1. Introduction
Human exposure to external chemical carcinogens is a well-defined cancer risk factor that accounts for about 40% of malignancies worldwide [
1]. Chemical carcinogens also include pharmaceuticals and their impurities, as per the International Agency for Research on Cancer Monographs; thus, for occupational and consumer safety protection, screening for drug carcinogenicity is internationally mandated by legal requirements. In the European Union, they are defined by the ICH S2 (R1) (“Genotoxicity testing and data interpretation for pharmaceuticals intended for human use”) [
2] and ICH M3 (R2) (“Non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals”) guidelines [
3] pertaining to drug products as well as by the most recent ICH M7 (R1) (“Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk”) guideline [
4] dealing with drug mutagenic impurities. These regulations remain effective for new drug substances and new drug products during their clinical development, and subsequent applications for marketing, and only in a few circumstances for post-approval submissions of marketed products. Notably, the ICH M7 (R1) guideline was not implemented until 2014. Hence, a considerable number of pharmaceuticals have remained outside its scope, based on their well-established use, leaving their potential carcinogenic impurities unverified. The accidental discovery of mutagenic N-nitrosodimethylamine impurity in well-established valsartan and ranitidine-containing products, followed by their global recall in 2018, ultimately uncovered the insufficiency of the existing procedures, prompting an immediate scientific and institutional response [
5]. As a consequence, worldwide risk assessment and mutagenic impurity profiling of old pharmaceuticals has led to the withdrawal of over 1800 batches of various finished formulations, until now, including sartans, antidiabetics, antihistamines and antibiotics in the United States, only due to mutagenic contaminants [
6]. Even recently, in March 2022, all batches of propranolol extended-release capsules were recalled in Canada for their contamination with mutagenic N-nitroso-propranolol [
7]. This clearly illustrates the huge scale of drug carcinogenic impurities problem, warranting the need for constant safety monitoring. The postulate applies in particular to all pharmaceuticals indicated for chronic use, whose lifetime cumulative doses are the highest.
Mutagenic N-nitroso compounds, besides being potential drug impurities, can also be endogenously formed from N-nitrosable drug precursors treated with nitrite in the strongly acidic environment of gastric juice. Possible sources of nitroso metabolites are amine, amide, cyanamide, guanidine, hydroxylamine, amidine, hydrazine, hydrazide, piperazine and diketopiperazine structures, which are present in a large portion of drugs, making them theoretically nitrosable [
6,
8]. Drug-nitrite interaction products can easily form DNA adducts, either in the nucleus or in the mitochondria, via electrophilic interactions, which translates into their multidirectional carcinogenic potency [
6]. Indeed, in a recent animal models study, reactive nitrosamines caused mutations in both nuclear and mitochondrial DNA, leading to upregulation of the beta-2-adrenergic-receptors-cholinergic-receptor-nicotinic-alpha-7-subunit-dependent nitrosamine canonical signaling, resulting in lung tumor growth. This was mediated by a malfunction of mitochondrial-reactive oxygen species [
9]. Nitrosamines also caused aberrations in the mitochondrial electron transport chain, subunits I, II and IV, consequently affecting transmembrane electric potential and increasing oxidative stress, as an alternative cancerogenic mechanism [
10]. Additionally, nitrosamines can reversively inhibit gap junction intercellular communication leading to cancer, recently confirmed by Tschernig [
11]. Therefore, the World Health Organization experts board developed “Nitrosation Assay Procedure” (NAP test) to screen for the liability of pharmaceuticals to endogenous nitrosation. The conditions of the NAP test involve a fourfold excess of nitrite in an acidic solution which mimics in vivo nitrosation in the stomach. Unfortunately, this procedure has never been included in registration dossiers. Hence, the potential for endogenous nitrosation of pharmaceuticals remains unknown unless verified by scientific research [
6]. It is therefore evident that the area of carcinogenic drug impurities and metabolites safety profiling still remains insufficiently supported by experimental data, which obviously poses a safety concern and explains the need for relevant toxicological studies, mainly for well-established pharmaceuticals of key clinical relevance and widespread use.
Ramipril (RAM) is a dicarboxylate-containing angiotensin-converting enzyme inhibitor (ACE-I) of major clinical importance due to its indications for long-term treatment of severe cardiovascular conditions [
12,
13]. It was patented in 1981 and approved in 1989 [
14]. Its impurity assessment was not required by the ICH M7 (R1) guideline during the post-approval period, and therefore it is hardly to be expected from the industry. In 2019, its total number of prescriptions in the United States was nearly 4 million, while the estimated number of treated individuals was 854,000, indicating its broad patient exposure and significant impact on public health [
15]. Concurrently, there has been a fair number of epidemiological studies demonstrating clear associations between ACE-Is (including RAM) and the development of malignancy, such as multiple myeloma, breast and lung cancer [
16,
17,
18,
19,
20,
21,
22]. Furthermore, in lung cancer, high cumulative ACE-Is doses were associated with modestly increased odds of disease, while lower doses only showed neutral associations. The mechanism behind these effects has never been elucidated [
20]. Based on the above-discussed insufficiency of safety profiling procedures in the group of well-established pharmaceuticals, a possible interpretation of these observations could be the carcinogenic impact of drug degradation impurities or N-nitroso metabolites, which have evaded the regulatory safety screening prior to drug approval, as was the case with valsartan and ranitidine. Such cause-effect relationships would be, however, hard to observe in the setting of observational case-control studies due to distant endpoints and multiple distracting factors. Therefore, the suggested correlation between RAM stability and its oncological safety must be verified by means of experimental methods.
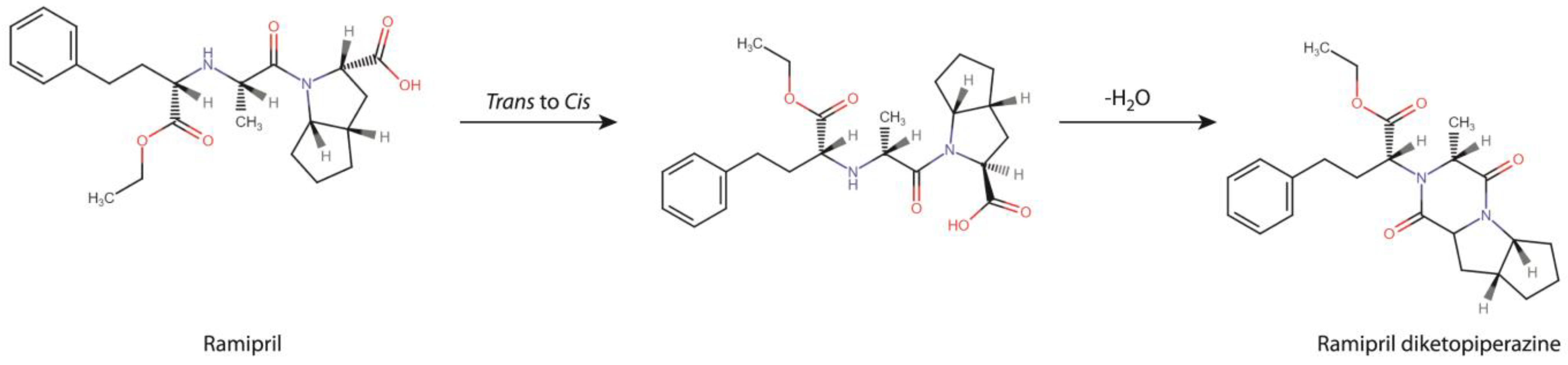
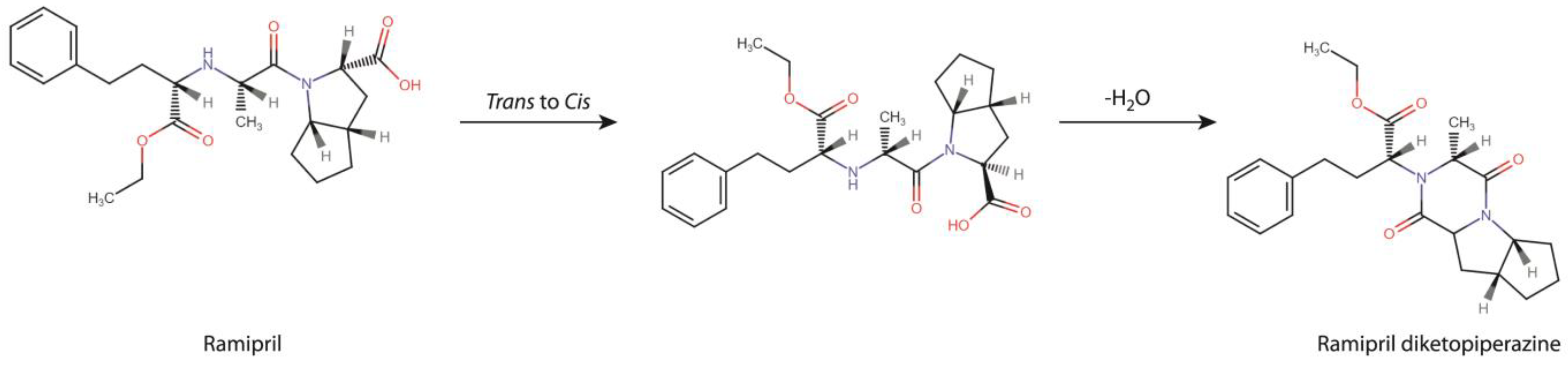
According to our previous report, RAM is chemically unstable as its degradation is rapid, and within the pharmacological class of ACE-Is it is the most vulnerable to temperature and humidity changes. Its degradation impurities formed under humid conditions include: biologically-active ramiprilat and a diketopiperazine derivative (DKP) [
15]. The available stability data for RAM are, however, incomplete. Its mechanism of degradation under dry air still remains unknown. Probably, the only degradation impurity formed under such conditions is DKP, as with other structurally-related ACE-Is [
23,
24]. This degradation pathway, in turn, is highly unfavorable from the clinical point of view, since DKP is pharmacologically inactive against RAM biological targets, its presence is of no clinical benefit and its toxicity to humans is unknown [
15]. DKP also contains a diketopiperazine structural alert that is potentially susceptible to N-nitrosation in vivo, with further mutagenic hazard to patients, as discussed above [
8]. Thus, we hypothesized that DKP formation in RAM dosage forms could be a possible reason for the reported increased cancer incidence, associated with ACE-I use. To verify this, we decided to perform this multistage study correlating the stability and safety features of RAM.
Taking all the above into consideration, the aim of our research was to check whether: (1) the degradation of RAM under dry air leads to the formation of DKP impurity; (2) RAM degradation impurity formed under dry air exerts a direct carcinogenic, genotoxic or mutagenic effect; (3) RAM degradation impurity formed under dry air exerts an indirect mutagenic effect by the formation of N-nitroso compounds in vivo. Our research plan was: (1) the detailed description of the kinetic mechanism of RAM degradation under dry air by appropriate qualitative and quantitative parameters; (2) the identification of the emerging degradation products; (3) a structure-based assessment of the identified impurity’s cancer risk by in silico QSAR model, employing three endpoints: carcinogenicity, genotoxicity and mutagenicity; (4) the verification of the QSAR simulation for genotoxicity by in vitro micronucleus assay; (5) the verification of the QSAR simulation for mutagenicity by Ames test; (6) the verification of the vulnerability of the investigated impurity to form mutagenic N-nitroso metabolites by NAP test and subsequent Ames test.
Our intention was to check various potential mechanisms of carcinogenicity, involving direct genotoxicity, direct mutagenicity or indirect mutagenicity by forming mutagenic N-nitroso metabolites. We designed our studies so as to mimic in vivo conditions and reflect the environment of human body. Accordingly, we assumed that genotoxic agents act via threshold mechanisms. Thus, for direct genotoxicity assessment, by in vitro micronucleus assay, we employed two experimental series of a studied compound: a screening series at high concentrations to detect the exact mechanism of genotoxicity with high sensitivity, and the series with typical blood concentrations to check the real-life impact of RAM degradant after standard dosing. On the other hand, for mutagenicity assessment by Ames test, we assumed that mutagens, unlike genotoxic agents, act via direct modification of DNA nucleotide sequence at any concentration, so there is no safe level of exposure to such substances. For this reason, for the evaluation of mutagenic activity of RAM degradant and its nitroso-metabolite, only one series at high concentrations was involved, which increased the sensitivity of our assay [
4].
2. Materials and Methods
2.1. Kinetic Studies
Pure RAM (100%) was purchased from Rolabo (Zaragoza, Spain, batch n◦: 11.PT24.01.02). HPLC-grade methanol, acetonitrile, formaldehyde and potassium dihydrogen phosphate were purchased from Merck, Darmstadt, Germany.
Samples of RAM were weighted on analytical scale Satorius BP 2105. They were heated in thermal chambers WAMED KBC–125 W with automatic temperature and RH control (Wamed, Warsaw, Poland).
The kinetic analysis was performed using a Shimadzu liquid chromatograph (Shimadzu Corporation, Kyoto, Japan) consisting of a Rheodyne 7125, 100 µL fixed loop injector, a UV-VIS SPO-6AV detector, an LC-6A pump and a C-RGA chromatopac integrator. The following operating conditions were applied: mobile phase consisting of acetonitrile–aqueous phosphate buffer, pH 2.0; 0.035 mol/L (65:35 v/v) and stationary phase consisting of LiChrospher 100 RP-18 (size 5 µm) 250 mm × 4 mm I.D column. The chromatographic separation was isocratically performed at ambient temperature at a flow rate of 1.2 mL/min with the detector wavelength set at 215 nm. The injection volume was 20 µL. The mobile phase had been filtered through a filter (0.22 µm) and degassed by ultrasound prior to use.
The employed HPLC method was revalidated in order to confirm its applicability for the assessment of the RAM stability profile under dry air conditions. The following validation parameters were determined: selectivity, linearity, sensitivity, precision and accuracy according to procedures described in our previous publication [
15]. They were in agreement with ICH guideline Validation of analytical procedures: Text and methodology Q2 (R1) [
25]. For that purpose, a calibration curve was constructed; limit of detection (DL), limit of quantification (QL), coefficient of variation (CV) and recovery were calculated. The procedures for revalidation are provided in the
Supplementary Material (Supplement S1: HPLC validation procedures).
The kinetic evaluation of RAM degradation was then conducted under stress conditions of elevated temperature, ranging from 353 K to 373 K and dry air (RH 0%). Samples of pure RAM (0.010 g) were analytically weighed into glass vials, placed into a sand bath and heated for different time intervals so as to induce degradation. Then, respective samples were dissolved in methanol to a total volume of 25.0 mL and filtered. The chromatograms were achieved. Basing on the chromatographic peaks areas, the contents of RAM and its degradation product were calculated (c) from the calibration curve. These results were plotted versus time (t) so as to construct kinetic curves: c = f(t) and kinetic equations. The kinetic model of the degradation was established by fitting the experimental data to the following theoretical equations: nucleation (power-law, Avrami–Erofeev, Prout–Tompkins), geometrical contraction (contracting area, contracting volume), diffusion (1D diffusion, 2D diffusion, 3D diffusion) and reaction-order (zero-order, first-order, second-order and third-order). The best fit, defined by the highest correlation coefficient, was identified. The degradation rate constants (k) were then calculated using least square method. Furthermore, thermodynamic parameters were evaluated based on the procedure described in our previous publication [
15]. The relevant equations for the kinetic study procedures and thermodynamic calculations [
26] are provided in
Supplement S2: Thermodynamic parameters calculation.
2.2. Identification of RAM Degradation Product
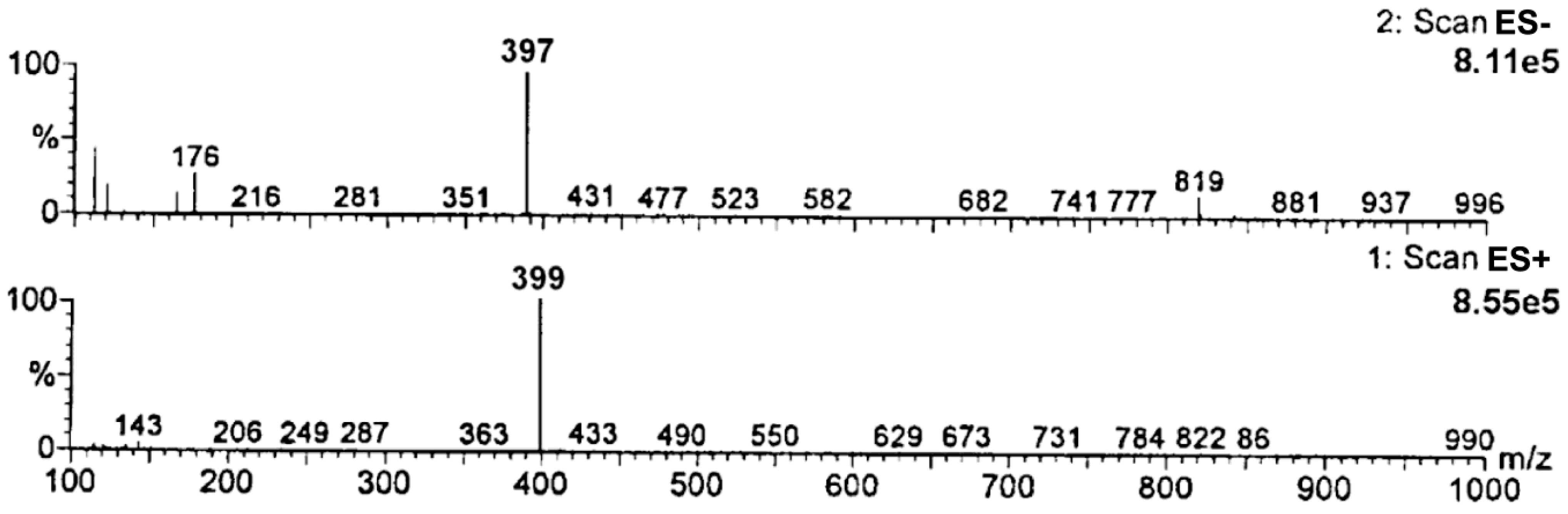
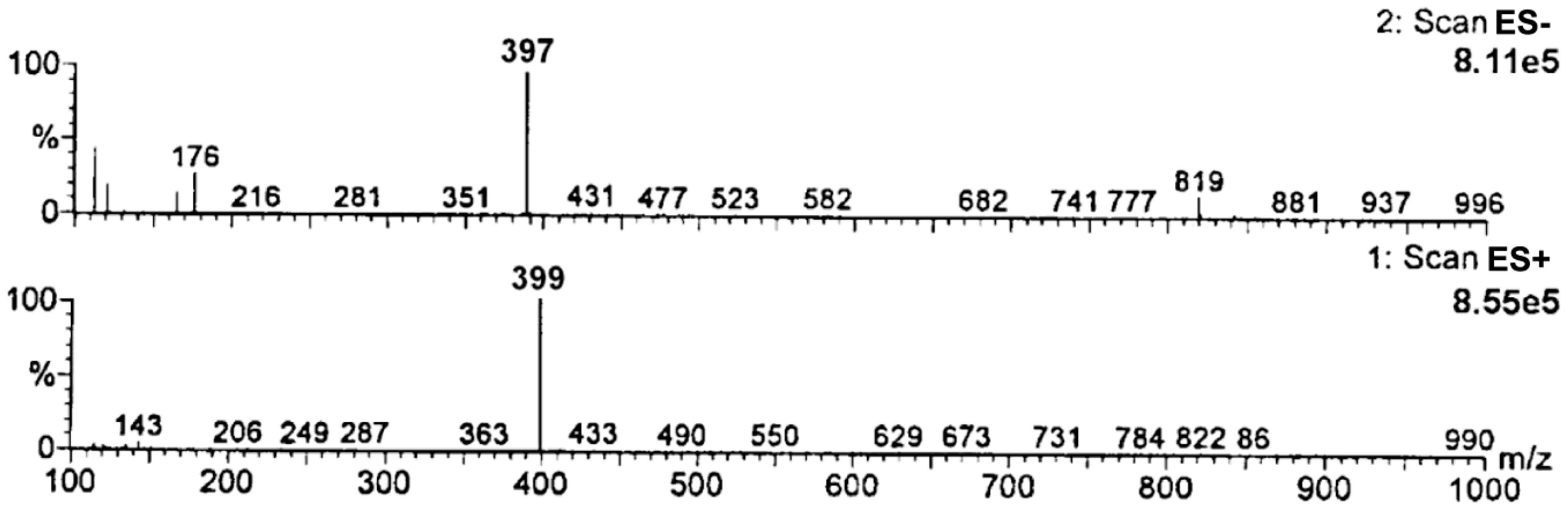
To confirm the identity of the emerging degradation product, a sample of fully degraded RAM under the following experimental conditions: t = 90 h; 373 K; RH 0%, was used. The product of RAM degradation was identified using a Liquid Chromatography/Electrospray Ionization-Mass Spectrometry System HPLC-MS (Waters ZQ with photodiode array detector (Waters 996 ZQ, Waters Corporation, Milford, MA, USA). LC separations were made on Hypersil MOS column, 5 µm particle size, 250 mm × 4 mm at 35 °C (308 K). The mobile phase consisted of methanol-water-formaldehyde (49:50:1 v/v/v). Its flow rate was 0.5 mL/min. The mobile phase was filtered through a 0.22 µm filter and degassed by ultrasound. The injection volume was 100 μL. The recorded mass range was from m/z 100 to 1000. Soft ionization technique—electrospray (ESI) was applied and the mass spectrometer was run in a negative (ES−) and positive (ES+) ionization mode. The spectral range was 200–400 nm. A molecular ion was identified. Its mass-to-charge ratio (m/z) was compared to the molecular mass of the predicted degradation impurity in order to confirm its identity.
2.3. In Silico Prediction of Genotoxicity, Mutagenicity and Carcinogenicity
The kinetic study was followed by an in silico safety evaluation of the identified RAM degradation impurity in order to set the scope of further toxicological research in vitro. An open access software VEGA-GUI version 1.2.0 from
www.vegahub.eu (accessed on 4 January 2023) was used for carcinogenicity, mutagenicity and genotoxicity prediction. The structure of the IMD derivative (a structural analogue of RAM) was tested in parallel in order to make a comparative analysis.
The employed VEGA platform accessed QSAR models, which correlated its internal dataset of structurally-related chemicals with target molecules. In the version 1.2.0, it has four models for mutagenicity, ten models for carcinogenicity, one model for chromosomal aberration and two for micronucleus activity. The models available for the mutagenicity endpoint (ISS, SARpy, CAESAR, Mutagenicity Read-Across/KNN) were built based on experimental data derived from in vitro studies (e.g., the Ames Test in
Salmonella typhimurium strains), while the carcinogenicity models (ISS, ISSCAN-CGX, CAESAR, ANTARES) used data from in vivo studies in different species, mainly mice and rats. All the VEGA models differ in their analytical approach (rule-based expert system, correlation between relevant fragments with toxicological endpoint, regression models, hybrid models), all of which had been validated prior to their commercialisation and none of which was considered inferior [
27]. Thus, in this study, all of them were used to increase the predictive power of the stimulation.
The identifiers of the tested molecules in SMILES format were inserted into the task list of the analysis tool. Then, the QSAR models for toxicity endpoints were selected and the prediction reports for each model were computed. The VEGA output was exported in .pdf file with ‘yes’ or ‘no’ prediction relative to each endpoint. The results were supported by the information on the reliability of prediction, measured by VEGA by the use of an in-built applicability domain evaluation tool. It was expressed as the Applicability Domain Index (ADI), which was computed to compare the tested molecules with the model training set. For ADI > 0.85, the prediction was considered strong. For ADI < 0.65, the prediction was considered weak. The values in between were considered as moderate. This parameter was automatically analyzed by VEGA and it was integrated into a final wording result expressed as “low reliability”, “moderate reliability”, “good reliability” [
27]. To compare the results obtained for different QSAR models, the scoring approach developed by Glück et al. [
28] was employed. It translated VEGA wording results into numeric values (endpoint scores, ES) ranging from 0 to 1 where “1” represents high toxicity and “0” is low toxicity with respect to model endpoint. Then, the average ES
av was calculated and expressed as the arithmetical mean of the ES values for respective endpoints. To interpret these data, the cut-off values were adopted as follows: ES
av > 0.66 for true-toxicity claim; ES
av < 0.33 for true non-toxicity claim. Scores between 0.33 and 0.66 were considered as inconclusive.
2.4. Micronucleus Assay Procesdure
The degradation impurity of RAM was then subjected to in vitro micronucleus assay based on the results of the computed in silico simulations. To that end, DMEM (Dulbecco’s Modified Eagle’s Medium), FBS (Fetal Bovine Serum), L-glutamine, amphotericin B, penicillin-streptomycin, Aroclor 1254-induced male Sprague Dawley rat liver S9, mitomycin C, cyclophosphamide, colchicine and cytochalasin B were used. They were purchased from Sigma-Aldrich (St. Louis, MO, USA). HCS CellMask™ Green Stain and Hoechst 33342 were supplied by Thermo Fisher Scientific (Waltham, MA, USA).
L929 cell line (ECACC 85011425, mouse C3H/An connective tissue) was cultured in DMEM supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 2.5 µg/mL amphotericin B, 100 U/mL penicillin and 100 µg/mL streptomycin. Cells were maintained at 37 °C (310 K) and 5% CO2 in a humidified atmosphere. Cell cultures were passaged before they reached 90% confluency to maintain exponential growth. They were regularly checked for mycoplasma contamination. For the cytokinesis-block in micronucleus assay 24 h before treatment, cells were seeded into 6-well plates at a density of 2.2 × 105 cells/well and incubated at 37 °C (310 K), 5% CO2 in a humidified atmosphere. After the incubation time, cells were treated with tested compounds in the same 6-well plates.
The in vitro micronucleus test was performed in accordance with the guideline 487: In Vitro Mammalian Cell Micronucleus Test, from the Organization for Economic Co-operation and Development (OECD, 2016) [
29]. In this study, two experimental series were carried out. The first one comprised three concentrations tested in duplicate: 2, 0.67, 0.22 mg/mL, for screening purposes. The concentrations and the number of replicates were selected basing on the recommendations of guideline 487 from OECD. In this setting, for each concentration, three experimental conditions were applied: short treatment with metabolic activation (6 h) and both, short (6 h) and long treatments (30 h) without metabolic activation, to investigate potential clastogenic and aneugenic activity. Metabolic activation was applied to detect genotoxic agents formed secondary to modifications by liver cytochromes. Hence, for each condition, different medium was required. For short treatment with metabolic activation, a culture medium with a 2% S9 mix to achieve metabolic activation was used. For short treatment without metabolic activation, only culture medium was necessary. For long treatment, medium with 2 µg/mL cytochalasin B as a cytokinesis inhibitor was applied.
In the second experimental series, three physiological concentrations of the tested compound were investigated. This experiment was designed in order to mimic a clinical scenario and check the activity of RAM degradation impurity after the administration of a standard RAM dose to a patient. The concentrations were the following: 1, 10 and 100 nM (which correspond to: 3.98 × 10
−7 mg/mL, 3.98 × 10
−6 mg/mL, 3.98 × 10
−5 mg/mL). They reflected blood levels of DKP, measured in blood samples derived from healthy individuals which were subjected to pharmacokinetic study [
30]. Only the extended treatment conditions (30 h of treatment in the culture medium with cytochalasin B) without metabolic activation were applied, relying on the results obtained in the screening series.
Stock solutions were prepared in DMSO, and appropriate solvent vehicle controls were also contained in the assay. The concentration of DMSO did not exceed 1% of the final culture volume. Additionally, positive controls were used, including: mitomycin C (0.25 µg/mL), cyclophosphamide (0.25 μM) and colchicine (0.025 µg/mL), depending on the type of exposure. The type and the concentration of the controls were selected based on the recommendations of the OECD guideline 487 [
29]. For cells with short treatment, after 6 h of incubation, medium was gently aspired, cells were washed with PBS (Phosphate-Buffered Saline) and medium with 2 µg/mL cytochalasin B was added. Following 24-h incubation, cells were fixed with a 1:1 ethanol-methanol mix 2 mL per well. Fixed cells were co-stained with HCS CellMask™ Green Stain (300 µL per well, conc. 2 µg/mL) and Hoechst 33342 (300 µL per well, conc. 5 µg/mL) according to the manufacturer’s protocol to reveal nuclei, micronuclei and surrounding cytoplasm. At least 500 cells were scored per one experimental condition. Among them, 100 binucleated cells were counted (if possible). The proportion of mono, bi- and multinucleated cells was also obtained to calculate the replication index (RI) and cytokinesis-block proliferation index (CBPI) to evaluate potential cytotoxicity, according to the formula below.
where T = treated and C = control.
where T = treated and C = control.
Then, the percentage of micronuclei found in binucleated cells relative to the control was established (% MN/BC). Concurrent negative controls (solvents only) were used for each experimental setting as indicators for the background frequency of micronuclei. Positive controls were used to demonstrate the ability to induce micronuclei formation. The percentage of induced micronuclei was assessed for binucleated cells.
2.5. NAP-Test Procedure
To verify the suspected indirect mutagenic activity of RAM degradation impurity, its potential to form mutagenic N-nitroso compounds in vivo was investigated using the NAP test.
Pepsin [n
o: 9001-75-6] was bought from Pol-Aura, Poland. Analytical grade hydrochloric acid 1.0 mol/L, sodium nitrite (NaNO
2) and ammonium sulfamate (NH
4SO
3NH
2) were obtained from Merck, Darmstadt, Germany. Ultrapure water was used. Simulated gastric juice (SGJ) (pH 1.2) was prepared as per the Polish Pharmacopoeia (ed. XII, 2020) by dissolving 2 g of sodium chloride and 3.2 g of pepsin in 80 mL HCl (1 mol/L) and sufficient water to make 1000 mL [
31].
The sample of RAM degradation product was dissolved in DMSO, adjusted to pH 1.2 using SGJ, and then treated with sodium nitrite (in a molar ration of 10:40 mM). Then, the pH was readjusted to 1.2. The obtained nitrosation mixture was incubated at a temperature of 37 °C (310 K) for 60 min in the dark on a shaker. The solvent with nitrite treatment was incubated under similar conditions as a negative control for the mutagenicity assay. The reaction was subsequently stopped by the addition of ammonium sulfamate (molar ratio NaNO2 vs. NH4SO3NO2 was 4:4). The obtained reaction mixture was subjected to mutagenicity evaluation. The concentrations of the tested nitrosation product were expressed as concentrations of the parent compound prior to nitrosation, i.e., 4.5, 2.25, 1.125, 0.56, 0.28 mg/dL. This evidenced the level of degradation impurity exposure that may pose a safety concern.
2.6. Mutagenicity Assay-Bacterial Reverse Mutation Test Procedure
Ames test was performed to verify the results of QSAR assessment for mutagenicity and to check the mutagenic activity of the N-nitroso metabolite of the tested compound. This procedure was chosen since it is the first choice option for mutagenicity assessment of pharmaceuticals according to the ICH M7 (R1) guideline. A commercial Ames MPF 98/100 microplate format mutagenicity assay kit (Xenometrix, Allschwil, Switzerland) with Salmonella typhimurium strains TA98 (containing frameshifts mutation hisD3052, rfa uvrB, pKM101) and TA100 (containing base-pair substitution mutation hisG46, rfa uvrB, pKM101) was used. It contained 2-nitrofluorene, 4-nitroquinolone-N-oxide (4-NQO), 2-aminoanthracene as positive controls, Aroclor 1254-induced rat liver fraction S9 as the activation system, sterile ampicillin (50 mg/mL), ready-to-use growth medium, exposure medium and indicator medium. For preparing the S9 mix, a ready-to-use kit from Xenometrix (Switzerland) containing Buffer Salts solution (phosphate buffer, MgCl2, KCl, NADP solution, G-6-P solution) was employed. The tested samples were dissolved in sterile DMSO (Merck, Darmstadt, Germany).
A completely degraded RAM sample (RAM content ~0%) and the nitrosation mixture of the RAM degradation product (
c = 112.6 mg/mL) were subjected to mutagenicity analysis. The degradation product was dissolved in DMSO to obtain a concentration of 125.0 mg/mL. Then, the test procedure provided by Ames MPF Instruction for use was followed [
32,
33]. The
Salmonella typhimurium strains TA98 and TA100 were grown overnight (15 h) in growth medium at 37 °C (310 K) on a shaker set at 250 rpm. The exposure concentrations were selected based on the pre-screening procedure for cytotoxicity and solubility using the TA98 strain, as per kit instructions [
33]. The
Salmonella typhimurium strains TA98 and TA100 were then exposed to the tested substances in the presence and absence of rat liver S9 fraction. In the experiments without a metabolic activation system, the following positive controls were used: 2-nitrofluorene (2.0 µg/mL) for TA98 and 4-nitroquinolone-N-oxide (0.1 µg/mL) for TA100. In the experiments with a metabolic activation system, 2-aminoanthracene was used as a positive control at a concentration of 1.0 µg/mL for TA98 and 2.5 µg/mL for TA100. For the experiment with the degradation product, the baseline concentration was 125.0 mg/mL. For the experiments with the nitrosation mixture, the baseline concentration of the parent compound was 112.6 mg/mL. A serial ½-log dilution was performed on a 96-well plate. To that end, in the experiments without metabolic activation, the standard solutions were diluted with histidine-rich exposure medium. In the experiments with a metabolic activation system, the standard solutions were diluted with histidine-rich exposure medium and microsomal S9 fraction mix. Six stock concentrations were achieved. Approximately 10
7 bacteria for each strain were treated in triplicate with the following test concentrations: 5.0 mg/mL, 2.5 mg/mL, 1.25 mg/mL, 0.63 mg/mL, 0.31 mg/mL, 0.16 mg/mL for pure degradation product and 4.5 mg/mL, 2.25 mg/mL, 1.125 mg/mL, 0.56 mg/mL, 0.28 mg/mL, 0.14 mg/mL for the nitrosation mixture, in a 24-well plate. Treated bacteria were incubated at 37 °C (310 K) on a shaker (250 rpm) for 90 min. Then, pH indicator medium without histidine was used to dilute the exposure cultures. They were aliquoted into 48 wells of a 384-well plate (50 µL per well). This was followed by 48 h of incubation at 37 °C (310 K) to allow revertant bacteria to grow. The indicator medium contained a pH indicator dye, which changed colour from purple to yellow due to bacterial metabolism, if the mutation occurred. The scoring of positive wells (revernants) was visually performed. Mutagenicity was confirmed if at least a two-fold increase over the baseline was observed (FIB > 2). The baseline was defined as the mean number of positive wells in the negative controls plus one standard deviation (SD).
For statistical analysis of the obtained results, the cumulative binominal test was applied and binominal B-value was calculated, as recommended by Xenometrix, Switzerland.
4. Discussion
These studies were designed and performed as a follow-up to the existing reports that suggested a possible carcinogenic effect of ACE-Is on humans, caused by the unknown mechanism. In this context, we intended to verify whether these actions could have been associated with the formation of toxic degradation impurities in the dosage forms of RAM, routinely administered to patients. We considered so, because the correlation between ACE-Is stability and toxicity, despite the existing theoretical rationale, still remains unexplored. We selected RAM as a model molecule due to its widespread clinical use, poor stability profile and incomplete stability information. Hence, to fulfill our research plan, we had to supplement the lack of data on RAM degradation kinetics under dry air and to then perform a well-structured toxicological assessment.
The first step of our project aimed at providing a detailed description of RAM degradation behavior under the conditions of dry air, combined with the identification of the emerging degradation products. For that purpose, we developed and validated a HPLC stability-indicating protocol, which can now be also successfully applied for similar assays by manufacturers. Using this method, we managed to establish the kinetic parameters of RAM degradation under dry air and then to compare them with the corresponding ones, previously reported for RAM at humid conditions (RH 76%). Here, we found that, at RH0%, the degradation of RAM follows first-order kinetics, meaning that its rate solely depends on the concentration of the parent compound. First-order reactions are preferred for drugs, due to their predictable and easy-to-control progress. On heating, the degradation accelerates, as evidenced by increasing k values with temperature, yet its kinetic order remains unaltered (
Table 1). For RAM under humid conditions, similar observations have been previously made. However, with the lack of environmental moisture, the rate of RAM degradation and its thermodynamics changed, as well as its degradation products’ yield when compared to RH 76%. In detail, at RH 0%, the reaction progressed more rapidly when compared to RH 76%, as shown a by higher degradation rate constant and shorter half-life. Simultaneously, the E
a of RAM degradation under dry air was increased relative to that reported for humid conditions. This means that, under dry air, the initiation of RAM degradation requires a higher energy input. Finally, the RAM degradation pathway was changed, as in the present study, it was found to follow a single intramolecular cyclization with the formation of DKP. On the contrary, the degradation of RAM in the presence of moisture involved two parallel reactions, i.e., hydrolysis and cyclization, with the former one being dominant. Hence, unlike dry air conditions, under humid conditions, the major degradation impurity was RAM diacid. The main reason for these variations between dry and humid environments is the obvious lack of water molecules to participate in hydrolytic bond cleavage in the environment of dry air. Thus, at RH0%, intramolecular cyclization is the only possible degradation pathway. Under humid conditions, in turn, although cyclization actually occurs, it is very limited in scope. This is probably caused by the fact that it requires a higher E
a than hydrolysis. Hence, it is not favored. As a consequence, in the presence of moisture, DKP is only minimally formed, while under dry air its yield equals 100%, as shown in our experiments (
Figure 1C). Furthermore, despite a high energy barrier between substrates and the activated complex under dry air (high E
a), once initiated RAM cyclization proceeds very fast. Probably, this is caused by the two-stage nature of this process, as shown in
Figure 5. Firstly, the adoption of an appropriate molecular conformation by RAM occurs. The second stage is deprotonation of the reacting amine, the addition of neutral nitrogen to the carbonyl of the neighboring carboxylic acid to form a tetrahedral intermediate, the escape of water molecules and a final new bond formation. Here, the rate-limiting step of this process is the
cis/
trans transformation of RAM, associated with multiple bond rotations and a consequent high energy consumption, defined by high E
a (
Table 1, ΔE
a = 174.12 kJ/mol). The subsequent water loss is thermodynamically favorable (as supported by positive ΔS,
Table 1); hence, its rapid progression translates into a high value of the reaction rate constant. At RH 76%, no conformational changes are necessary for water molecules to access the ester bond in RAM; thus, the E
a for RAM hydrolysis under humid conditions is relatively low. Our findings are significant from a manufacturing point of view. Employing dry formulation methods would lead to compromised RAM stability, secondary to dry air degradation, which would be faster than that at humid conditions. Furthermore, the impurity profile would be affected, with DKP becoming a major degradant instead of RAM diacid. As a result, on heating and dry-processing, RAM would rapidly cyclize to DKP with all downstream consequences on its clinical and toxicological performance. Therefore, the application of dry procedures for RAM in the industry should be avoided.
In the next stage of this study, we decided to assess the impact of the RAM degradation mechanism on its safety. We only focused our interest on DKP since the toxicological data for both RAM and RAM diacid are available in the registration dossiers of commercially available RAM dosage forms. Based on the literature background, which suggested a possible carcinogenic activity of ACE-Is, we performed a preliminary in silico QSAR simulation assessing various oncologic endpoints, i.e., the general carcinogenicity, mutagenicity and genotoxicity of DKP. Mutagenicity refers to the capacity of chemicals to cause changes in DNA sequences, leading to mutations, while genotoxicity causes damage to genomes, i.e., DNA or chromosomes. The genotoxic agents that cause structural chromosomal aberrations are clastogens, while those that cause numerical chromosomal aberrations are aneugens. The main practical difference between mutagenicity and genotoxicity is that mutagens have no threshold level and any exposure to them is hazardous. Thus, they are not allowed in final dosage forms. Genotoxic agents, in turn, act via threshold mechanisms and their low exposure is not always associated with cancer outcome. Thus, their safe concentration must be established and maintained in drug formulations. The results obtained from our QSAR simulations allowed us to classify DKP as a non-mutagen, as the calculated endpoint score (ESav = 0.2) fell within the non-mutagenicity criteria. The reliability of this prediction was high. Despite this, the adopted in silico model suggested other mechanisms of toxicity, i.e., the carcinogenic and genotoxic activity of DKP, but these predictions were not sufficiently reliable (ESav = 0.5 and 0.6, respectively). On this basis, we assumed that DKP is a potential carcinogen that acts via a mechanism unrelated to direct DNA damage (non-mutagen), probably via chromosome damage (genotoxic agent). However, due to the insufficient reliability of the QSAR simulation for the genotoxicity endpoint, follow-up experiments were necessary, either by in vitro micronucleus or by chromosome aberration assay.
The in vitro mammalian cell micronucleus test for the genotoxicity assessment of DKP was selected as a follow-up to our QSAR simulations. The study was designed so as to screen for both aneugenic (aneuploidy-inducing) or clastogenic (chromosome-damaging) activity of the investigated compound. To that end, different treatment modes were applied (a short one for clastogens and an extended one for aneugens). Because genotoxic substances usually have a threshold mechanism, two experimental series of concentrations were conducted. The first series involved high concentrations of DKP, and it was employed to screen for the exact mechanism of genotoxicity. The second series covered physiological concentrations of DKP that corresponded to the level ordinarily present in the blood of patients treated with RAM due to cardiological indications. The second series enabled us to show the real-life activity of DKP after standard RAM dosing.
Firstly, the cytotoxicity of DKP was established, as this effect is a known confounding factor for micronuclei scoring. Notably, under the conditions of our test, the investigated degradation impurity exhibited a significant cytotoxic activity, exceeding 55.5%, as shown in
Table 4, especially in the extended treatment setting in the screening series. Hence, only one screening concentration (0.22 mg/mL) was subjected to micronuclei scoring. For this concentration, in both short treatments, the percentage of micronuclei relative to the control was not increased, suggesting no clastogenic potential of DKP (
p > 0.05,
Table 4). On the contrary, based on the statistically significant, three-fold increase in micronuclei formation observed in the extended treatment mode, DKP was suggested to act as an aneugen (% MN/BMN = 33.33,
p = 0.0184). This means that DKP could interact with proteins involved in the segregation of chromosomes during mitotic cell division, rather than DNA, leading to chromosome loss via micronuclei formation. Notably, it is typical for aneugenic substances that their effect only occurs in a narrow range of concentrations with a sublinear dose-response relationship; thus, their threshold dose can be estimated, as suggested by Lynch et al. [
34]. As a consequence, low doses of aneugens induce zero effect, which was actually evidenced for DKP in our study, as shown in
Table 4; at its physiological (micromolar) concentrations, the micronuclei were not formed (
p < 0.05). This means that standard dosing of RAM should not pose a carcinogenic risk in humans with respect to the suspected genotoxicity of DKP. However, the threshold level of DKP, above which the aneugenic effect is manifested, remains unknown. It falls between the tested series, and it should be established, preferably in follow-up in vivo studies; for example, using an in vivo micronucleus assay. This knowledge is necessary to set the safe level of human exposure to this impurity. Furthermore, the observed significant cytotoxicity of DKP is also representative of a typical aneugen [
34]. In fact, high doses of aneugens cause severe genomic imbalance and launch cell death mechanisms, leading to cell elimination, as was the case in our study. The high cytotoxic potency of DKP can be also explained by its structural features. Its changeable chiral and rigid skeleton favors its easier binding to molecular targets; for example, Aurora kinases or tubulin, both of which participate in chemical-induced aneugenicity in vitro [
35]. Interestingly, this property of other DKP analogs, i.e., unsaturated 2,5-diketopiperazine derivatives, was examined in a number of human tumor cell lines, in which a 2,5-diketopiperazine ring was set as an optimal scaffold for exploring anticancer drug candidates [
36]. Nonetheless, in the context of our studies, it must be emphasized that aneuploidy caused by aneugens is an important, but not independent, contributor to cancer development [
37]. For this reason, a direct genotoxic/aneugenic activity of excessive doses of DKP combined with other carcinogenic factors is necessary to induce cancer in humans. Consequently, we further investigated other mechanisms of carcinogenicity of DKP; i.e., its mutagenic activity and the mutagenic activity of its N-nitrsoso metabolite. Obviously, our conclusions need confirmatory, in vivo experiments because the cytotoxicity of DKP could have disturbed the micronuclei scoring in our studies, while our QSAR model predicting DKP carcinogenicity/genotoxicity was not sufficiently reliable to provide unquestionable conclusions. In addition to this, no other DKP derivatives of ACE-Is had been subjected to in vitro micronucleus assay before, thus no comparative analysis was possible. Despite these limitations, we believe that the evidence from our investigations is sufficient to set follow-up in vivo experiments, without further confirmatory in vitro protocols [
38]. Therefore, we strongly suggest that the aneugenicity threshold of DKP and its impact on living organisms should to be evaluated under in vivo conditions by marketing authorization holders in order to validate the acceptable limits of this impurity in formulated drugs.
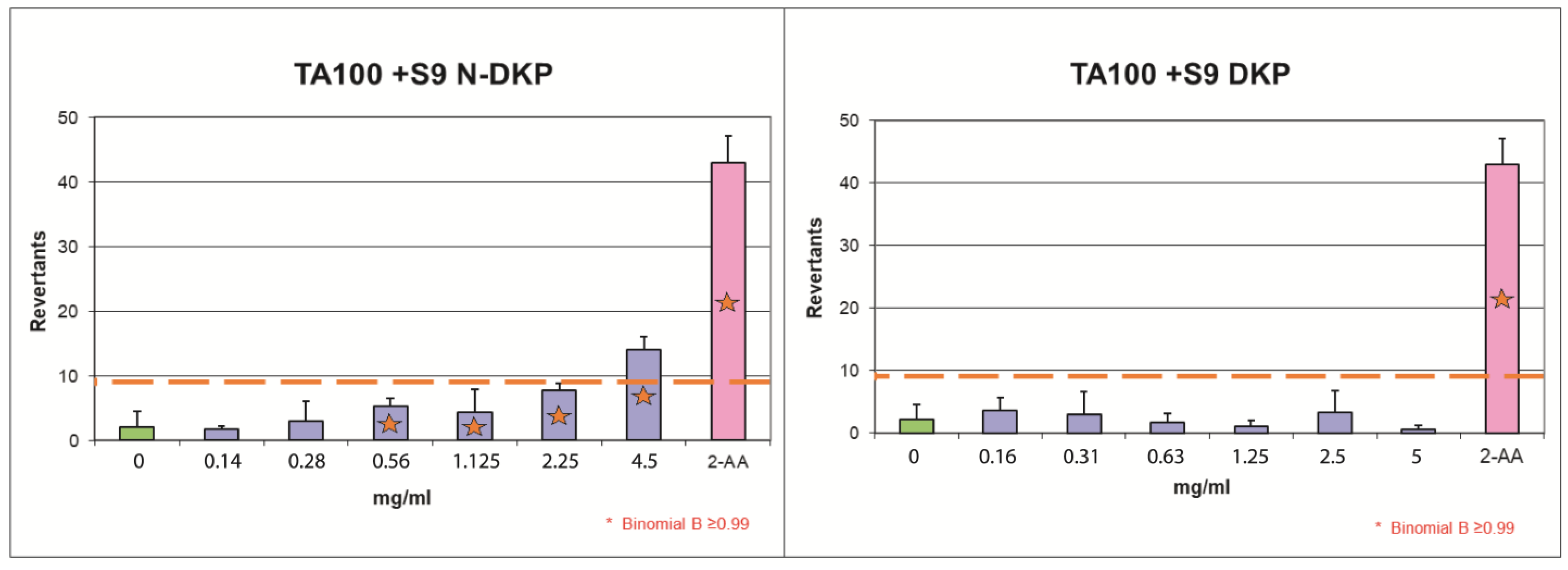
Finally, due to the presence of a diketopiperazine structural alert in the RAM degradation product, potentially leading to the formation of mutagenic nitroso compounds, we performed mutagenicity evaluation for pure DKP and its nitrosation product using the Ames test. To that end, the NAP test was first carried out using SGJ, which served as an in vitro model of DKP nitrosation in the stomach. Then, pure DKP and the product of its nitrosation (N-DKP) were subjected to mutagenicity evaluation. Both compounds were tested only at high concentrations, since mutagens have no threshold mechanism and their exposure is hazardous at any level. High concentrations were therefore applied to increase the sensitivity of our assay. The obtained results (
Table 5) showed that pure DKP was not mutagenic, which was consistent with our QSAR predictions. In other words, pure DKP is not reactive against DNA. On the other hand, for N-DKP, a mutagenic effect was observed in the
Salmonella typhimurium T100 system, with S9 fraction at 4.5 mg/mL (FIB = 3.09; B = 1.00,
Table 6). This indicates that DKP nitrosation products cause mutations by base substitutions upon activation by liver cytochromes. The possible products of DKP-nitrite interaction could involve the N-nitrosoramipril formed by the DKP ring opening or N-nitrosodiketopiperazine derivative, formed secondary to the loss of carboxyphenylalanine moiety. Our result corresponds with the reported mechanism of mutagenic action of N-nitrosamines. In fact, their emerging electrophilic metabolites, produced by cytochrome catalytic activity, easily alkylate nuclear DNA and thus, contribute to cancer initiation [
39]. This justifies the fact that the mutagenic effect of N-DKP occurred in the experiment with metabolic activation. This also means that there is no safe level of N-DKP and its presence in human blood at any concentration could induce cancer. Given that the only source of N-DKP in humans is DKP formed secondary to RAM degradation, the content of this impurity in drug formulation must be minimized.
Based on our results, it can be therefore concluded that the carcinogenic effect of DKP in humans is probable and it may be manifested via two independent mechanisms: direct aneugenic activity at excessive doses as a secondary factor, and indirect mutagenic action secondary to in vivo transformation to nitroso-metabolites. Here, it must be also noted that independent aneugenicity of pure DKP at standard blood levels is rather unlikely. The nitroso-metabolites of DKP, in turn, are mutagenic irrespective of blood level, and they could be the reason for increased cancer incidence among RAM users independently. The relevance of these in vitro findings must be verified in in vivo settings. Until this is done, one strategy to minimize the risk of human exposure to mutagenic DKP-nitrite metabolites might be employing additional stabilization methods by manufacturers so that DKP is not formed. Furthermore, antioxidants such as ascorbic acid or α-tocopherol added to final RAM formulation could serve as a mitigation strategy to eliminate electrophilic nitroso-radicals endogenously formed from DKP. Antioxidants can also be introduced as a supportive therapy, for example, by using quercetin-loaded nanostructures lipid carriers, which were effective in preventing oxidative stress induced by paraquat in vitro [
40].
At this point, it is also important to emphasize that our studies are pioneering in the field of genotoxicity, mutagenicity and nitrosation screening for drug impurities. In fact, the only similar report is available for IMD degradation impurities, and it was prepared well before the worldwide nitrosamine crisis by our research team [
8,
33]. Other toxicological studies regard either final drug products or active pharmaceutical ingredients. Drug degradation impurities, in turn, have been paid minimal attention until now, even though they are also present in drug formulations, and hence, their cancer risk needs evaluation as well. As evidenced recently by Schmidtsdorff et al. [
6], nitrosamine metabolites are common among different groups of pharmaceuticals, since, in their study, a total of 33 out of 67 prodrugs were found to produce nitrosamine derivatives using the NAP test. Furthermore, in the study by Ozahn et al. [
41], 22 drugs (including enalapril) out of 28 examined formed mutagenic nitroso-compounds, verified with
Salmonella typhimurium TA 1535/pSK1002 in vitro assay. We believe that screening for the nitrosation potential of drug degradation impurities would provide similar proportions.
A final remark on our project is that a structural analog of RAM-DKP, IMD-DKP was not mutagenic in the Ames test, neither in pure nor in nitosation mixtures [
8,
33]. Hence, it can be stated that IMD-DKP remains non-reactive in the presence of nitrite, or else its nitrosation products induce no mutations. Furthermore, VEGA-GUI simulations assessing the oncological endpoints of IMD-DKP were favorable and reliable. Only the predictions for non-genotoxicity were insufficiently supported by an applicability domain of the corresponding models. However, since IMD-DKP is clearly non-carcinogenic, its genotoxicity is rather improbable. Furthermore, IMD-DKP is less likely to appear in final dosage forms, as IMD is more stable than RAM (t
0.5 RH 76.4% T363K = 177 h and t
0.5 RH 76.4% T363K = 14 h, respectively) [
15,
42], indicating its better safety profile. Therefore, we hypothesized that IMD should receive more clinical attention over RAM, especially in patients with high risk of cancer development.
Our study has several limitations. In fact, the employed in vitro and in silico systems are not fully representative of real-life conditions. In the process of drug research and development, they serve as screening procedures to provide rationale for further invasive studies. Therefore, our findings need confirmatory experiments in animal models for carcinogenicity. This postulate is extremely important, since RAM is a valuable cardiology drug, and for this reason it is crucial to both guarantee its safety and assess its benefit-to-risk ratio without any confounding factors.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}