the Cu2+ concentration. Moreover, below a threshold Cu2+ concentration large disordered HS-Cu2+ agglomerates were formed proportionally to the Cu2+ addition.
Evidences of fractal structures of Suwannee River fulvic acid (SRFA) in D2O characterized by a lower limit for their radii of gyration (Rg > 242 nm) were reported by Diallo et al. [178] at different values of solution pH, concentration and background electrolyte (NaCl). The authors used the fractal model developed by Chen et al. [161] to fit the SANS curves. However, owing to the absence of both well-defined Guiner (low ) and Porod (high ) regions in the experimental SANS curves, the authors were unable to extract meaningful values of df, particle-particle correlation length ξ and particle radius Rp for SRFA aggregates.
3.3.2. Ultrasonic Velocimetry
Over past years, it became clear that the research of DOM is closely connected with its interaction with water [2] but the studies on solvation of DOM [69] and understanding of interaction of water with DOM and its structural stabilization role [179] are rare. In this respect the combination of old approaches and introduction of new methodologies is necessary to understand all the aspects of DOM environmental relevance [4,14].
One of the methods that can analyze the properties of water in various materials is ultrasonic velocimetry. The sound wave is a wave of oscillating pressure and associated longitudinal deformation. When low-intensity ultrasonic wave propagates through the sample containing dispersed particles, it is scattered, diffracted and refracted. Change in ultrasonic velocity and wave attenuation are related to system composition, physical state, microstructure and molecular relaxation phenomena [180].
The amplitudes of deformations in the ultrasonic waves employed in analytical ultrasound are extremely small, making ultrasonic analysis a non-destructive technique. The resolution of modern devices, allowing precise determination of ultrasonic velocity, is down to ±0.2 mm/s [181]. The speed of sound in liquids can be expressed by the Laplace equation
where stands for sound velocity, for density and for adiabatic compressibility. Virtually, all changes in physical and chemical state of solutions, suspensions or emulsions are associated with changes in density and compressibility, therefore, measurement of speed velocity in high resolution allows their monitoring. The adiabatic compressibility is the fractional decrease of volume per unit increase in pressure, when no heat flows in or out,
where and are the changes in the volume V and density at fixed entropy S. Compressibility is a measurable parameter and it is sensitive to solute-solvent interactions, i.e., it can be utilized for characterization of hydration of a solute or for interaction of two solutes. In the study of hydration, apparent molar or specific compressibility of the solute is used. It is influenced by (i) interatomic interactions within the solute itself (intrinsic compressibility of a solute molecule), (ii) solute-solute interaction if any occur under experimental conditions and (iii) solute-solvent interactions (hydration in the case of aqueous solutions) [182]. The effect “(ii)” diminishes with decreasing concentration and at infinite dilution become equal to partial compressibility of the solute. In such case the partial compressibility of solute reflects only contribution of intrinsic compressibility and the hydration of solute [183]. Various functionalities and moieties differ in their hydration and thus their contribution to partial compressibility of the solvent/solute system. Since they are temperature dependent, their role differs with respect to the system temperature [184].
Differentiating Equation (2) with respect to molar concentration of solute C, and performing simple arithmetical rearrangements, for infinitely diluted solutions Equation (4) can be derived:
where is the coefficient of adiabatic compressibility of the solvent, M is molecular weight of the solute, is the partial molar volume of a solute; is the relative molar sound velocity increment of a solute and and are the sound velocities in the solution and solvent, respectively. The partial volume of a solute can be calculated based on differential solution vs. solvent density measurement [185].
Therefore, the partial molar adiabatic compressibility of a solute can be calculated from precise density and sound velocity in solutions. Measurement of sound velocity, however, requires a strict temperature control such as thermal stability of the order 10−3 °C. Therefore, the measuring of a relative change in a physical characteristic per unit of solute concentration rather than its absolute value is often more important, more precise and easier. That is the reason why the modern devices use the differential arrangement: two identical resonator cells are placed in one termostated volume and the difference in ultrasonic velocities is measured [186]. In this way the increment of ultrasonic velocity is easily and precisely determined under the thermal stability of 10−2 °C.
The arrangement of modern devices, i.e., using the so-called resonator method [182] enables using of two modes such as ultrasonic velocimetry and ultrasonic spectroscopy. Spectroscopic approach utilizes measurements of the attenuation of ultrasound over a wide range of frequencies in order to determine data about the kinetics and thermodynamics of fast molecular equilibrium. The ultrasonic velocimetry is based on the accurate measurement of the ultrasonic velocity, optionally in combination with density measurement to determine the compressibility and volume of a molecule or aggregate in solution [187]. The results of investigation of physical structure of HS using ultrasonic velocimetry demonstrated the progressive aggregation of various humates from 10−3 to 10 g/L under various conditions such as neutral and alkaline pH and at increased salinity of the solution [47]. It has also been demonstrated the difference in aggregation of HS and classical surfactants monomers such sodium dodecyl sulphate, (CMC = 2.3 g/L) [47] and polymers (Triton) [179] (Figure 7a). The increment of ultrasonic velocity (defined as a difference between ultrasonic velocity in HS () and water () divided by , (see Figure 7a) was used. The increment is constant when no interaction between sample molecules occurs (below CMC) and when CMC is reached, it progressively decreases. HS in solution showed a steep decrease already at concentration hundreds to thousand times lower that the CMC reported for classical surfactants (Figure 7a). This confirmed the view that HA form rather premicellar aggregates than classical micelle with a precisely defined CMC. Similar experiments conducted in 1M NaCl and in NaOH at pH 12 showed similar, but less intensive, aggregation of HS in solutions [47].
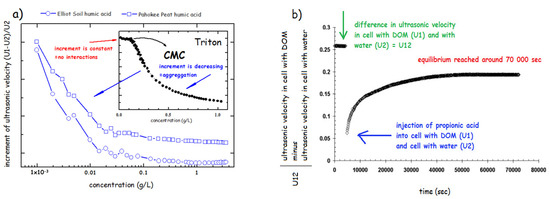
Figure 7.
(a) Comparison of aggregation behavior of DOM and triton using increment of ultrasonic velocity (based on the results of ref. [180]). (b) Kinetic curve—ultrasonic velocity corresponding to the reconformation of DOM after addition of propionic acid as indicated by the change in , which is the difference between cell 1 with DOM sample and cell 2 filled with water (based on the results of ref. [47]).
The rearrangement kinetics of the HS aggregates was tested by injection of various molecules including HCl, propionic acid and propanol into HS solution (Figure 7b). The results showed the specificity in interactions with molecules of various chemical properties. Whereas HCl and propanol showed a rearrangement within several minutes, the propionic acid treatment caused the long-term reconformation taking several hours. This supported the previously reported view [52] that organic acid can cause a reversible disruption of hydrophobic interactions stabilizing HS molecules at neutral pH and shift to HS structure to a new equilibrium at pH = 3.5 in which HS structure is stabilized via H-bonds.
In order to test the nature and stability of HS, the salts of HA and FA from various sources in the concentration range from 0.001 to 10 g/L were repeatedly heated from 5 to 90 °C [69,179]. The change in ultrasonic velocity in heating run indicated no transition above 4 g/L; below this concentration existence of transitions was demonstrated as breaks on the curve (Figure 8a). Decreasing concentration caused the weakening of the stabilizing interactions and several transitions could be observed (breaks in Figure 8a).
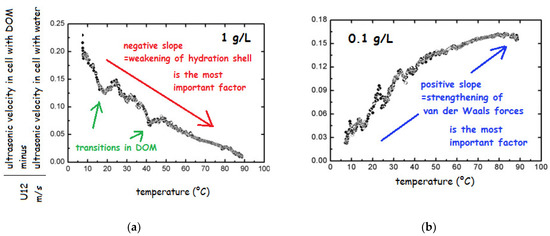
Figure 8.
Temperature dependence of ultrasonic velocity of sodium fulvates Suwannee River at concentrations (a) 1 and (b) 0.1 g/L (based on the results of ref. [69]).
The most important change could be seen in the slope of the record. In particular, below 1 g/L the slope changed from negative to positive (see Figure 8b), which was attributed to the change in prevailing role of different types of interactions influencing the adiabatic compressibility. Specifically, below 1 g/L the most important factor was change in structural compressibility, which is caused by strengthening of van der Waals forces upon heating while hydration component of compressibility plays only minor role. In contrast, at higher HS concentration, only weakening of hydration shell plays an important role and the structural compressibility could be neglected since the structure is stabilized by weak interactions such as H-bonds, charge-charge interactions and possibly hydrophobic interactions. The observations about a change in the HS structure at 1 g/L were later confirmed by other authors [153,188]. Results obtained by FFC NMR relaxometry showed that also at extremely high concentration of DOM (30 g/L) in the structure occur changes, which are, however, not visible by velocimetry under applied conditions, i.e., around 6 MHz in the temperature range from 25 to 60 °C [71].
The heat-cool experiments were also used for testing of solutions of HS modified by either HCl or propionic acids. The results revealed a great difference in physical structure between resulted structures at pH 3.5. In case of HCl the structure resembled the original system with increasing strength of H-bonds, reflected by the increase in the transition temperatures. The addition of propionic acid induced a complete reconformation and strengthening of the HS structure [70]. The comparison of the heating and cooling cycles highlighted the structural hysteresis of HS, proving that the physical structure of HS also depends on the thermal history (Figure 9a) [69].
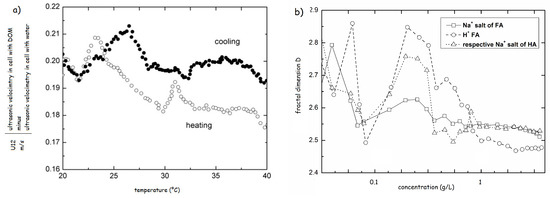
Figure 9.
(a) Hysteresis in ultrasonic velocity of Elliot Soil sodium fulvate 0.01 g/L (based on the results of ref. [69]). (b) Fractal dimensions of Na+ salts of Elliot soil HA and FA and H+ form of Elliot soil FA (based on the results of ref. [70]).
This problem was in detail investigated by Řezáčová et al. [189], who found that the increase of the heating/cooling amplitude caused an increase of the relaxation time needed for HS to reach the equilibrium. In addition, the authors confirmed that in the thermally-induced hysteresis were not involved aromatic structures whereas alkyl moieties, carboxylic and carbonyl carbons influenced the hysteresis positively. On the contrary, negative effect was observed for peptide and O-alkyl groups. The structural hysteresis was confirmed by FFC NMR. In addition, the method also unraveled that correlation time of water in DOM is lower after than before heating. Decreasing of correlation time indicates lower restriction of rotational and translational motions of water in DOM thereby indicating the delay in re-formation of DOM structure or potentially less rigid structure upon cooling. The ultrasonic velocimetry showed a fast reconformation dynamics of HS aggregates in water (up to 10 min) which was in contrast to processes taking part at the water/air interface (10 h) [190].
Ultrasonic velocimetry data allowed calculation of fractal dimensions of DOM aggregates [191] and calculation of changes in thermodynamical parameters caused by aggregation during progressive increase in concentration such as entropy of the DOM/water system [179]. As illustrated in the section Light/X-ray/Neutron Scattering Studies and Fractal Architecture of HS Aggregates, most approaches for analysis of fractal geometry of HS provide all only 2D arrangement. On the contrary, the approach based on ultrasonic velocimetry allowed calculation of 3D dimension of humic aggregates. Data confirmed fluctuations in conformation of HS below 1 g/L (Figure 9b).
Ultrasonic velocity in combination with the density measurement allowed for the determination of adiabatic compressibility, which was used to determine the hydration water in a set of chemically treated lignite HA. The determined values ranged from 0.45 to 0.95 g of water per 1 g of DOM at the concentration of HS 0.25 g/L [192]. Interestingly, these values are similar to those reported by Kučerík et al. [193] in a study on IHSS standard HS using differential scanning calorimetry. Other work [153] combining the density and velocity measurements confirmed the earlier observations [70] about concentration 1 g/L at which an abrupt change in conformation occurred.
4. Conclusions
Most of the scientific reports reviewed in literature seem to indicate that humic materials from different sources have different aggregate morphologies (though the aggregation mechanisms seem to be similar), indirectly underscoring the importance of specific chemistry to humic material geometry. So far, the vast physicochemical heterogeneity of HS, mainly due to the extensive variability of the sources from which they are extracted, justifies the current absence of a unified model capable of describing and predicting their dynamic properties and fate both in the aqueous phase and in the solid state.
Table 1 is an attempt to gather most of the scientific reports cited in this review and classified according to the various colloidal or fractal models that inspired them.

Table 1.
Models on the structure of Humic Substances, experimental methods and cited references.
Several questions still remain open on the level of comprehension of HS aggregation: (i) How this aspect may impact on the capacity of soil humic aggregates to be dissolved and then transported to the surface freshwater or as groundwater colloidal dispersions? (ii) If the water carrying the humic material percolates through the soil, would the interaction with metal cations on the soil interfaces help to change the shape and degree of aggregation?
Still debated seems to be also the influence of the concentration of the suspension on the pH dependence of the HS configuration. Indeed, several studies demonstrate that at least some types of HS are strongly pH sensitive, in contrast with studies of other humic material, where far less pH sensitivity has been observed. Another very real possibility is that the pH configuration dependence is a function of the chemistry of the HS involved.
Last, but not least, (iii) do the aggregates and dissolved molecules of HS influence the H-bonds and 3D structure of surrounding water? Consequently, do the HS aggregates influence also other fundamental water properties similarly as shown for water structural hysteresis? And vice versa, how the water anomalies (i.e., hysteresis) influence HS properties? Is the fate, stability and biological activity of HS aggregates influenced by fluctuation of the environmental conditions, e.g., temperature, pH and ionic strength?
Answering these questions may help to create a concept enabling to model the role of HS in biogeochemical cycles. Such a concept is missing and for this reason there are not carbon sequestration strategies that include also HS and their dynamics. Currently, the fundamental strategy is application of HS from various sources as soil amendments or fertilizers to support the yield of agricultural plants. This strategy, however, leads only to C redistribution as HS are relatively fast metabolized or mineralized. Apparently, this is not a meaningful strategy leading to a long-term fixation of C in soils and its protection thereby supporting fundamental soil ecosystem functions.
The advent of high-resolution techniques such as electron microscopy and NMR have provided mechanisms to conduct highly detailed studies of the interconnection between HS form and function. The technique of small-angle scattering, capable of elucidating numerous aspects of macromolecular morphology, continues to be the primary experimental tool for determining the fractal dimension df for a variety of colloidal systems.
Using scattering and related techniques to investigate the fractal behavior of NOM continues the process of developing means to quantify important distinguishing features of these materials. Initial experiments have shown that this can be a powerful tool for interpreting humic responses in a variety of situations. The following conjectures attempt to extract from this information possible future directions suggested by these early results.
In particular, an attempt is made to extrapolate from results with humic materials in solution to implications about humic interactions with inorganic surfaces. The suggestion from fractal analysis is that cationic complexes need not necessarily be collapsed structures. It will be of interest to determine whether this openness in solution is translated to the formation of relatively open networks upon sorbing onto inorganic surfaces. HS with high df might be expected to form denser surface coatings; similarly, those forming larger clusters in solution may form deep coatings on surfaces. The tendency in the past has been to think in terms of small versus large aggregates. The mass fractal model encourages thinking in terms of open versus compact, as well. Variations in source material, temperature, and/or pH can all affect aggregating properties, which in turn affect adsorbing and cosorbing behaviors.
The degree of irregularity of the HS surfaces is hypothesized to be related to the number, type and availability reactive sites, on the catalytic activity and adsorption capacity, and on the interactions that may occur between HS and mineral surfaces, ions metals, organic chemicals, plant roots and microorganisms in the soil.
Within the fractal formalism, several open questions about the genesis, structural nature, surface reactivity, and aggregation-dispersion phenomena of HS in the environment, may be unraveled. Some issues should be addressed such as: (i) the appropriate use of methodologies for interpreting the non-fractal, surface fractal and mass fractal nature of HS; (ii) a clear definition of the experimental conditions used in the fractal analysis of HS, including the state (solid, solution or suspension) and concentration of the sample, as well as the pH, temperature and ionic strength of the medium; and (iii) the consistency of fractal dimension values obtained through different techniques such as scattering, turbidimetry and microscopy.
The intensity of the interactions that determine the size of HS aggregates affects the fate and transport of nutrients and pollutants. Considering these situations, the study of the forces responsible for the aggregation phenomena in different environmental conditions will be one of the challenges of the research on humic substances in the near future.
Author Contributions
Conceptualization, R.A. and C.C.; methodology, P.C.; validation, E.D.I., M.B. and J.F.; formal analysis, E.D.I.; investigation, M.B.; resources, J.F.; data curation, C.C.; writing—original draft preparation, R.A., C.C. and P.C.; writing—review and editing, R.A., C.C. and P.C.; visualization, C.C. and P.C.; supervision, R.A.; project administration, R.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AFM | Atomic Force Microscopy |
| AHA | Aldrich Humic Acid |
| CMC | Critical Micellar Concentration |
| CP | Cross Polarization |
| CZE | Capillary Zone Electrophoresis |
| df | fractal dimension |
| Dh | hydrodynamic particle diameter |
| DLS | Dynamic Light Scattering |
| DSC | Differential Scanning Calorimetry |
| DOC | Dissolved Organic Carbon |
| DOM | Dissolved Organic Matter |
| DOSY | Diffusion Ordered Spectroscopy |
| EHA | Elliot soil Humic Acid |
| FA | Fulvic Acid |
| FCS | Fluorescence Correlation Spectroscopy |
| FFC | Fast Field Cycling |
| GPC | Gel Permeation Chromatography |
| HA | Humic Acid |
| HPSEC | High-Performance Size Exclusion Chromatography |
| HRMAS | High-Resolution Magic Angle Spinning |
| HS | Humic Substances |
| IHSS | International Humic Substances Society |
| LDE | Laser Doppler Electrophoresis |
| MALLS | Multi-Angle Laser Light Scattering |
| MAOM | Mineral-Associated Organic Matter |
| MAS | Magic Angle Spinning |
| Mw | weight-averaged molar mass |
| NOM | Natural Organic Matter |
| NMR | Nuclear Magnetic Resonance |
| PCS | Photon Correlation Spectroscopy |
| PdI | Polydispersity Index |
| PSD | Particle Size Distribution |
| POM | Particulate Organic Matter |
| Rp | Particle Radius |
| Rg | Radius of gyration |
| RLHA | Rendzic Leptosol Humic Acid |
| SANS | Small-Angle Neutron Scattering |
| SAXS | Small-Angle X-ray Scattering |
| SEC | Size Exclusion Chromatography |
| SOM | Soil Organic Matter |
| SRHA | Suwannee River HA |
| SRFA | Suwannee River FA |
| TOC | Total Organic Carbon |
| UF | Ultrafiltration |
References
- Cambardella, C.A.; Elliott, E.T. Particulate Soil Organic-Matter Changes across a Grassland Cultivation Sequence. Soil Sci. Soc. Am. J. 1992, 56, 777–783. [Google Scholar] [CrossRef]
- Zsolnay, A. Dissolved organic matter: Artefacts, definitions, and functions. Geoderma 2003, 113, 187–209. [Google Scholar] [CrossRef]
- Dafner, E.V.; Wangersky, P.J. A brief overview of modern directions in marine DOC studies—Part I. Methodological aspects. J. Environ. Monit. 2002, 4, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Bolan, N.S.; Adriano, D.C.; Kunhikrishnan, A.; James, T.; McDowell, R.; Senesi, N. Dissolved Organic Matter: Biogeochemistry, Dynamics, and Environmental Significance in Soils. Adv. Agron. 2011, 110, 1–75. [Google Scholar] [CrossRef]
- Blaser, P. The role of natural organic matter in the dynamics of metals in forest soils. In Humic Substances in the Global Environment and Implications on Human Health; Senesi, N., Miano, T.M., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; pp. 943–960. [Google Scholar]
- Chiou, C.T.; Malcolm, R.L.; Brinton, T.I.; Kile, D.E. Water solubility enhancement of some organic pollutants and pesticides by dissolved humic and fulvic acids. Environ. Sci. Technol. 1986, 20, 502–508. [Google Scholar] [CrossRef]
- Quagliotto, P.; Montoneri, E.; Tambone, F.; Adani, F.; Gobetto, R.; Viscardi, G. Chemicals from wastes: Compost-derived humic acid-like matter as surfactant. Environ. Sci. Technol. 2006, 40, 1686–1692. [Google Scholar] [CrossRef]
- Guggenberger, G.; Kaiser, K. Dissolved organic matter in soil: Challenging the paradigm of sorptive preservation. Geoderma 2003, 113, 293–310. [Google Scholar] [CrossRef]
- Raulund-Rasmussen, K.; Borggaard, O.K.; Hansen, H.C.B.; Olsson, M. Effect of natural organic soil solutes on weathering rates of soil minerals. Eur. J. Soil Sci. 1998, 49, 397–406. [Google Scholar] [CrossRef]
- Lundström, U.S.; van Breemen, N.; Iongmans, A.G. Evidence for microbial decomposition of organic acids during podzolization. Eur. J. Soil Sci. 1995, 46, 489–496. [Google Scholar] [CrossRef]
- Lu, Y.H.; Wassmann, R.; Neue, H.U.; Huang, C.Y. Dynamics of dissolved organic carbon and methane emissions in a flooded rice soil. Soil Sci. Soc. Am. J. 2000, 64, 2011–2017. [Google Scholar] [CrossRef]
- McDowell, W.H. Dissolved organic matter in soils—Future directions and unanswered questions. Geoderma 2003, 113, 179–186. [Google Scholar] [CrossRef]
- Marschner, B.; Kalbitz, K. Controls of bioavailability and biodegradability of dissolved organic matter in soils. Geoderma 2003, 113, 211–235. [Google Scholar] [CrossRef]
- Kalbitz, K.; Solinger, S.; Park, J.H.; Michalzik, B.; Matzner, E. Controls on the dynamics of dissolved organic matter in soils: A review. Soil Sci. 2000, 165, 277–304. [Google Scholar] [CrossRef]
- Kleber, M.; Lehmann, J. Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems. J. Environ. Qual. 2019, 48, 207–216. [Google Scholar] [CrossRef]
- Schmidt, M.W.I.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kögel-Knabner, I.; Lehmann, J.; Manning, D.A.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef]
- Li, Y.; Fang, F.; Wei, J.; Wu, X.; Cui, R.; Li, G.; Zheng, F.; Tan, D. Humic acid fertilizer improved soil properties and soil microbial diversity of continuous cropping peanut: A three-year experiment. Sci. Rep. 2019, 9, 12014. [Google Scholar] [CrossRef]
- Rullkötter, J. Geochemistry Organic. In Encyclopedia of Physical Science and Technology; Meyers, R.A., Ed.; Academic Press: New York, NY, USA, 2003; pp. 549–574. [Google Scholar] [CrossRef]
- Knicker, H. How does fire affect the nature and stability of soil organic nitrogen and carbon? A review. Biogeochemistry 2007, 85, 91–118. [Google Scholar] [CrossRef]
- Gerke, J. Concepts and misconceptions of humic substances as the stable part of soil organic matter: A review. Agronomy 2018, 8, 76–91. [Google Scholar] [CrossRef]
- Di Iorio, E.; Circelli, L.; Angelico, R.; Torrent, J.; Tan, W.; Colombo, C. Environmental implications of interaction between humic substances and iron oxide nanoparticles: A review. Chemosphere 2022, 303, 135172–135183. [Google Scholar] [CrossRef]
- Cotrufo, M.F.; Ranalli, M.G.; Haddix, M.L.; Six, J.; Lugato, E. Soil carbon storage informed by particulate and mineral-associated organic matter. Nat. Geosci. 2019, 12, 989–994. [Google Scholar] [CrossRef]
- Sanderman, J.; Maddern, T.; Baldock, J. Similar composition but differential stability of mineral retained organic matter across four classes of clay minerals. Biogeochemistry 2014, 121, 409–424. [Google Scholar] [CrossRef]
- Lehmann, J.; Kinyangi, J.; Solomon, D. Organic matter stabilization in soil microaggregates: Implications from spatial heterogeneity of organic carbon contents and carbon forms. Biogeochemistry 2007, 85, 45–57. [Google Scholar] [CrossRef]
- Keiluweit, M.; Bougoure, J.J.; Nico, P.S.; Pett-Ridge, J.; Weber, P.K.; Kleber, M. Mineral protection of soil carbon counteracted by root exudates. Nat. Climate Change 2015, 5, 588–595. [Google Scholar] [CrossRef]
- Lehmann, J.; Kleber, M. The contentious nature of soil organic matter. Nature 2015, 528, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Woolf, D.; Lehmann, J. Microbial models with minimal mineral protection can explain long-term soil organic carbon persistence. Sci. Rep. 2019, 9, 6522. [Google Scholar] [CrossRef]
- Baveye, P.C.; Wander, M. The (bio)chemistry of soil humus and humic substances: Why is the “new view” still considered novel after more than 80 years? Front. Environ. Sci. 2019, 7, 27–32. [Google Scholar] [CrossRef]
- Janzen, H. The Future of Humic Substances Research: Preface to a Debate. J. Environ. Qual. 2019, 48, 205–206. [Google Scholar] [CrossRef]
- Olk, D.C.; Bloom, P.R.; De Nobili, M.; Chen, Y.; McKnight, D.M.; Wells, M.J.M.; Weber, J. Using Humic Fractions to Understand Natural Organic Matter Processes in Soil and Water: Selected Studies and Applications. J. Environ. Qual. 2019, 48, 1633–1643. [Google Scholar] [CrossRef]
- Nobili, M.; Bravo, C.; Chen, Y. The spontaneous secondary synthesis of soil organic matter components: A critical examination of the soil continuum model theory. Appl. Soil Ecol. 2020, 154, 103655. [Google Scholar] [CrossRef]
- Ghosh, K.; Schnitzer, M. Macromolecular structures of humic substances. Soil Sci. 1980, 129, 266–276. [Google Scholar] [CrossRef]
- Marinsky, J.A.; Reddy, M.M. Vapor-pressure osmometric study of the molecular weight and aggregation tendency of a reference-soil fulvic acid. Anal. Chim. Acta 1990, 232, 123–130. [Google Scholar] [CrossRef]
- Swift, R.S.; Posner, A.M. Gel chromatography of humic acids. J. Soil Sci. 1971, 22, 237–249. [Google Scholar] [CrossRef]
- Swift, R.S. Macromolecular properties of soil humic substances: Fact, fiction, and opinion. Soil Sci. 1999, 164, 790–802. [Google Scholar] [CrossRef]
- Chin, W.C.; Orellana, M.; Verdugo, P. Spontaneous assembly of marine dissolved organic matter into polymer gels. Nature 1998, 391, 568–572. [Google Scholar] [CrossRef]
- Wershaw, R.L. A new model for humic materials and their interactions with hydrophobic organic chemicals in soil-water or sediment-water systems. J. Contam. Hydrol. 1986, 1, 29–45. [Google Scholar] [CrossRef]
- Wershaw, R.L. Model for humus in soils and sediments. Environ. Sci. Technol. 1993, 27, 814–817. [Google Scholar] [CrossRef]
- Wershaw, R.L. Molecular aggregation of humic substances. Soil Sci. 1999, 164, 803–813. [Google Scholar] [CrossRef]
- Guetzloff, T.F.; Rice, J.A. Does humic acid form a micelle? Sci. Tot. Environ. 1994, 152, 31–35. [Google Scholar] [CrossRef]
- Guetzloff, T.F.; Rice, J.A. Micellar nature of humic colloids. In Humic and Fulvic Acids: Isolation, Structure, and Environmental Role; Chapter 2; Gaffney, J.S., Marley, N.A., Clark, S.B., Eds.; ACS Symposium Series 651; American Chemical Society: Washington, DC, USA, 1996. [Google Scholar] [CrossRef]
- Engebretson, R.R.; von Wandruszka, R. Microorganization in dissolved humic acids. Environ. Sci. Technol. 1994, 28, 1934–1941. [Google Scholar] [CrossRef]
- Puchalskl, M.M.; Morra, M.J.; von Wandruszka, R. Fluorescence quenching of synthetic organic compounds by humic materials. Environ. Sci. Technol. 1992, 26, 1787–1792. [Google Scholar] [CrossRef]
- von Wandruszka, R.; Ragle, C.; Engebretson, R. The role of selected cations in the formation of pseudomicelles in aqueous humic acid. Talanta 1997, 44, 805–809. [Google Scholar] [CrossRef]
- von Wandruszka, R. The micellar model of humic acid: Evidence from pyrene fluorescence measurements. Soil Sci. 1998, 163, 921–930. [Google Scholar] [CrossRef]
- von Wandruszka, R.; Engebretson, R. Kinetics of humic acid associations. In Humic Substances and Chemical Contaminants; Clapp, C.E., Hayes, M.H.B., Senesi, N., Bloom, P.R., Jardine, P.M., Eds.; Soil Science Society of America: Madison, WI, USA, 2001. [Google Scholar] [CrossRef]
- Kučerík, J.; Šmejkalová, D.; Čechlovská, H.; Pekař, M. New insights into aggregation and conformational behaviour of humic substances: Application of high resolution ultrasonic spectroscopy. Org. Geochem. 2007, 38, 2098–2110. [Google Scholar] [CrossRef]
- Piccolo, A.; Nardi, S.; Concheri, G. Micelle-like conformation of humic substances as revealed by size exclusion chromatography. Chemosphere 1996, 33, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Peuravuori, J.; Pihlaja, K. Molecular size distribution and spectroscopic properties of aquatic humic substances. Anal. Chim. Acta 1997, 337, 133–149. [Google Scholar] [CrossRef]
- Fetsch, D.; Hradilová, M.; Peña-Méndez, E.M.; Havel, J. Capillary zone electrophoresis study of humic substances aggregation. J. Chromatogr. A 1998, 817, 313–323. [Google Scholar] [CrossRef]
- Jones, M.N.; Bryan, N.D. Colloidal properties of humic substances. Adv. Colloid. Interface Sci. 1998, 78, 1–48. [Google Scholar] [CrossRef]
- Conte, P.; Piccolo, A. Conformational arrangement of dissolved humic substances. Influence of solution composition on association of humic molecules. Environ. Sci. Technol. 1999, 33, 1682–1690. [Google Scholar] [CrossRef]
- Piccolo, A.; Conte, P.; Cozzolino, A. Effects of mineral and monocarboxylic acids on the molecular association of dissolved humic substances. Eur. J. Soil Sci. 1999, 50, 687–694. [Google Scholar] [CrossRef]
- Varga, B.; Kiss, G.; Galambos, I.; Gelencser, A.; Hlavay, J.; Krivacsy, Z. Secondary structure of humic acids. Can micelle-like conformation be proved by aqueous size exclusion chromatography? Environ. Sci. Technol. 2000, 34, 3303–3306. [Google Scholar] [CrossRef]
- Young, C.; von Wandruszka, R. A comparison of aggregation behavior in aqueous humic acids. Geochem. Trans. 2001, 2, 16–20. [Google Scholar] [CrossRef]
- Piccolo, A. The supramolecular structure of humic substances. Soil Sci. 2001, 166, 810–832. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular structure in soil humic substances: The new view. Environ. Sci. Technol. 2005, 39, 9009–9015. [Google Scholar] [CrossRef]
- Yates, L.M.; von Wandruska, R. Effects of pH and metals on the surface tension of aqueous humic materials. J. Soil Sci. Soc. Am. 1999, 63, 1645–1649. [Google Scholar] [CrossRef]
- Senesi, N.; Lorusso, G.F.; Miano, T.M.; Maggipinto, G.; Rizzi, F.R.; Capozzi, V. The fractal dimension of humic substances as function of pH by turbidity measurements. In Humic Substances in the Global Environment and Implications on Human Health; Senesi, N., Miano, T.M., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1994; pp. 121–126. [Google Scholar]
- Piccolo, A. The supramolecular structure of humic substances: A novel understanding of humus chemistry and implications in soil science. Adv. Agron. 2002, 75, 57–133. [Google Scholar] [CrossRef]
- Piccolo, A. In memoriam Prof. F.J. Stevenson and the question of humic substances in soil. Chem. Biol. Technol. Agric. 2016, 3, 23–25. [Google Scholar] [CrossRef]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M. Analysis of molecular aggregation in humic substances in solution. Colloids Surf. A Physicochem. Eng. Asp. 2007, 302, 301–306. [Google Scholar] [CrossRef]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M. Simultaneous presence of diverse molecular patterns in humic substances in solution. J. Phys. Chem. B 2007, 111, 10577–10582. [Google Scholar] [CrossRef]
- Nebbioso, A.; Piccolo, A. Basis of a humeomics science: Chemical fractionation and molecular characterization of humic biosuprastructures. Biomacromolecules 2011, 12, 1187–1199. [Google Scholar] [CrossRef]
- Chilom, G.; Bruns, A.S.; Rice, J.A. Aggregation of humic acid in solution: Contributions of different fractions. Org. Geochem. 2009, 40, 455–460. [Google Scholar] [CrossRef]
- Chilom, G.; Rice, J.A. Structural organization of humic acid in the solid state. Langmuir 2009, 25, 9012–9015. [Google Scholar] [CrossRef] [PubMed]
- Chilom, G.; Baglieri, A.; Johnson-Edler, C.A.; Rice, J.A. Hierarchical self-assembling properties of natural organic matter’s components. Org. Geochem. 2013, 57, 119–126. [Google Scholar] [CrossRef]
- Zheng, G.; Price, W.S. Direct hydrodynamic radius measurement on dissolved organic matter in natural waters using diffusion NMR. Environ. Sci. Technol. 2012, 46, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Drastík, M.; Novak, F.; Kučerík, J. Origin of heat-induced structural changes in dissolved organic matter. Chemosphere 2013, 90, 789–795. [Google Scholar] [CrossRef]
- Kučerík, J.; Čechlovská, H.; Bursáková, P.; Pekař, M. Lignite humic acids aggregates studied by high resolution ultrasonic spectroscopy: Thermodynamic stability and molecular feature. J. Therm. Anal. Calorim. 2009, 96, 637–643. [Google Scholar] [CrossRef]
- Conte, P.; Kučerík, J. Water dynamics and its role in structural hysteresis of dissolved organic matter. Environ. Sci. Technol. 2016, 50, 2210–2216. [Google Scholar] [CrossRef]
- Wells, M.J.M. Supramolecular answers to the organic matter controversy. J. Environ. Qual. 2019, 48, 1644–1651. [Google Scholar] [CrossRef]
- Wells, M.J.M.; Stretz, H.A. Supramolecular architectures of natural organic matter. Sci. Tot. Environ. 2019, 671, 1125–1133. [Google Scholar] [CrossRef]
- Esfahani, M.R.; Stretz, H.A.; Wells, M.J.M. Abiotic reversible self-assembly of fulvic and humic acid aggregates in low electrolytic conductivity solutions by dynamic light scattering and zeta potential investigation. Sci. Total Environ. 2015, 537, 81–92. [Google Scholar] [CrossRef]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions, 2nd ed.; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Wang, H.; Adeleye, A.S.; Huang, Y.; Li, F.; Keller, A.A. Heteroaggregation of nanoparticles with biocolloids and geocolloids. Adv. Colloid Interface Sci. 2015, 226, 24–36. [Google Scholar] [CrossRef]
- Leenheer, J.A.; Croué, J.-P. Peer reviewed: Characterizing aquatic dissolved organic matter. Environ. Sci. Technol. 2003, 37, 18A–26A. [Google Scholar] [CrossRef]
- Ohno, T.; Hess, N.J.; Qafoku, N.P. Current understanding of the use of alkaline extractions of soils to investigate soil organic matter and environmental processes. J. Environ. Qual. 2019, 48, 1561–1564. [Google Scholar] [CrossRef]
- Hatcher, P.G.; Waggoner, D.C.; Chen, H. Evidence for the existence of humic acids in peat soils based on solid-state 13C NMR. J. Environ. Qual. 2019, 48, 1571–1577. [Google Scholar] [CrossRef]
- Olk, D.C.; Bloom, P.R.; Perdue, E.M.; McKnight, D.M.; Chen, Y.; Farenhorst, A.; Senesi, N.; Chin, Y.-P.; Schmitt-Kopplin, P.; Hertkorn, N.; et al. Environmental and agricultural relevance of humic fractions extracted by alkali from soils and natural waters. J. Environ. Qual. 2019, 48, 217–232. [Google Scholar] [CrossRef]
- Seaman, J.C. Thin-foil SEM analysis of soil and groundwater colloids: Reducing instrument and operator bias. Environ. Sci. Tech. 2000, 34, 187–191. [Google Scholar] [CrossRef]
- Lead, J.R.; Muirhead, D.; Gibson, C.T. Characterization of freshwater natural aquatic colloids by Atomic Force Microscopy (AFM). Environ. Sci. Technol. 2005, 39, 6930–6936. [Google Scholar] [CrossRef]
- Chen, Y.; Schnitzer, M. Sizes and shapes of humic substances by electron microscopy. In Humic Substances II. In Search of Structure; Hayes, M.H.B., MacCarthy, P., Malcolm, R.L., Swift, R.S., Eds.; Wiley-Interscience: Chichester, UK, 1989; pp. 621–638. [Google Scholar]
- Senesi, N.; Rizzi, F.R.; Dellino, P.; Acquafredda, P. Fractal dimension of humic acids in aqueous suspension as a function of pH and time. J. Soil. Sci. Soc. Am. 1996, 60, 1773–1780. [Google Scholar] [CrossRef]
- Senesi, N.; Rizzi, F.R.; Dellino, P.; Acquafredda, P. Fractal humic acids in aqueous suspensions at various concentrations, ionic strengths, and pH values. Colloids Surf. A 1997, 127, 57–68. [Google Scholar] [CrossRef]
- Rizzi, F.R.; Stoll, S.; Senesi, N.; Buffle, J. A transmission electron microscopy study of the fractal properties and aggregation processes of humic acids. Soil Sci. 2004, 169, 765–775. [Google Scholar] [CrossRef]
- Shevchenko, S.M.; Bailey, G.W.; Akim, L.G. The conformational dynamics of humic polyanions in model organic and organo-mineral aggregates. J. Mol. Struct. 1999, 460, 179–190. [Google Scholar] [CrossRef]
- Chen, Y.; Schnitzer, M. Scanning electron microscopy of a humic acid and its metal and clay complexes. Soil Sci. Soc. Am. J. 1976, 40, 682–686. [Google Scholar] [CrossRef]
- Ikai, A.; Österberg, R. Atomic force microscopy of humic acids. Scanning Microsc. 1996, 10, 947–951. [Google Scholar]
- Baalousha, M.; Motelica-Heino, M.; Galaup, S.; Coustumer, P. Supramolecular structure of humic acids by TEM with improved sample preparation and staining. Microsc. Res. Tech. 2005, 66, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Heath, G.R.; Scheuring, S. High-speed AFM height spectroscopy reveals μs-dynamics of unlabeled biomolecules. Nat. Commun. 2018, 9, 4983–4993. [Google Scholar] [CrossRef]
- Baalousha, M.; Prasad, A.; Lead, J.R. Quantitative measurement of the nanoparticle size and number concentration from liquid suspensions by atomic force microscopy. Environ. Sci. Processes Impacts 2014, 16, 1338–1347. [Google Scholar] [CrossRef]
- Heath, G.R.; Kots, E.; Robertson, J.L.; Lansky, S.; Khelashvili, G.; Weinstein, H.; Scheuring, S. Localization atomic force microscopy. Nature 2021, 594, 385–390. [Google Scholar] [CrossRef]
- Cheng, S.; Bryant, R.; Doerr, S.H.; Williams, P.R.; Wright, C.J. Application of atomic force microscopy to the study of natural and model soil particles. J. Microsc. 2008, 231, 384–394. [Google Scholar] [CrossRef]
- Copello, F.D.R.; Lizarraga, L.; Orsetti, S.; Molina, F.V. Swelling and aggregation of Leonardite upon pH change and Pb II binding: An AFM study. Environ. Chem. 2018, 15, 162–170. [Google Scholar] [CrossRef]
- Xu, C.-Y.; Zhou, T.-T.; Wang, C.-l.; Liu, H.-Y.; Zhang, C.-T.; Hu, F.-N.; Zhao, S.-W.; Geng, Z.-C. Aggregation of polydisperse soil colloidal particles: Dependence of Hamaker constant on particle size. Geoderma 2020, 359, 113999–114007. [Google Scholar] [CrossRef]
- Colombo, C.; Palumbo, G.; Angelico, R.; Cho, H.G.; Francioso, O.; Ertani, A.; Nardi, S. Spontaneous aggregation of humic acid observed with AFM at different pH. Chemosphere 2015, 138, 821–828. [Google Scholar] [CrossRef]
- Balnois, E.; Wilkinson, K.J.; Lead, J.; Buffle, J. Atomic force microscopy of humic substances: Effects of pH and ionic strength. Environ. Sci. Technol. 1999, 33, 3911–3917. [Google Scholar] [CrossRef]
- Chen, C.L.; Wang, X.K.; Jiang, H.W.P.; Hu, W.P. Direct observation of macromolecular structures of humic acid by AFM and SEM. Colloid Surf. A 2007, 302, 121–125. [Google Scholar] [CrossRef]
- Liu, A.G.; Wu, R.C.; Eschenazi, E.; Papadopoulos, K. AFM on humic acid adsorption on mica. Colloid Surf. A Physicochem. Eng. Asp. 2000, 174, 245–252. [Google Scholar] [CrossRef]
- Schaumann, G.E.; Thiele-Bruhn, S. Molecular modeling of soil organic matter: Squaring the circle? Geoderma 2011, 166, 1–14. [Google Scholar] [CrossRef]
- Plaschke, M.; Römer, J.; Klenze, R.; Kim, J.I. In situ AFM study of sorbed humic acid colloids at different pH. Colloids Surf. A 1999, 160, 269–279. [Google Scholar] [CrossRef]
- Plaschke, M.; Romer, J.; Kim, J.I. Characterization of Gorleben groundwater colloids by atomic force microscopy. Environ. Sci. Technol. 2002, 36, 4483–4488. [Google Scholar] [CrossRef]
- Conte, P.; Spaccini, R.; Piccolo, A. State of the art of CPMAS 13C-NMR spectroscopy applied to natural organic matter. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 215–223. [Google Scholar] [CrossRef]
- Dais, P.; Spyros, A. Nuclear magnetic resonance. In Chemical Analysis of Food: Techniques and Applications; Picó, Y., Ed.; Academic Press-Elsevier: Waltham, MA, USA, 2012; pp. 91–115. [Google Scholar]
- Simpson, A.J.; Simpson, M.J.; Soong, R. Nuclear magnetic resonance spectroscopy and its key role in environmental research. Environ. Sci. Technol. 2012, 46, 11488–11496. [Google Scholar] [CrossRef]
- Farooq, H.; Courtier-Murias, D.; Soong, R.; Bermel, W.; Kingery, W.; Simpson, A. HR-MAS NMR spectroscopy: A practical guide for natural samples. Curr. Org. Chem. 2013, 17, 3013–3031. [Google Scholar] [CrossRef]
- Conte, P.; Lo Meo, P. Nuclear magnetic resonance with fast field-cycling setup: A valid tool for soil quality investigation. Agronomy 2020, 10, 1040–1072. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: New York, NY, USA, 1983. [Google Scholar]
- Rinaldi, P.L. Three-dimensional solution NMR spectroscopy of complex structures and mixtures. Analyst 2004, 129, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Fuloria, N.K.; Fuloria, S. Structural elucidation of small organic molecules by 1D, 2D and multi-dimensional-solution NMR spectroscopy. J. Anal. Bioanal. Tech. 2013, 4, 1–8. [Google Scholar] [CrossRef]
- Berns, A.E.; Conte, P. Effect of rf field inhomogeneity and sample restriction on spectral resolution of CP/MAS- 13C NMR spectra of natural organic matter. Open Magn. Reson. J. 2010, 3, 75–83. [Google Scholar] [CrossRef]
- Berns, A.E.; Conte, P. Effect of ramp size and sample spinning speed on CP/MAS- 13C NMR spectra of soil organic matter. Org. Geochem. 2011, 42, 926–935. [Google Scholar] [CrossRef]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Uniform-penalty inversion of multiexponential decay data. J. Magn. Reson. 1998, 132, 65–77. [Google Scholar] [CrossRef]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Uniform-penalty inversion of multiexponential decay data: II. Data spacing, T2 data, systematic data errors, and diagnostics. J. Magn. Reson. 2000, 147, 273–285. [Google Scholar] [CrossRef]
- Borgia, G.C.; Brown, R.J.S.; Fantazzini, P. Examples of marginal resolution of NMR relaxation peaks using UPEN and diagnostics. Magn. Reson. Imaging 2001, 19, 473–475. [Google Scholar] [CrossRef]
- Bortolotti, V.; Brown, R.J.S.; Fantazzini, P.; Landi, G.; Zama, F. Uniform Penalty inversion of two-dimensional NMR relaxation data. Inverse Probl. 2016, 33, 015003–015021. [Google Scholar] [CrossRef]
- Bortolotti, V.; Brown, R.J.S.; Fantazzini, P.; Landi, G.; Zama, F. I2DUPEN: Improved 2DUPEN algorithm for inversion of two-dimensional NMR data. Microporous Mesoporous Mater. 2018, 269, 195–198. [Google Scholar] [CrossRef]
- Conte, P. Applications of fast field cycling NMR relaxometry. Annu. Rep. NMR Spectrosc. 2021, 104, 141–188. [Google Scholar] [CrossRef]
- Anoardo, E.; Galli, G.; Ferrante, G. Fast-Field-Cycling NMR: Applications and Instrumentation. Appl. Magn. Reson. 2001, 20, 365–404. [Google Scholar] [CrossRef]
- Conte, P.; Piccolo, A.; van Lagen, B.; Buurman, P.; de Jager, P.A. Quantitative differences in evaluating soil humic substances by liquid- and solid-state 13C-NMR spectroscopy. Geoderma 1997, 80, 339–352. [Google Scholar] [CrossRef]
- Simpson, A.J.; Kingery, W.L.; Hayes, M.H.B.; Spraul, M.; Humpfer, E.; Dvortsak, P.; Kerssebaum, R.; Godejohann, M.; Hofman, M. Molecular structure and associations of humic substances in the terrestrial environment. Naturwissenschaften 2002, 89, 84–88. [Google Scholar] [CrossRef]
- Hakim, A.; Suzuki, T.; Kobayashi, M. Strength of humic acid aggregates: Effects of divalent cations and solution pH. ACS Omega 2019, 4, 8559−8567. [Google Scholar] [CrossRef]
- Mao, J.; Chen, N.; Cao, X. Characterization of humic substances by advanced solid state NMR spectroscopy: Demonstration of a systematic approach. Org. Geochem. 2011, 42, 891–902. [Google Scholar] [CrossRef]
- Conte, P.; Berns, A.E. Dynamics of cross polarization in solid state nuclear magnetic resonance experiments of amorphous and heterogeneous natural organic substances. Anal. Sci. 2008, 24, 1183–1188. [Google Scholar] [CrossRef]
- Simpson, A.J.; Simpson, M.J. Nuclear Magnetic Resonance Analysis of Natural Organic Matter. In Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems; Senesi, N., Xing, B., Huang, P.M., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2009; pp. 589–650. [Google Scholar]
- Hu, Y.F.; Cheng, K.; He, L.C.; Zhang, X.; Jiang, B.; Jiang, L.; Li, C.G.; Wang, G.; Yang, Y.H.; Liu, M.L. NMR-Based Methods for Protein Analysis. Anal. Chem. 2021, 93, 1866–1879. [Google Scholar] [CrossRef]
- Yao, H.Y.; Wang, J.Q.; Yin, J.Y.; Nie, S.P.; Xie, M.Y. A review of NMR analysis in polysaccharide structure and conformation: Progress, challenge and perspective. Food Res. Int. 2021, 143, 110290–110308. [Google Scholar] [CrossRef]
- Lehmann, J.; Solomon, D.; Kinyangi, J.; Dathe, L.; Wirick, S.; Jacobsen, C. Spatial complexity of soil organic matter forms at nanometre scales. Nat. Geosci. 2008, 1, 238–242. [Google Scholar] [CrossRef]
- Zhou, J.L. Sampling of humic and colloidal phases in liquid samples. In Comprehensive Sampling and Sample Preparation: Analytical Techniques for Scientists; Pawliszyn, J., Ed.; Academic Press: Waltham, MA, USA, 2012; Volume 1, pp. 335–348. [Google Scholar] [CrossRef]
- Goldburg, W.I. Dynamic light scattering. Am. J. Phys. 1999, 67, 1152–1160. [Google Scholar] [CrossRef]
- Braun, J.; Renggli, K.; Razumovitch, J.; Vebert, C. Dynamic light scattering in supramolecular materials chemistry. In Supramolecular Chemistry: From Molecules to Nanomaterials, 1st ed.; Gale, P.A., Steed, J.W., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp. 1–14. [Google Scholar] [CrossRef]
- Hassan, P.A.; Rana, S.; Verma, G. Making sense of Brownian motion: Colloid characterization by dynamic light scattering. Langmuir 2015, 31, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Reid, P.M.; Wilkinson, A.E.; Tipping, E.; Jones, M.N. Aggregation of humic substances in aqueous media as determined by light-scattering methods. Soil Sci. 1991, 42, 259–270. [Google Scholar] [CrossRef]
- Pinheiro, J.P.; Mota, A.M.; d’Oliveira, J.M.R.; Martinho, J.M.G. Dynamic properties of humic matter by dynamic light scattering and voltammetry. Anal. Chim. Acta 1996, 329, 15–24. [Google Scholar] [CrossRef]
- Ren, S.-Z.; Tombàcz, E.; Rice, J.A. Dynamic light scattering from fractals in solution: Application of dynamic scaling theory to humic acid. Phys. Rev. E 1996, 53, 2980–2983. [Google Scholar] [CrossRef]
- Martin, J.E.; Leyvraz, F. Quasielastic-scattering linewidths and relaxation times for surface and mass fractals. Phys. Rev. A 1986, 34, 2346–2350. [Google Scholar] [CrossRef]
- International Humic Substances Society. IHSS. Available online: www.humicsubstances.org (accessed on 29 January 2023).
- Palmer, N.E.; von Wandruszka, R. Dynamic light scattering measurements of particle size development in aqueous humic materials. Fresenius J. Anal. Chem. 2001, 371, 951–954. [Google Scholar] [CrossRef]
- Hosse, M.; Wilkinson, K.J. Determination of electrophoretic mobilities and hydrodynamic radii of three humic substances as a function of pH and ionic Strength. Environ. Sci. Technol. 2001, 35, 4301–4306. [Google Scholar] [CrossRef]
- Avena, M.J.; Vermeer, A.W.P.; Koopal, L.K. Volume and structure of humic acids studied by viscometry pH and electrolyte concentration effects. Colloids Surf. A 1999, 151, 213–224. [Google Scholar] [CrossRef]
- Alvarez-Puebla, R.A.; Garrido, J. Effect of pH on the aggregation of a gray humic acid in colloidal and solid states. Chemosphere 2005, 59, 659–667. [Google Scholar] [CrossRef]
- Baalousha, M.; Motelica-Heino, M.; Coustumer, P.L. Conformation and size of humic substances: Effects of major cation concentration and type, pH, salinity, and residence time. Colloids Surf. A Physicochem. Eng. Asp. 2006, 272, 48–55. [Google Scholar] [CrossRef]
- Verdugo, P.; Santschi, P.H. Polymer dynamics of DOC networks and gel formation in seawater. Deep-Sea Res. II 2010, 57, 1486–1493. [Google Scholar] [CrossRef]
- Verdugo, P. Marine microgels. Annu. Rev. Mar. Sci. 2012, 4, 375–400. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, U.D.; Marković, M.M.; Cupać, S.B.; Tomic, Z.P. Soil humic acid aggregation by dynamic light scattering and laser Doppler electrophoresis. J. Plant Nutr. Soil Sci. 2013, 176, 674–679. [Google Scholar] [CrossRef]
- Angelico, R.; Ceglie, A.; He, J.-Z.; Liu, Y.-R.; Palumbo, G.; Colombo, C. Particle size, charge and colloidal stability of humic acids coprecipitated with ferrihydrite. Chemosphere 2014, 99, 239–247. [Google Scholar] [CrossRef]
- Tarasevich, Y.I.; Tryfonova, M.Y.; Dolenko, S.A.; Aksenenko, E.V. Adsorption-based approach to determine the size and mass of humic acids molecules. Adsorpt. Sci. Technol. 2016, 34, 125–133. [Google Scholar] [CrossRef]
- Dolenko, S.A.; Trifonova, M.Y.; Tarasevich, Y.I. Aqueous solutions of humic acids as self-organizing dissipative systems. J. Water Chem. Technol. 2017, 39, 360–367. [Google Scholar] [CrossRef]
- Klučáková, M.; Kalina, M. Composition, particle size, charge and colloidal stability of pH-fractionated humic acids. J. Soil Sediment 2015, 15, 1900–1908. [Google Scholar] [CrossRef]
- Klučáková, M. Characterization of pH-fractionated humic acids with respect to their dissociation behaviour. Environ. Sci. Pollut. Res. 2016, 23, 7722–7731. [Google Scholar] [CrossRef]
- Klučáková, M.; Věžníková, K. The role of concentration and solvent character in the molecular organization of humic acids. Molecules 2016, 21, 1410–1418. [Google Scholar] [CrossRef]
- Klučáková, M.; Věžníková, K. Micro-organization of humic acids in aqueous solutions. J. Mol. Struct. 2017, 1144, 33–40. [Google Scholar] [CrossRef]
- Klučáková, M. Size and charge evaluation of standard humic and fulvic acids as crucial factors to determine their environmental behavior and impact. Front. Chem. 2018, 6, 235–242. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, R.; Tang, J.; Li, H. Specific ion effect of H+ on variably charged soil colloid aggregation. Pedosphere 2020, 30, 844–852. [Google Scholar] [CrossRef]
- Tian, R.; Liu, X.; Gao, X.; Li, R.; Li, H. Observation of specific ion effects in humus aggregation process. Pedosphere 2021, 31, 736–745. [Google Scholar] [CrossRef]
- Hu, F.; Li, H.; Liu, X.; Li, S.; Ding, W.; Xu, C.; Li, Y.; Zhu, L. Quantitative characterization of non-classic polarization of cations on clay aggregate stability. PLoS ONE 2015, 10, e0122460. [Google Scholar] [CrossRef]
- Meakin, P. Fractal aggregates in geophysics. Rev. Geophys. 1991, 29, 317–354. [Google Scholar] [CrossRef]
- Senesi, N. Aggregation patterns and macromolecular morphology of humic substances: Fractal approach. Soil Sci. 1999, 164, 841–856. [Google Scholar] [CrossRef]
- Chen, S.-H.; Teixeira, J. Structure and fractal dimension of protein-detergent complexes. Phys. Rev. Lett. 1986, 57, 2583–2586. [Google Scholar] [CrossRef]
- Anitas, E.M. Small-Angle Scattering (Neutrons, X-rays, Light) from Complex Systems; Springer Briefs in Physics; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Kawahigashi, M.; Fujitake, N.; Azuma, J.; Takahashi, T.; Kajiwara, K.; Urakawa, H. The shape of humic acid in solution as observed by small-angle X-ray scattering. Soil Sci. Plant Nutr. 1995, 41, 363–366. [Google Scholar] [CrossRef]
- Rice, J.A.; Tombàcz, E.; Malekani, K. Applications of light and X-ray scattering to characterize the fractal properties of soil organic matter. Geoderma 1999, 88, 251–264. [Google Scholar] [CrossRef]
- Mandelbrot, B.B. The Fractal Geometry of Nature; W.H. Freeman: New York, NY, USA, 1982. [Google Scholar]
- Oh, C.; Sorensen, C. The Effect of Overlap between Monomers on the Determination of Fractal Cluster Morphology. J. Colloid Interface Sci. 1997, 193, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Anitas, E.M. Small-angle scattering from fractals: Differentiating between various types of structures. Simmetry 2020, 12, 65–95. [Google Scholar] [CrossRef]
- Rice, J.A. Applications of fractals in the study of humic materials. In Biophysical Chemistry of Fractal Structures in Environmental Systems; Senesi, N., Wilkinson, K.J., Eds.; John Wiley & Sons: Chichester, UK, 2008; Volume 11, pp. 221–238. [Google Scholar]
- Rice, J.A.; Lin, J.S. Fractal nature of humic materials. Environ. Sci. Technol. 1993, 27, 413–414. [Google Scholar] [CrossRef]
- Österberg, R.; Mortensen, K. Fractal dimension of humic acids—A small angle neutron scattering study. Eur. Biophys. J. 1992, 21, 163–167. [Google Scholar] [CrossRef]
- Österberg, R.; Mortensen, K. The growth of fractal humic acids: Cluster correlation and gel formation. Radiat. Environ. Biophys. 1994, 33, 269–276. [Google Scholar] [CrossRef]
- Österberg, R.; Mortensen, K.; Ikai, A. Direct observation of humic acid clusters, a nonequilibrium system with a fractal structure. Naturwissenschaften 1995, 82, 137–139. [Google Scholar] [CrossRef]
- Wagoner, D.B.; Christman, R.F.; Cauchon, G.; Paulson, R. Molar mass and size of Suwannee river natural organic matter using multi-angle laser light scattering. Environ. Sci. Technol. 1997, 31, 937–941. [Google Scholar] [CrossRef]
- Tombàcz, E.; Rice, J.A.; Ren, S.-Z. Fractal structure of polydisperse humic acid particles in solution studied by scattering methods. ACH Models Chem. 1997, 134, 877–888. [Google Scholar]
- Manning, T.J.; Bennett, T.; Milton, D. Aggregation studies of humic acid using multiangle laser light scattering. Sci. Total Environ. 2000, 257, 171–176. [Google Scholar] [CrossRef]
- Myneni, S.C.B.; Brown, J.T.; Martinez, G.A.; Meyer-Ilse, W. Imaging of humic substance macromolecular structures in water and soils. Science 1999, 286, 1335–1337. [Google Scholar] [CrossRef]
- Pranzas, P.K.; Willumeit, R.; Gehrke, R.; Thieme, J.; Knöchel, A. Characterisation of structure and aggregation processes of aquatic humic substances using small-angle scattering and X-ray microscopy. Anal. Bioanal. Chem. 2003, 376, 618–625. [Google Scholar] [CrossRef]
- Diallo, M.S.; Glinka, C.J.; Goddard, W.A.; Johnson J.H., J.H., Jr. Characterization of nanoparticles and colloids in aquatic systems 1. Small angle neutron scattering investigations of Suwannee River fulvic acid aggregates in aqueous solutions. J. Nanoparticle Res. 2005, 7, 435–448. [Google Scholar] [CrossRef]
- Kučerík, J.; Drastík, M.; Zmeškal, O.; Čtvrtníčková, A. Ultrasonic spectroscopy and fractal analysis in the study on progressive aggregation of humic substances in diluted solutions. WSEAS Trans. Environ. Dev. 2009, 5, 705–715. [Google Scholar]
- Taylor, T.M.; Davidson, P.M.; Bruce, B.D.; Weiss, J. Ultrasonic spectroscopy and differential scanning calorimetry of liposomal-encapsulated nisin. J. Agric. Food Chem. 2005, 53, 8722–8728. [Google Scholar] [CrossRef]
- Buckin, V.; Kudryashov, E.; Morrissey, S. High resolution ultrasonic spectroscopy for analysis in biocolloids. Int. Labmate 2002, 27, 23–24. [Google Scholar]
- Sarvazyan, A.P. Ultrasonic velocity of biological compounds. Annu. Rev. Biophys. Biophys. Chem. 1991, 20, 321–342. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Sarvazyan, A.P.; Funck, T.; Cain, C.A.; Breslauer, K.J. Partial Molar Characteristics of Glycine and Alanine in Aqueous-Solutions at High-Pressures Calculated from Ultrasonic Velocity Data. J. Phys. Chem. 1994, 98, 321–328. [Google Scholar] [CrossRef]
- Buckin, V.A. Hydration of nucleic bases in dilute aqueous solutions. Apparent molar adiabatic and isothermal compressibilities, apparent molar volumes and their temperature slopes at 25 degrees C. Biophys. Chem. 1988, 29, 283–292. [Google Scholar] [CrossRef]
- Taulier, N.; Chalikian, T.V. Compressibility of protein transitions. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2002, 1595, 48–70. [Google Scholar] [CrossRef]
- Sarvazyan, A.P. Development of methods of precise ultrasonic measurements in small volumes of liquids. Ultrasonics 1982, 20, 151–154. [Google Scholar] [CrossRef]
- Pavlovskaya, G.; McClements, D.J.; Povey, M.J.W. Ultrasonic investigation of aqueous solutions of a globular protein. Food Hydrocoll. 1992, 6, 253–262. [Google Scholar] [CrossRef]
- Klučáková, M.; Kargerová, A.; Nováčková, K. Conformational changes in humic acids in aqueous solutions. Chem. Pap. 2012, 66, 875–880. [Google Scholar] [CrossRef]
- Řezáčová, V.; Conte, P.; Komendová, R.; Novák, F.; Repková, M.; Kučerík, J. Factors influencing structural heat-induced structural relaxation of dissolved organic matter. Ecotoxicol. Environ. Saf. 2019, 167, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Čtvrtníčková, A.; Drastík, M.; Vlčková, Z.; Kučerík, J. Surface tension of regenerated humic acids salts. Chem. List. 2008, 102, s1142–s1143. [Google Scholar]
- Drastík, M.; Čtvrtníčková, A.; Zmeskal, O.; Kučerík, J. Aggregation of humic and fulvic acids in diluted solutions. In Energy, Environment, Ecosystems, Development and Landscape Architecture; Mastorakis, N., Helmis, C., Papageorgiou, C.D., Bulucea, C.A., Panagopoulos, T., Eds.; WSEAS Press: Corfu, Greece, 2009; pp. 163–168. [Google Scholar]
- David, J.; Šmejkalová, D.; Hudecová, Š.; Zmeškal, O.; von Wandruszka, R.; Gregor, T.; Kučerík, J. The physico-chemical properties and biostimulative activities of humic substances regenerated from lignite. SpringerPlus 2014, 3, 156–171. [Google Scholar] [CrossRef]
- Kučerík, J.; Bursáková, P.; Průšová, A.; Grebíková, L.; Schaumann, G.E. Hydration of humic and fulvic acids studied by DSC. J. Therm. Anal. Calorim. 2012, 110, 451–459. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).