
Adapting the GACT-X Aligner to Accelerate Minimap2 in an FPGA Cloud Instance

Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Research Approach
2.2. Collecting Read-Assembly Data
2.2.1. Collecting and Preparing the Human Genome Reference
2.2.2. Collecting Long-Read Datasets
2.3. Profiling Minimap2’s Execution Time
2.4. Adapting GACT-X for Minimap2 on an FPGA Platform
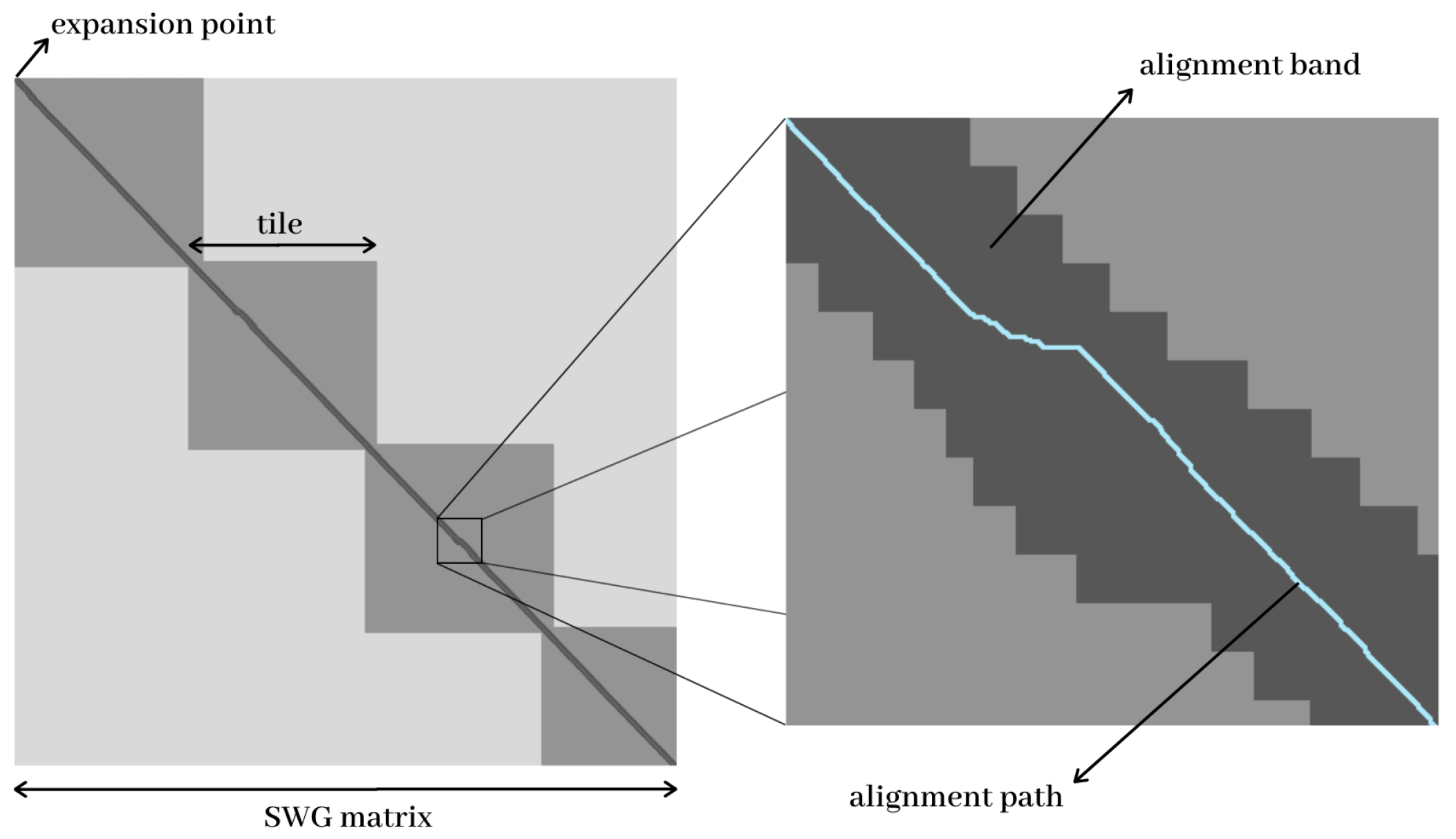
2.4.1. The Tile Approach
2.4.2. The FPGA Cloud Platform
2.5. GACT-X in Minimap2: Performance Assessment and Optimization
2.6. Acquiring Mapping and Alignment Accuracy
2.7. Integrating GACT-X into Minimap2
3. Results
3.1. Minimap2’s Throughput and Profile
3.2. GACT-X and Minimap2’s ksw Alignment Accuracy
3.3. Hybrid Minimap2 with GACT-X and Software Minimap2’s Alignment Speed
4. Discussion
4.1. Analyzing Minimap2’s Throughput and Profile
4.2. Comparing Alignment Accuracy
4.3. Comparing Alignment Speed for Software Minimap2 with the ksw Function and Hybrid Minimap2 wit GACT-X
4.4. The Transfer Channel Issue
4.5. Future Work
- A multi-FPGA design could sustain the acceleration of more threads of Minimap2 with more kernels. For this, the implementation has to be updated to the Vitis environment since SDAccel instances have been deprecated during the length of this work, and a new multi-FPGA AWS instance has to be created with an additional cost. All of these must be evaluated since, according to the findings in [32], the best performance/cost instance is the single FPGA one, and, for memory and communication intensive applications, the increase in the number of FPGAs may not bring a proportional performance increase, unless careful management of the virtual machine is executed.
- Trace-back support could be added to the design to increase the acceleration, mainly by reducing the size of the transferred data (from trace-back pointers to CIGAR strings). This implies changing the Verilog RTL description with a probable increase in the hardware area in the programmable logic, which has to be evaluated with the current design.
- A second affine function could be incorporated into the GACT-X’s hardware design to properly mirror Minimap2’s alignment scores and improve the compared accuracy, especially for datasets with many long gaps.
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASIC | Application Specific Integrated Circuit |
| AMI | Amazon Machine Image |
| AWS | Amazon Web Services |
| BRAM | Block Random-Access Memory |
| CIGAR | Concise Idiosyncratic Gapped Alignment Report |
| COVID-19 | coronavirus disease 2019 |
| CPU | Central Processing Unit |
| CUDA | Compute Unified Device Architecture |
| DNA | deoxyribonucleic acid |
| DRAM | Dynamic Random Access Memory |
| DSA | domain-specific architecture |
| ENA | European Nucleotide Archive |
| FPGA | Field Programmable Gate Array |
| GPU | Graphic Processing Unit |
| GRCh38.p14 | Genome Reference Consortium Human Build 38 patch release 14 |
| indel | insertion/deletion |
| I/O | Input/Output |
| KNL | Knights Landing |
| MDPI | Multidisciplinary Digital Publishing Institute |
| NCBI | National Center for Biotechnology Information |
| ONT | Oxford Nanopore Technologies |
| OpenCL | Open Computing Language |
| PacBio | Pacific Biosciences |
| PCIe | Peripheral Component Interconnect Express |
| PE | processing element |
| SSD | Solid-state drive |
| SIMD | Single Instruction, Multiple Data |
| SK | Suzuki–Kasahara |
| SSE | Streaming SIMD Extensions |
| SV | structural variant |
| SWG | Smith–Waterman–Gotoh |
References
- Kushnick, T. Thompson & Thompson Genetics in Medicine. JAMA 1992, 267, 2115. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef] [PubMed]
- Orteu, A.; Jiggins, C.D. The genomics of coloration provides insights into adaptive evolution. Nat. Rev. Genet. 2020, 21, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Alser, M.; Bingol, Z.; Cali, D.S.; Kim, J.; Ghose, S.; Alkan, C.; Mutlu, O. Accelerating Genome Analysis: A Primer on an Ongoing Journey. IEEE Micro 2020, 40, 65–75. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, J.L.; Patterson, D.A. A New Golden Age for Computer Architecture. Commun. ACM 2019, 62, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Samura, O. Update on noninvasive prenatal testing: A review based on current worldwide research. J. Obstet. Gynaecol. Res. 2020, 46, 1246–1254. [Google Scholar] [CrossRef]
- Gadsbøll, K.; Petersen, O.B.; Gatinois, V.; Strange, H.; Jacobsson, B.; Wapner, R.; Vermeesch, J.R.; NIPT-map Study Group; Vogel, I. Current use of noninvasive prenatal testing in Europe, Australia and the USA: A graphical presentation. Acta Obstet. Et Gynecol. Scand. 2020, 99, 722–730. [Google Scholar] [CrossRef]
- Liu, T.; Chen, Z.; Chen, W.; Chen, X.; Hosseini, M.; Yang, Z.; Li, J.; Ho, D.; Turay, D.; Gheorghe, C.P.; et al. A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. iScience 2021, 24, 102892. [Google Scholar] [CrossRef]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- GRCh38.p14. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.40 (accessed on 18 February 2023).
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Burrows-Wheeler Aligner. Available online: http://bio-bwa.sourceforge.net/ (accessed on 18 February 2023).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, O. Optimal sequence alignment allowing for long gaps. Bull. Math. Biol. 1990, 52, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, D.; Wu, S.; Ozog, N.; Goliya, K.; Blaauw, D.; Narayanasamy, S.; Das, R. SeedEx: A Genome Sequencing Accelerator for Optimal Alignments in Subminimal Space. In Proceedings of the 2020 53rd Annual IEEE/ACM International Symposium on Microarchitecture (MICRO), Athens, Greece, 17–21 October 2020; pp. 937–950. [Google Scholar] [CrossRef]
- Liao, Y.L.; Li, Y.C.; Chen, N.C.; Lu, Y.C. Adaptively Banded Smith-Waterman Algorithm for Long Reads and Its Hardware Accelerator. In Proceedings of the 2018 IEEE 29th International Conference on Application-Specific Systems, Architectures and Processors (ASAP), Milano, Italy, 10–12 July 2018; pp. 1–9. [Google Scholar] [CrossRef]
- Adewale, B.A. Will long-read sequencing technologies replace short-read sequencing technologies in the next 10 years? Afr. J. Lab. Med. 2020, 9, 1–5. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2015, 32, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Kasahara, M. Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC Bioinform. 2018, 19, 33–47. [Google Scholar] [CrossRef]
- Goyal, A.; Kwon, H.; Lee, K.; Garg, R.; Yun, S.; Kim, Y.; Lee, S.; Lee, M. Ultra-Fast Next Generation Human Genome Sequencing Data Processing Using DRAGENTM Bio-IT Processor for Precision Medicine. Open J. Genet. 2017, 7, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Lau, J.; Ruan, Z.; Wei, P.; Cong, J. Hardware Acceleration of Long Read Pairwise Overlapping in Genome Sequencing: A Race Between FPGA and GPU. In Proceedings of the 2019 IEEE 27th Annual International Symposium on Field-Programmable Custom Computing Machines (FCCM), San Diego, CA, USA, 28 April–1 May 2019; pp. 127–135. [Google Scholar] [CrossRef]
- Kaplan, R.; Yavits, L.; Ginosar, R. RASSA: Resistive Prealignment Accelerator for Approximate DNA Long Read Mapping. IEEE Micro 2019, 39, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Qiu, S.; Wang, L.; Luo, Q. Accelerating Long Read Alignment on Three Processors. In Proceedings of the Proceedings of the 48th International Conference on Parallel Processing, Kyoto, Japan, 5–8 August 2019; Association for Computing Machinery: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Koliogeorgi, K.; Voss, N.; Fytraki, S.; Xydis, S.; Gaydadjiev, G.; Soudris, D. Dataflow Acceleration of Smith-Waterman with Traceback for High Throughput Next Generation Sequencing. In Proceedings of the 2019 29th International Conference on Field Programmable Logic and Applications (FPL), Barcelona, Spain, 8–12 September 2019; pp. 74–80. [Google Scholar] [CrossRef] [Green Version]
- Turakhia, Y.; Goenka, S.D.; Bejerano, G.; Dally, W.J. Darwin-WGA: A Co-processor Provides Increased Sensitivity in Whole Genome Alignments with High Speedup. In Proceedings of the 2019 IEEE International Symposium on High Performance Computer Architecture (HPCA), Washington, DC, USA, 16–20 February 2019; pp. 359–372. [Google Scholar] [CrossRef]
- Amazon EC2 F1 Instances. Available online: https://aws.amazon.com/ec2/instance-types/f1/?nc1=h_ls (accessed on 18 February 2023).
- Wang, X.; Niu, Y.; Liu, F.; Xu, Z. When FPGA Meets Cloud: A First Look at Performance. IEEE Trans. Cloud Comput. 2020, 10, 1344–1357. [Google Scholar] [CrossRef]
- GRCh38. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26 (accessed on 18 February 2023).
- Run: ERR2585114. Available online: https://www.ebi.ac.uk/ena/browser/view/ERR2585114 (accessed on 18 February 2023).
- Coster, W.D.; Rijk, P.D.; Roeck, A.D.; Pooter, T.D.; D’Hert, S.; Strazisar, M.; Sleegers, K.; Broeckhoven, C.V. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- SRX9063500: PacBio SMRT Whole Genome Sequencing of Sri Lankan Tamil H. sapiens. Available online: https://www.ncbi.nlm.nih.gov/sra/SRX9063500[accn] (accessed on 18 February 2023).
- Ono, Y.; Asai, K.; Hamada, M. PBSIM: PacBio reads simulator—toward accurate genome assembly. Bioinformatics 2013, 29, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Human 54x Dataset. Available online: http://datasets.pacb.com/2014/Human54x/fast.html (accessed on 18 February 2023).
- GNU Gprof. Available online: https://ftp.gnu.org/old-gnu/Manuals/gprof-2.9.1/html_mono/gprof.html (accessed on 18 February 2023).
- FPGA Developer AMI. Available online: https://aws.amazon.com/marketplace/pp/prodview-gimv3gqbpe57k (accessed on 18 February 2023).
- tmux. Available online: https://github.com/tmux/tmux (accessed on 18 February 2023).
- Minimap2-2.18. Available online: https://github.com/lh3/minimap2/releases/tag/v2.18 (accessed on 18 February 2023).
- aws-fpga. Available online: https://github.com/aws/aws-fpga (accessed on 18 February 2023).
- Darwin-WGA. Available online: https://github.com/gsneha26/Darwin-WGA (accessed on 18 February 2023).
- Teng, C.; Achjian, R.W.; Braga, C.C.; Zuffo, M.K.; Chau, W.J. Accelerating the base-level alignment step of DNA assembling in Minimap2 Algorithm using FPGA. In Proceedings of the 2021 IEEE 12th Latin America Symposium on Circuits and System (LASCAS), Arequipa, Peru, 22–25 February 2021; pp. 1–4. [Google Scholar] [CrossRef]
- Adapting-the-GACT-X-Aligner-to-Accelerate-Minimap2-in-an-FPGA-Cloud-Instance. Available online: https://github.com/carolina-teng/Adapting-the-GACT-X-Aligner-to-Accelerate-Minimap2-in-an-FPGA-Cloud-Instance (accessed on 19 March 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Reads | Coverage Depth | Read Length | Read Accuracy | Variant Rates | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | s.d. | min. | max. | Mean | s.d. | Substitution | Insertion | Deletion | ||
| 7,460,510 | 20 | 8310 | 106.26 | 100 | 24,988 | 0.85 | 0.00017 | 0.015 | 0.090 | 0.046 |
| Match Score | Mismatch Score | 1st Affine Function | 2nd Affine Function | X-Drop | ||||
|---|---|---|---|---|---|---|---|---|
| Gap Open | Gap Extend | Gap Open | Gap Extend | |||||
| Minimap2 | 2 | −4 | −4 | −2 | −24 | −1 | - | |
| GACT-X | Original | 91 to 100 | −31 to −125 | −430 | −30 | - | - | 9430 |
| Adapted | 10 | −20 | −30 | −10 | - | - | 943 | |
| Average Length (nt) | Real Time (Hours) * | CPU Time (Hours) * | Dataset Size (Gbases) | Throughput (kbases/s) | Chaining Time ** | Extending Time ** | |
|---|---|---|---|---|---|---|---|
| Simulated PacBio Reads | 8300 | 2:46 | 42:41 | 61.99 | 403.46 | 8% | 49% |
| Real PacBio Reads | 13,300 | 4:55 | 88:21 | 52.20 | 164.11 | 50% | 25% |
| Real ONT Reads | 16,900 | 2:14 | 34:27 | 28.53 | 230.00 | 27% | 42% |
| GACT-X ksw Score Difference | <0 | 0 | >0 |
| Simulated PacBio | 3.90% | 95.42% | 0.68% |
| Real ONT | 41.21% | 47.79% | 11.00% |
| Real PacBio | 14.90% | 66.83% | 18.28% |
| Percentage of Aligned Bases | 0–80 | 80–90 | 90–100 | 100 | |
|---|---|---|---|---|---|
| Simulated PacBio | Minimap2’s ksw | 1.06% | 0.22% | 60.97% | 37.62% |
| GACT-X 4000 | 1.52% | 0.23% | 55.21% | 43.04% | |
| Real ONT | Minimap2’s ksw | 21.56% | 2.59% | 73.57% | 2.29% |
| GACT-X 4000 | 36.24% | 3.64% | 58.47% | 1.64% | |
| Real PacBio | Minimap2’s ksw | 18.42% | 0.76% | 13.15% | 67.67% |
| GACT-X 4000 | 12.01% | 2.71% | 19.59% | 65.69% |
| Number of Threads (sw) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Simulated PacBio | software | total execution (s) | 759.157 | 390.198 | 266.926 | 206.255 | 197.863 | 186.727 | 177.508 | 169.927 |
| 1 kernel | total execution (s) | 539.56 | 299.76 | 233.88 | 208.93 | 203.74 | 253.84 | 409.03 | 453.26 | |
| acceleration | 1.41 | 1.30 | 1.14 | 0.99 | 0.97 | 0.74 | 0.43 | 0.37 | ||
| thread acceleration | 1.00 | 1.80 | 2.31 | 2.58 | 2.65 | 2.13 | 1.32 | 1.19 | ||
| 2 kernels | total execution (s) | 538.996 | 289.557 | 211.837 | 198.700 | 203.366 | 228.516 | 273.066 | 313.150 | |
| acceleration | 1.41 | 1.35 | 1.26 | 1.04 | 0.97 | 0.82 | 0.65 | 0.54 | ||
| thread acceleration | 1.00 | 1.86 | 2.54 | 2.71 | 2.65 | 2.36 | 1.97 | 1.72 | ||
| Real ONT | software | total execution (s) | 3105.50 | 1559.59 | 1056.77 | 806.37 | 774.30 | 719.29 | 684.98 | 646.35 |
| 1 kernel | total execution (s) | 2243.69 | 1,168.48 | 837.14 | 712.91 | 653.68 | 660.44 | 937.68 | 1258.13 | |
| acceleration | 1.38 | 1.33 | 1.26 | 1.13 | 1.18 | 1.09 | 0.73 | 0.51 | ||
| thread acceleration | 1.00 | 1.92 | 2.68 | 3.15 | 3.43 | 3.40 | 2.39 | 1.78 | ||
| 2 kernels | total execution (s) | 2197.265 | 1126.070 | 794.728 | 638.109 | 586.025 | 576.393 | 672.990 | 858.846 | |
| acceleration | 1.41 | 1.38 | 1.33 | 1.26 | 1.32 | 1.25 | 1.02 | 0.75 | ||
| thread acceleration | 1.00 | 1.95 | 2.76 | 3.44 | 3.75 | 3.81 | 3.26 | 2.56 | ||
| Real PacBio | software | total execution (s) | 4255.21 | 2132.71 | 1431.38 | 1083.29 | 1037.82 | 965.57 | 904.33 | 858.31 |
| 1 kernel | total execution (s) | 3570.25 | 1,813.39 | 1243.76 | 1003.40 | 919.39 | 889.68 | 1077.63 | 1575.97 | |
| acceleration | 1.19 | 1.18 | 1.15 | 1.08 | 1.13 | 1.09 | 0.84 | 0.54 | ||
| thread acceleration | 1.00 | 1.97 | 2.87 | 3.56 | 3.88 | 4.01 | 3.31 | 2.27 | ||
| 2 kernels | total execution (s) | 3470.799 | 1765.341 | 1239.144 | 994.346 | 885.107 | 831.761 | 855.454 | 1117.282 | |
| acceleration | 1.23 | 1.21 | 1.16 | 1.09 | 1.17 | 1.16 | 1.06 | 0.77 | ||
| thread acceleration | 1.00 | 1.97 | 2.80 | 3.49 | 3.92 | 4.17 | 4.06 | 3.11 | ||
| Number of Threads (sw) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Simulated PacBio | 1 kernel time (s) | processing | 94.14 | 94.62 | 95.16 | 96.16 | 96.71 | 98.11 | 132.26 | 150.40 |
| data transfer | 26.90 | 28.81 | 32.07 | 33.24 | 34.13 | 37.59 | 152.30 | 150.51 | ||
| time in line | 0.21 | 83.11 | 318.66 | 845.77 | 1789.56 | 3377.48 | 13,990.11 | 21,404.43 | ||
| 2 kernels time (s) | processing | 94.20 | 94.99 | 95.43 | 95.89 | 96.49 | 110.83 | 129.61 | 141.75 | |
| data transfer | 27.00 | 34.77 | 37.87 | 37.25 | 47.99 | 146.70 | 165.48 | 166.65 | ||
| time in line | 0.15 | 0.24 | 44.49 | 186.04 | 619.47 | 3813.98 | 7592.47 | 12,094.00 | ||
| Real ONT | 1 kernel time (s) | processing | 322.29 | 323.02 | 324.33 | 326.42 | 327.82 | 331.18 | 388.75 | 476.91 |
| data transfer | 12.20 | 48.54 | 51.66 | 53.72 | 59.88 | 75.27 | 361.88 | 503.51 | ||
| time in line | 0.15 | 148.91 | 544.15 | 1310.52 | 2548.16 | 4926.27 | 24,567.76 | 50,482.29 | ||
| 2 kernels time (s) | processing | 322.66 | 324.16 | 324.98 | 326.04 | 327.69 | 346.86 | 376.96 | 420.51 | |
| data transfer | 42.80 | 53.15 | 61.25 | 60.11 | 86.25 | 227.04 | 347.46 | 476.46 | ||
| time in line | 0.24 | 0.27 | 54.48 | 242.43 | 802.66 | 4210.88 | 11,777.99 | 25,100.46 | ||
| Real PacBio | 1 kernel time (s) | processing | 255.12 | 255.52 | 257.62 | 259.56 | 261.43 | 264.64 | 324.07 | 465.84 |
| data transfer | 53.28 | 57.38 | 61.37 | 65.27 | 74.19 | 91.04 | 383.26 | 723.37 | ||
| time in line | 0.20 | 63.13 | 219.84 | 499.16 | 962.39 | 1909.10 | 16,175.72 | 53,765.80 | ||
| 2 kernels time (s) | processing | 255.79 | 257.62 | 258.97 | 259.90 | 261.93 | 284.62 | 333.82 | 388.27 | |
| data transfer | 53.64 | 64.29 | 70.02 | 70.59 | 91.89 | 275.96 | 452.71 | 683.43 | ||
| time in line | 0.10 | 0.41 | 15.78 | 68.70 | 228.42 | 2063.87 | 8240.65 | 23,054.56 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, C.; Achjian, R.W.; Wang, J.C.; Fonseca, F.J. Adapting the GACT-X Aligner to Accelerate Minimap2 in an FPGA Cloud Instance. Appl. Sci. 2023, 13, 4385. https://doi.org/10.3390/app13074385
Teng C, Achjian RW, Wang JC, Fonseca FJ. Adapting the GACT-X Aligner to Accelerate Minimap2 in an FPGA Cloud Instance. Applied Sciences. 2023; 13(7):4385. https://doi.org/10.3390/app13074385
Chicago/Turabian StyleTeng, Carolina, Renan Weege Achjian, Jiang Chau Wang, and Fernando Josepetti Fonseca. 2023. "Adapting the GACT-X Aligner to Accelerate Minimap2 in an FPGA Cloud Instance" Applied Sciences 13, no. 7: 4385. https://doi.org/10.3390/app13074385