Abstract
Whole-genome alignment (WGA) is a critical process in comparative genomics, facilitating the detection of genetic variants and aiding our understanding of evolution. This paper offers a detailed overview and categorization of WGA techniques, encompassing suffix tree-based, hash-based, anchor-based, and graph-based methods. It elaborates on the algorithmic properties of these tools, focusing on performance and methodological aspects. This paper underscores the latest progress in WGA, emphasizing the increasing capacity to manage the growing intricacy and volume of genomic data. However, the field still grapples with computational and biological hurdles affecting the precision and speed of WGA. We explore these challenges and potential future solutions. This paper aims to provide a comprehensive resource for researchers, deepening our understanding of WGA tools and their applications, constraints, and prospects.
Keywords:
anchors; graphs; high-throughput sequencing; suffix trees; hashing; whole-genome alignment 1. Introduction
The genomics era, heralded by the availability of whole-genome sequences for a wide range of organisms, has created unprecedented opportunities for researchers to understand evolutionary relationships, genetic variation, and functional elements of genomes [1]. A critical step towards this understanding is whole-genome alignment (WGA), a cornerstone of bioinformatics that aligns entire genomes from different species or individuals within the same species. WGAs provide a global perspective on genomic similarity and variation, yielding insights into species’ evolution, gene function, and genetic diseases [2].
In the context of sequence alignment, classical algorithms such as the Smith–Waterman algorithm (local) and the Needleman–Wunsch algorithm (global) have played pivotal roles in bioinformatics [3]. The Smith–Waterman algorithm, introduced by Temple F. Smith and Michael S. Waterman in 1981, is renowned for its ability to perform local sequence alignments by identifying the optimal local alignment between two sequences [4]. This algorithm is particularly useful for detecting regions of similarity or homology within larger sequences, thereby facilitating the identification of conserved functional domains or motifs. Similarly, the Needleman–Wunsch algorithm, developed by Saul B. Needleman and Christian D. Wunsch in 1970, revolutionized global sequence alignment by efficiently computing the optimal alignment between two sequences based on their entire lengths, regardless of gaps or mismatches Through considering the entire length of the sequences, the Needleman–Wunsch algorithm enables researchers to identify regions of similarity between sequences, facilitating comparative analysis and evolutionary studies.
These classical algorithms serve as the foundation upon which modern whole-genome alignment techniques are built. They provided fundamental insights into the principles of sequence alignment and have paved the way for the development of more sophisticated alignment algorithms tailored to the challenges of aligning entire genomes. In the subsequent sections of this review, we explore the evolution of whole-genome alignment methodologies, highlighting the contributions of classical algorithms and modern computational tools in advancing our understanding of genomic structure and function.
Next-generation sequencing (NGS) technologies have revolutionized genomics through delivering vast amounts of data with unparalleled speed and cost-efficiency. These technologies yield two principal types of sequence outputs: short reads and long reads. Short reads, typically ranging from 100 to 600 base pairs, have been lauded for their high precision but pose challenges in genome assembly and alignment due to their propensity for multiple mappings on the same genome [5]. This can result in fragmented alignments and complicate the reconstruction of complex or repetitive genomic regions. Tools such as BOWTIE2 and BWA have been optimized for short reads, excelling in processing large volumes of data and pinpointing small-scale genetic variations with high accuracy [6]. Conversely, long reads, which can extend to several thousand base pairs, hold considerable promise for enhancing genome assembly and facilitating the alignment of challenging regions. However, they are often marred by higher error rates, introducing new challenges in terms of alignment precision. Tools like PacBio’s SMRT Analysis and Oxford Nanopore’s MinION-based Minimap2 have been specifically designed for long reads and are adept at untangling complex genomic architectures and reducing gaps in assemblies [7].
The distinction between short and long reads significantly influences the selection of algorithms and tools for genome alignment. This dichotomy highlights the necessity for a meticulously balanced approach and a deep comprehension of the unique advantages and drawbacks associated with each read type in whole-genome alignment. Understanding these differences is crucial for researchers in choosing the most appropriate tools tailored to their specific genomic research needs.
Despite the importance of WGA, aligning whole genomes is non-trivial due to the sheer size of genomes, their complex evolutionary histories, and the computational demands of alignment algorithms [8]. For instance, the human genome consists of approximately 3 billion base pairs, posing significant computational challenges such as execution time, memory usage, and management of genomic rearrangements [9]. A multitude of algorithms have been developed over the years to address these challenges [10]. Each algorithm offers a unique approach to WGA and has its specific strengths and weaknesses in terms of computational efficiency, scalability, and alignment accuracy [11]. Therefore, a comprehensive understanding of these algorithms is crucial for researchers when choosing the most suitable tool for their specific tasks [3].
This study aims to provide a comprehensive review of prevalent WGA algorithms, highlighting their algorithmic aspects, methodological underpinnings, and the current challenges they face. We focus on three primary classes of algorithms: suffix tree-based methods, anchor-based methods, and graph-based methods, with an emphasis on recent advancements, including algorithms such as SibeliaZ and BubbZ [4] and innovative methods like PlusV [12] and MAGOT [13].
Our goal is to offer a balanced, comprehensive, and critical view of the current landscape of WGA algorithms, assisting researchers in their choice of suitable algorithms for specific applications.
In the following sections, we delve into the classification of WGA algorithms, their algorithmic aspects, recent advancements, and the challenges in whole-genome alignment. We hope that this review will serve as a valuable guide for researchers and practitioners in the field of bioinformatics, genomics, and computational biology [14].
2. Classification of Whole-Genome Alignment Algorithms
Whole-genome alignment, a foundational task in genomics, relies on a variety of sophisticated algorithms to compare entire genomes. These algorithms can be broadly classified into three categories: suffix tree-based methods, anchor-based methods, hash-based methods, and graph-based methods. Each of these categories embodies different alignment strategies, offering unique advantages and facing distinct challenges.
2.1. Suffix Tree-Based Alignment Methods
Suffix trees are particularly efficient for pattern-matching tasks and fundamental for data structures used in string processing. A suffix tree is a compressed data tree structure that represents all the suffixes of a given string (Figure 1). The tree saves their positions in the text as well as their values, allowing efficient pattern matching and substring search operations [15]. They can be constructed for a string of length (n) that typically requires O (n) time and space complexity. There are several algorithms for constructing suffix trees, such as Ukkonen’s algorithm and McCreight’s algorithm [16]. While many operations on suffix trees are efficient, it is essential to understand the time complexities associated with various operations, especially for large input strings.
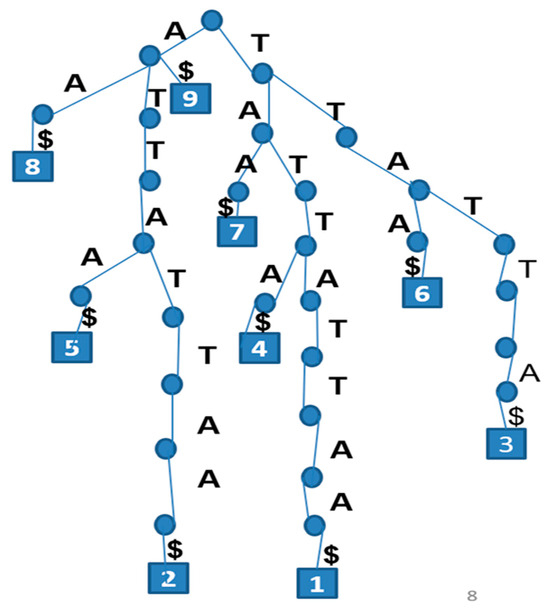
Figure 1.
Suffix Tree for the Sequence TATTATTAA with 9 Suffixes and $ marking the termination of each suffix.
Data structured in this way provide fast implementation for string operations. The main advantage is the fast computational time to detect an exact match with user-defined inputs [14]. Despite the existence of some bioinformatics applications, such as MUMmer, to efficiently evaluate queries on biological sequence data, suffix tree-based are not widely used because of their high cost of construction. However, various techniques such as suffix array, compressed suffix trees, or using specialized data structures like suffix links can be employed to reduce memory usage.
2.1.1. MUMmer Technique
MUMmer uses a suffix tree-based algorithm called a “Maximal Unique Match” (MUM) finding algorithm, which is an alignment algorithm based on suffix tree representation, being a suite of bioinformatics tools designed for whole-genome alignment and comparison tasks. The main idea of this algorithm is to find all distinct matches for two given genomes [17]. To better align the two given genomes, it is assumed that they are closely homologous. MUMmer is widely used in bioinformatics for tasks such as genome alignment, genome assembly validation, sequence annotation, and comparative genomics studies. Its suffix tree-based MUM finding algorithm provides an efficient and accurate method for identifying conserved regions and similarities between large genomic sequences [18]. This accuracy is crucial in NGS alignment tasks where even small errors can lead to misinterpretation of genetic variations or structural variations. While newer alignment methods continue to emerge, suffix tree-based methods remain a key method in NGS alignment due to their accuracy, efficiency, and ability to handle large-scale genomic data effectively. Researchers and bioinformaticians often choose these methods based on their specific alignment requirements, data characteristics, and the need for accurate and reliable alignment results.
MUMmer constructs a suffix tree for each input genome sequence. Its algorithm is based on the following four main steps:
- (i)
- Perform a maximal unique match (MUM) decomposition of the two genomes. The algorithm identifies all MUMs between pairs of genomes, which are subsequences that occurs exactly once in genome A and once in genome B. This decomposition identifies all maximal unique matches between the two genomes. To detect these MUMs, the two genomes are represented by a suffix tree. The common substrings detected on the tree represent all the MUMs between the two genomes (Figure 2). MUMs represent regions of high similarity or conserved regions between genomes;
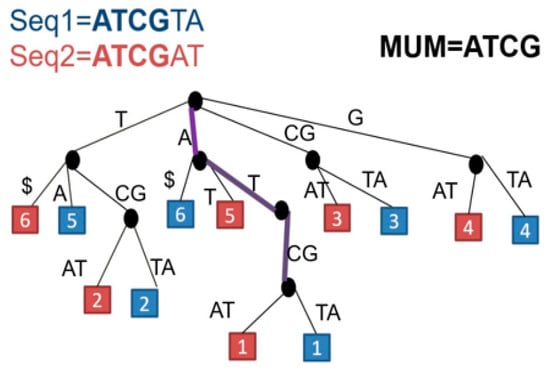 Figure 2. MUMs detected between Seq1 and Seq2. The maximal exact match is ATCG. $ is marking the termination of each suffix.
Figure 2. MUMs detected between Seq1 and Seq2. The maximal exact match is ATCG. $ is marking the termination of each suffix. - (ii)
- Apply filtering techniques to remove spurious matches and improve the accuracy of the alignment results. Organize the MUMs and identify the most extended sequence of matches found in both genomes, maintaining their original order;
- (iii)
- Fill in the spaces within the alignment by detecting significant insertions, repetitions, altered sections, and single nucleotide variations (SNVs);
- (iv)
- Perform a Smith–Waterman alignment for the regions between the MUMs and construct the final alignment;
- (v)
- Output alignments, allowing visualization and analysis to understand the evolutionary relationships and structural similarities between the input genomes.
MUMmer 2.1
MUMmer 1.0 was first used to detect large-scale inversions in bacterial genomes. The bacterial genomes’ sizes did not exceed a few Mbp. This means that MUMmer 1.0 would require powerful computational resources to align genomes comprising billions of nucleotides. In addition, when the human genome was sequenced, it was necessary to implement a new algorithm able to align entire human chromosomes rapidly and accurately.
For that reason, the authors proposed a new method that required less memory and ran the four steps faster [13]. In addition, a new parameter k was introduced. This parameter represents the maximum gap length allowed between MUMs. If the gap is lower than k, the two adjacent MUMs are regrouped in the same cluster (Figure 3).
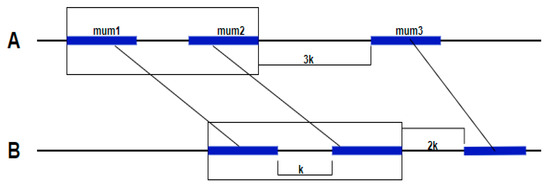
Figure 3.
Cluster detected with MUMmer 2.1. The algorithm detects the adjacent MUMs between Genome A and Genome B. If a gap between these MUMs is lower than a parameter k, the two adjacent MUMs are regrouped in the same cluster.
MUMmer 3.0
Unique MUMs occur exactly once in both genomes. However, in certain cases, the exact match may be duplicated within the subject genome. To overcome this problem, the new MUMmer 3.0 uses all maximal matches including non-unique ones to align the two genomes [19]. In addition, the execution time of MUMmer 3.0 and the memory usage have been improved. While using MUMmer 3.0, the query time for a whole-genome alignment for comparison between human chromosomes is about 300 min and can reach 600 min depending on the sizes of the chromosomes used for the alignment.
MUMmer 4.0
With large genomic data, MUMmer 3.0 has limitations and fails to perform whole-genome alignments. In 2018, the new version MUMmer 4.0 was launched, which includes an improved version of MUMmer algorithm and uses a 48-bit suffix array to overcome genome size constraints. In addition, MUMmer 4.0 introduces improved speed and memory usage through parallel processing of input query sequences [20].
With this improvement, a whole-genome alignment of human/chimpanzee genomes can be performed in 2897 min. This operation cannot be performed using MUMmer 3.0. Furthermore, with a theoretical limit on the input size of 141 Tbp, MUMmer 4.0 can perform alignment with input sequences of any biologically realistic length. The complexity of MUMmer 4.0 is O(n lg n) for constructing suffix trees and O(m + n) O(m + n) for aligning sequences, where n is the length of the reference genome and m is the length of the query sequence.
Table 1 provides a comprehensive overview of the differences between various versions of MUMmer, highlighting the evolution of the algorithm over time and major improvements such as enhanced suffix arrays in MUMmer 3.1 and optimized suffix tree utilization in MUMmer 4. Whole-genome alignment algorithm classification is based on various approaches to alignment. Table 2 provides a detailed overview of these classification approaches.

Table 1.
Overview of the differences between various versions of MUMmer software.
Table 2.
Whole-genome alignment (WGA) algorithm classification based on approaches to alignment.
2.1.2. Other Sequence Comparison Approaches Based on the Suffix Tree Method
Suffix tree approaches provide fast computational time to detect exact matches between sequences. Different algorithms rely on the construction of suffix trees between the two sequences to detect similar regions (words). The Lwords algorithm is a sequence comparison alignment-free approach based on the construction of a generalized suffix tree of all sequences. The Lwords algorithm aims to represent repeated substrings in a string using a dictionary of unique codes. This algorithm is primarily used for data compression, enabling efficient storage and transmission of repetitive data [21]. The algorithm scans the input string and builds a dictionary of unique substrings, assigning each substring a unique code. Following the construction of the suffix tree, the frequency of all possible words with length L, provided as a parameter, is calculated. Then, based on the L-words frequency profile for each sequence, a pairwise Euclidean distance is computed, producing a symmetrical genetic distance matrix between the sequences.
The efficiency of the Lwords algorithm depends on the frequency and length of repeated substrings in the input string. It performs well when there are significant repetitions in the input data; however, it can be ineffective for data with minimal redundancy [22]. The execution time of the Lwords algorithm is satisfactory when the algorithm is applied to complete intra/inter mitochondrial species’ genomes. It can perform its sequence comparison alignment-free approach for 29 primates’ genomes (ranging from 14 to 25 Mbp) in just 66 s.
Another algorithm based on the suffix tree method is the multiple sequence alignment algorithm (MSA) [23]. The MSA algorithm starts by detecting the similar substrings between the sequences. Then, it uses an approach called the center star strategy in order to calculate the pairwise optimal alignment distance between the set of inputs and select the central sequence which has the highest degree of similarity with other sequences in the set [24]. Finally, the algorithm runs a pairwise alignment between the aligned center and the other sequences to construct the final multiple alignment. This algorithm can perform an alignment of 67,200 strains, all longer than 10,000 bps, in 9 min, and its quality has been evaluated based on alignment accuracy, biological relevance, and statistical significance, through metrics or scoring schemes such as sum-of-pairs score (scores the sum of pairs of aligned residues), total column score (scores based on the similarity of residues to a consensus sequence), and consensus identity (scores based on concepts of information theory like entropy and mutual information [25].
This algorithm is a fundamental problem in bioinformatics, where the goal is to align three or more biological sequences (DNA, RNA, or protein) to identify conserved regions, functional motifs, and evolutionary relationships. Let us say, for a given number of sequences “N”, Seq1, Seq2, …, Seqn, the goal is to find an optimal alignment that maximizes the similarity of the sequences by inserting gaps to account for insertions, deletions, and substitutions. However, there are a number of important challenges regarding this algorithm, such as computationally expensive, accurate alignment and selecting a gap penalty that accurately reflects biological reality [26].
MSA algorithms play a crucial role in various fields of bioinformatics, allowing analysis of sequence similarities and evolutionary relationships among sequences. Understanding the principles and techniques used in MSA is essential for researchers working in these domains. Current research in MSA has focused on improving alignment accuracy, improving scalability to handle large datasets, and incorporating evolutionary models and structural information into the alignment process [26].
2.2. Anchor-Based Methods
Anchor-based methods use a set of anchor sequences, which are typically a subset of the input sequences, to guide the alignment of the remaining sequences in the dataset [27]. Through aligning sequences to the anchor sequences, the algorithm aims to preserve the overall structure and evolutionary relationships among the sequences. An anchor is a similar region of two or more genomes chosen based on their conservation, significance, or known biological relationships.
Anchor-based methods provide a framework for aligning the remaining sequences by anchoring them to known conserved regions or motifs present in the anchor sequences. These methods can improve the accuracy and efficiency of the alignment process via leveraging known conserved regions or motifs present in the anchor sequences. Overall, they may reduce the complexity of aligning large datasets and improve the scalability of the alignment algorithm. However, selecting appropriate anchor sequences is crucial for the success of anchor-based methods, considering that they cannot be too divergent or unrepresentative of a given dataset; otherwise, they may lead to suboptimal alignments and poor performance in cases where the anchor sequences do not adequately capture the diversity or complexity of the input dataset.
Some algorithms perform local alignments on each consecutive pair of anchors that are separated by a non-similar region smaller than a given length k. The algorithms join all the anchors and the non-similar regions together. Different algorithms use this method either to perform a local alignment on chromosomes or a specific DNA sequence or to perform a global genomic alignment. In this section, we enumerate some of the most used algorithms that involve anchor-based methods and explain their computational steps.
2.2.1. Lagan
The Lagan algorithm is based on dynamic programming as it uses anchoring to subdivide the alignment into small anchors [28]. The Lagan algorithm also assumes that the two sequences are relatively close. It starts by detecting the anchors and then performs a dynamic programming alignment algorithm on the limited area around the anchors. The Lagan algorithm’s complexity is O(n3), where n is the length of the input sequences.
Steps:
- (i)
- Generation of Local Alignments:The Lagan algorithm uses the CHAOS method [29] to detect local homologies between the two genomes and chains them into a rough global map.The first step of CHAOS is chaining short exact matches (seeds) which match between the two genomes. The seeds that are close are regrouped to the same anchors. The gaps between the seeds are aligned using a dynamic programming alignment method;
- (ii)
- Construction of a Rough Global Map:The Lagan algorithm uses local alignments to develop a rough global map. Each local alignment has a score of similarity. The optimal rough global map has the highest-scoring chain, which can be computed using sparse dynamic programming [30].
- (iii)
- Computation of Global Alignment:To compute the final global alignment, the Lagan method uses the Needleman–Wunsch algorithm to perform an alignment of the limited area between the anchors.
Multi-Lagan
Multi-Lagan aligns a set of genomes progressively. An alignment of n sequences is constructed after the n − 1 pairwise alignment operation. Multi-Lagan uses a phylogenetic tree of the sequence to choose, at each step, which genome will be used for the alignment. This similarity-based method performs an alignment with high similarity between sequences and can detect orthologous genomic regions between them. Multi-Lagan employs two primary stages to align N genomes with a phylogenetic binary tree connecting them:
- (i)
- Creation of initial global maps: the approximate global map between each sequence pair is determined using the Lagan algorithm;
- (ii)
- Progressive multiple alignment with anchors:
- (1)
- Alignment between the two closest sequences according to the binary phylogenetic tree is performed using the Lagan algorithm;
- (2)
- Each alignment is a new multi-sequence. The rough global maps of this multi-sequence and the closest sequence are identified;
- (3)
- Steps 2.1 and 2.2 are repeated, and global alignment is carried out between the multi-sequences and the closest sequence;
- (4)
- Step 2 is repeated until a multiple alignment of all the set of sequences has been performed (Figure 4).
 Figure 4. MUMmer and MUMmer 4 Pseudo-code.
Figure 4. MUMmer and MUMmer 4 Pseudo-code.
2.2.2. Mauve
Mauve is a genome alignment method which is able to identify entire similar regions between a set of genomes, such as rearrangements, translocation, and inversions, and the exact breakpoints of each rearrangement across multiple genomes [27]. Therefore, Mauve is primarily used for aligning and comparing multiple genomes from different species. It helps researchers identify conserved regions, rearrangements, and evolutionary events across genomes.
Mauve uses a progressive alignment algorithm, and the main originality of this algorithm is that unlike the other multiple genomes’ alignment methods, it uses a heuristic approach to predict whether the anchors represent a similar region between the genomes or only a similar region caused by random mutations. The experimental results made using the Progressive Mauve method shows that the algorithm was able to detect the pan-genome and the core genome of species from Enterobacteriaceae family. In addition to the Mauve alignment algorithm, a visualization tool has been developed to display the whole-genome alignment that shows the rearrangement between the genomes. Mauve offers several features to analyze and visualize the alignment, including the ability to zoom in and out of the alignment, rearrange genomes for better visualization, and export alignment data in various formats. Mauve is a valuable tool for comparative genomics research, providing insights into genome evolution and conservation across multiple organisms or strains. However, it may have limitations in handling extremely large genomes or genomes with high levels of divergence. Users should be aware of these limitations and consider alternative approaches for analyzing such datasets [31]. The algorithm constructs a similarity graph based on shared regions among genomes and then uses heuristics to identify and extend locally collinear blocks (LCBs) that represent conserved segments across genomes. Content and rearrangement are the two main techniques that are used in this method to better detect variable genes; the first scores all possible configurations of alignment anchors across the set of genomes [32], and the second applies the hidden Markov model (HMM) for homology to predict similar subsequences caused by random mutations and reject them, as in the following steps:
- (i)
- Finding multi-MUMs: While the algorithm detects anchors across multiple genomes comparison, some repetitive regions can occur several times in each genome as a duplication of those regions. The more aligned genomes there are, the more difficult it becomes to place each anchor in the correct place within the global alignment. To resolve this problem, Mauve uses multiple maximal unique matches (multi-MUMs) with a minimal length k as anchors. Those multi-MUMs are the exact matching subchains shared by two or more genomes that occur only once in each genome and that are bounded on either side by mismatched nucleotides. In addition, to detect other anchors of a length less than k, Mauve uses an anchoring technique that reduces k while looking for smaller anchors in the remaining unmatched regions.
- (ii)
- Calculating a phylogenic guide tree: Mauve uses the genomes’ similar regions indicated by the subset of multi-MUMs as a binary distance metric to build a phylogenetic guide tree using neighbor joining [25].
- (iii)
- Selecting a set of anchors: This step consists in the detection of homologous subsequences. These regions are called locally collinear blocks (LCBs). Each locally collinear block is a homologous region shared by two or more genomes and does not contain any rearrangement of similar blocks.
- (iv)
- Recursive anchoring and gapped alignment: The previous step may not detect all the regions of homology between the genomes, as a minimum length k is required for a region to be considered as an LCB. To resolve this problem, two recursive anchoring techniques are performed. The first technique consists in the detection of similar regions outside of LCBs to extend the number of LCBs and identify new ones. The second technique consists of detecting unanchored regions within LCBs. For that reason, new LCBs of the minimum length k may be identified as outside LCB regions.
- (v)
- Regions that are not unique in the entire genome may be unique in regions outside it.
Finally, Mauve performs a CLUSTAL-W alignment using the set of anchors as well as the genome guide tree generated at step 2 [33]. The progressive alignment algorithm is executed once for each adjacent anchor pair in every LCB and performs a global alignment for each LCB. Its complexity is O(n3), where n is the length of the input sequences (Figure 5).

Figure 5.
Mauve algorithm pseudo-code.
2.2.3. BLASTZ
BLASTZ is a bioinformatics tool used for aligning DNA sequences, particularly large genomic sequences or whole genomes, making it suitable for comparing entire genomes of different species or strains. It is widely used for identifying conserved regions, homologous genes, and evolutionary relationships among genomes. Many research projects have analyzed similarities between the human and the mouse genome. Many programs have been implemented to align the mouse and human chromosomes, such as Blast2sequences [34] and Pattern Hunter [35]. The high percentage of similarity between the two genomes has led researchers to implement a new algorithm that is able to detect chromosomal synteny and orthologous regions. The first use of the algorithm was to asess the conservation of synteny between human and mouse chromosomes. The experimental results showed that human Chromosome 20 is partially homologous to mouse Chromosome 2. In addition, only 3.3% of human Chromosome 20 nucleotides align outside of the mouse chromosome 2.
Overall, BLASTZ is a powerful tool for whole-genome sequence alignment, providing insights into genome evolution and functional conservation across different organisms. BLASTZ can also be integrated into larger bioinformatics pipelines or used through web-based interfaces. Additionally, it is related to other alignment tools, and its chaining strategy is like that used in the Axt tools, such as AxtBest.
The BLASTZ method employs a local alignment algorithm, emphasizing the identification of conserved regions within sequences. It uses a strategy called “chaining” to link multiple short alignments into longer, contiguous alignments, which helps identify regions of similarity even in the presence of genomic rearrangements [31].
First, the algorithm started by detecting similar regions between the two genomes that occur in the same order and orientation. Via this approach, it is possible to detect exact orthologous regions [36].
Second, BLASTZ aligns the other regions to detect small matching subsequences (seeds) and extend them while limiting the gap length to k (Figure 6). This step is based on looking for identical words of eight or nineteen consecutive nucleotides in each sequence. To increase sensitivity, the algorithm allows transitions between the nucleotides. BLASTZ has a complexity of O(k × n × m), where k is the number of hits found and n and m are the lengths of the two sequences being compared.
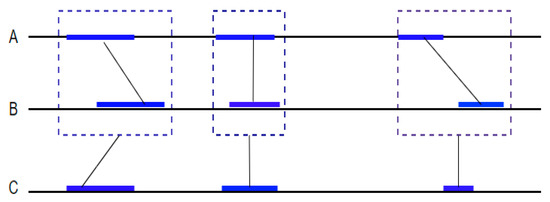
Figure 6.
Multi-sequence alignment using Lagan. The Lagan method performs pairwise alignment between the closest sequences A and B. Each alignment is considered a new multi-sequence to be compared with another sequence in the set which is C in this case.
2.2.4. LASTZ
LASTZ is a versatile alignment tool commonly used in bioinformatics for comparing DNA sequences, mainly used to perform alignment of complete chromosomes [37]. It is widely used in comparative genomics, evolutionary analysis, genome assembly, and sequence annotation. Additionally, it has been employed for tasks such as identifying conserved regions, detecting sequence variations, annotating genomic features, and studying genome rearrangements [38]. It is often integrated into bioinformatics pipelines and genome analysis workflows alongside other tools and resources.
LASTZ is known for its speed and sensitivity, making it suitable for aligning both closely related and distantly related sequences. It supports both global and local alignment modes, enabling users to align entire sequences (global) or identify regions of similarity (local), handling large genomes efficiently due to its memory-efficient indexing and alignment algorithms. Additionally, it is highly configurable, allowing users to specify parameters such as seed size, gap penalties, scoring matrix, and alignment sensitivity, which can be an advantage but also can be a disadvantage if its performance becomes sensitive to the choice of these parameters. Users need to carefully tune these parameters to balance sensitivity and specificity based on the specific characteristics of their sequences and the alignment task [39]. LASTZ has a complexity of O(k × n × m), where k is the number of hits found and n and m are the lengths of the two sequences being compared.
The LASTZ algorithm utilizes a seed-and-extend approach for DNA sequence alignment, as follows:
- (i)
- This algorithm starts by detecting short near matches (seeds) between target and query sequences;
- (ii)
- Next, these seeds are extended into longer alignments using a heuristic approach. An adaptive score threshold is calculated taking into account various factors such as the scoring matrix, gap penalties, and statistical significance;
- (iii)
- A dynamic programming approach is adopted to align the non-extended blocks and construct the final alignment result, similar to the Lagan method.
2.2.5. DIALLGN
DIALLGN (Differential Alignment of Gene Neighborhoods) is a powerful tool for comparative genomics research, enabling the identification and analysis of conserved gene neighborhoods across multiple genomes. It was designed for multiple sequence alignment and is particularly useful to detect local homologies in sequences with low overall similarities [40]. Researchers can use DIALLGN to gain insights into gene function, genome evolution, and regulatory mechanisms underlying the organization of genes in the genome.
Various versions of this algorithm have been implemented and their main innovation consists of the insertion of user-specified external information. The user can insert the two sequences’ anchors’ details, including the length of the anchor and its starting position in the sequences. DIALLGN’s performance may be affected by the quality of the input genomic sequences, the presence of genomic rearrangements or structural variations, and the parameters chosen for the analysis. Users should carefully interpret the results and validate the identified gene neighborhoods using additional experimental or computational methods.
The DIALLGN algorithm leverages sequence similarity and genomic context to identify conserved gene neighborhoods across multiple genomic sequences, through aligning and clustering genes with similar sequences and genomic positions [41].
- (i)
- The algorithm starts by identifying anchor genes that are conserved across the genomic sequences being compared. These anchor genes serve as reference points for aligning and extending the gene neighborhoods;
- (ii)
- Then, it extends the gene neighborhoods by searching for additional genes that are located nearby in the genomic sequences and exhibit sequence similarity to the anchor genes;
- (iii)
- It uses a scoring system to evaluate the similarity between genes and determine the quality of the alignments. The scoring criteria may include sequence identity, gap penalties, and other parameters that reflect the degree of conservation between genes;
- (iv)
- As the algorithm extends the gene neighborhoods and identifies conserved gene pairs, it clusters these genes together based on their proximity and sequence similarity.
The complexity of DIALLGN depends on the chosen parameters and the size of the input sequences. It typically has a worst-case time complexity of O(n3), where n is the length of the input sequences.
2.2.6. AnchorWave
AnchorWave stands as a versatile algorithm designed for whole-genome alignment, adept at efficiently aligning expansive genomic sequences while pinpointing areas of resemblance between them. It executes a process of whole-genome duplication-informed collinear anchor identification across genomes and conducts base-pair-resolved global alignment for collinear blocks, employing a two-piece affine gap cost strategy [42]. Compared with the nearest competitive approach, it accurately aligns up to three times more of the genome, encompassing position matches or indels, particularly notable when contrasting diverse genomes. Its ability to handle large datasets and produce high-quality alignments makes it valuable for comparative genomics studies and evolutionary analyses.
The algorithm starts by aligning the query genome with a reference genome annotation. Then, it uses a dynamic programming algorithm to find the collinear blocks between the two genomes. It may use the longest path to define these similar regions. The user can also decide to include paths containing inversions, rearrangements, and whole-genome duplication. The next step of the algorithm is optional. In order to reduce the size of non-similar blocks (inter-anchor-s blocks), the user can decide to try to identify new additional anchors. Finally, AnchorWave performs base-pair alignment using the two-piece affine gap cost strategy [43]. This pairwise alignment method reduces the error rate and assemble similar genomes up to 30 faster than existing alignment tools [44].
2.2.7. Minimap2
Minimap2 is a versatile alignment tool commonly used in bioinformatics and genomics research, being designed to efficiently align DNA sequences, typically whole genome sequences, against a reference genome or assembly [44] (Figure 7). It is widely used for tasks such as genome assembly, variant calling, structural variation detection, and transcriptome mapping. As an anchor-based approach, it uses a method based on hashing and chaining.

Figure 7.
Minimap2 algorithm pseudo-code.
This technique involves identifying “anchors” (similar or identical sequence regions) between the sequences being compared, and then chaining these anchors together to create an alignment. The efficiency and speed of Minimap2 largely stem from its ability to quickly identify and utilize these anchors for alignment [44]. The algorithm proceeds in two main stages: indexing and alignment. During indexing, Minimap2 preprocesses the reference genome to create an index structure that facilitates rapid sequence comparison. During alignment, it uses the index to efficiently locate and extend sequence matches between the query and reference genomes. Minimap2’s complexity depends on the size of the input sequences and the chosen parameters. It typically has a worst-case time complexity of O(n lg n), where n is the length of the reference sequence.
2.3. Graph-Based Homology Mapping Methods
The previous alignment methods have mainly been used for the detection of similarities at the DNA level. A new class of alignment methods has emerged to perform genome comparison in terms of gene composition and interspecies evolutionary relationships. These methods are graph-based methods that map the DNA/genetic composition of the genomes and, thus, highlight the evolutionary relationship between species, including syntenic, homologous, orthologous, and paralogous regions.
2.3.1. Mercator
Mercator is a multiple-whole-genome orthologous map construction algorithm. It is mainly used to identify the evolutionary relationships between multiple genomes [45]. First, the algorithm takes a set of exon annotation for each genome. Second, it compares all exons of all the genomes and builds an alignment between them. Third, the algorithm builds a graph where each vertex represents an exon and edges between vertexes represent the alignment score between exons.
Finally, the algorithm identifies cliques in this graph and joins neighboring cliques together to form similar anchors between the genomes. The cliques formed in each genome are used to highlight the orthologous blocks between them (Figure 8).
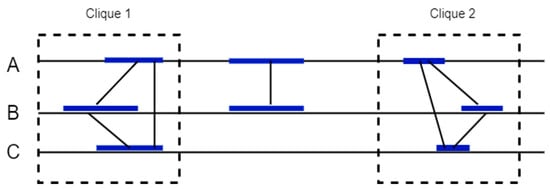
Figure 8.
Mercator orthologous gene detection between sequences A, B and C. Mercator builds a graph where each vertex represents an exon and edges between vertexes represent the alignment score between exons. The cliques formed are used to highlight the orthologous blocks between them.
2.3.2. Mugsy
Mugsy is a multiple-whole-genome alignment tool, primarily designed to align multiple genomes or genomic regions simultaneously, enabling researchers to compare and analyze evolutionary relationships, genomic rearrangements, and structural variations across different species or individuals. It is a graph-based algorithm that is mainly used for whole bacterial genome alignment and multiple human-chromosome alignment [46]. It first performs pairwise local alignment between sequences. Then, it constructs and uses an alignment graph to identify LCBs.
The main advantage of this algorithm is that it identifies genomic regions that are homologous, collinear, free of rearrangements, and suitable for multiple alignment. In addition, Mugsy can align 57 E. coli genomes (299 Mb) in <1 day on a single CPU. It can also perform an alignment of four assembled human chromosomes, completing the LCB identification and multiple alignment in <1 h. Muggsy’s complexity is O(n3), where nn is the length of the input sequences.
2.3.3. BubbZ
BubbZ is a bioinformatics algorithm designed for the identification and analysis of genomic structural variations (SVs), specifically large insertions and deletions (indels), in whole-genome sequencing data. It is a fast whole-genome homology mapper which detects pairwise chains in homologous blocks using a de Bruijn graph [47]. The algorithm was recently tested on closely related mammalian genomes and a large collection of bacterial genomes. The algorithm identifies bubbles in the de Bruijn graph, which represent regions of potential structural variation. A bubble occurs when there are multiple alternative paths (alleles) through the graph for a specific genomic region. Then, it expands each detected bubble by traversing alternative paths through the de Bruijn graph. It performs variant calling to classify the structural variations into different categories, such as insertions, deletions, duplications, or complex rearrangements. Based on the graph topology and read alignments, the algorithm determines the size, orientation, and sequence content of each variant and assigns confidence scores to the detected structural variants based on various factors, including read coverage, allele frequency, and consistency across sequencing reads [48]. The complexity of BubbZ depends on the size and complexity of the input sequences. It typically has a worst-case time complexity of O(n2), where n is the length of the input sequences.
2.3.4. SibeliaZ
SibeliaZ-LCB is suitable for detecting homologous sequences that have an evolutionary distance to the most recent common ancestor (MRCA) of at most 0.09 substitutions per site [39]. This algorithm is used to identify collinear blocks in closely related genomes based on the construction of a de Bruijn graph [39] to build collinear blocks (Figure 9). It globally aligns the collinear blocks to generate the whole-genome alignment. Thus, SibeliaZ-LCB performs pairwise sequence alignments between the input genomes to identify regions of similarity and homology. It uses efficient alignment algorithms to detect conserved regions and infer synteny relationships; based on the alignment, it constructs locally collinear blocks (LCBs) by grouping aligned regions that exhibit consistent gene order and orientation across the genomes. LCBs are represented as blocks of homologous sequences that are likely to have originated from a common ancestor.

Figure 9.
SibeliaZ algorithm pseudo-code.
On sixteen recently assembled mice genomes, SibeliaZ ran in less than 16 h on a single machine, while other tools did not run to completion for eight mice within a week. It also provided better performance in terms of time of execution compared with the Mercator tool. The complexity of SibeliaZ depends on the size of the input genomes and the chosen parameters. However, it typically has a worst-case time complexity of O(n2), where n is the total length of the input sequences.
2.3.5. Progressive Cactus
Progressive Cactus employs a graph-based method for whole-genome alignment, introducing a novel methodology through a progressive alignment strategy [49]. This approach systematically aligns multiple genomes by constructing and manipulating a graph structure that represents relationships between different genomic sequences.
Key Steps and Graph-Based Methodology:
- Initial Pairwise Alignment:
- The process begins with aligning two closely related genomes using a pairwise alignment algorithm;
- This initial alignment is used to create a baseline graph structure, where nodes represent sequences and edges represent alignment matches, identifying regions of sequence similarity and dissimilarity;
- Additional Genome Integration:
- As additional genomes are integrated, the alignment graph is expanded iteratively, one genome at a time;
- Each new genome is incorporated into the existing graph by identifying sequences and aligning these to the graph’s nodes, updating the graph structure to include new alignment paths;
- This step ensures optimal alignment quality across the entire set of genomes, leveraging the graph structure to maintain comprehensive genomic relationships;
- Progressive Refinement:
- The existing graph undergoes iterative refinement as new genomes are added, optimizing the graph structure based on sequence homologies and structural constraints;
- This refinement process adjusts the graph’s edges and nodes to enhance alignment accuracy, maintaining both sequence similarity and structural integrity across the graph;
- Iterative Interaction:
- Progressive Cactus iterates through the alignment process multiple times, refining the alignment graph at each step;
- Each interaction improves the graph’s accuracy and consistency, ensuring that the final graph represents the most accurate genomic relationships;
- Parallelization:
- To enhance computational efficiency, Progressive Cactus employs parallelization techniques;
- The graph manipulation and alignment processes are distributed across multiple computing nodes or cores, enabling faster processing of the alignment graph for large genomic datasets.
The use of a graph-based method allows Progressive Cactus to efficiently manage and process complex genomic alignments through representing sequences and their relationships in a dynamic, scalable structure. This graph-based approach facilitates the handling of large-scale genomic datasets and supports detailed analysis of genomic similarities and variations across multiple species.
The complexity of Progressive Cactus varies depending on the size and complexity of the input genomes. It generally has a time complexity of O(n2 lg n) for aligning n genomes.
2.3.6. GraphAligner
GraphAligner is designed to align long reads to a graph-based reference. The algorithm treats the reference genome as a graph where variations and alternative splicing events are represented as branches [50]. This allows the alignment to accommodate complex genomic structures that are not linearly represented.
Steps and Key Points:
- Graph Construction:
- The reference genome is first constructed into a graph where nodes represent sequence blocks and edges represent possible transitions between these blocks. This can include linear sequences as well as alternative paths representing variations or repeating elements;
- Seed Finding:
- The algorithm begins with identifying seed matches between the read and sub-sequences in the graph. Seeds are typically exact matches that are used as anchor points for more detailed alignment;
- Seed Extension:
- Using dynamic programming, seeds are extended along the paths of the graph. The extension process respects the graph’s topology, exploring different paths where the read might align with segments of the reference;
- Path Scoring and Selection:
- After extension, paths are scored based on the alignment’s quality, and the best-scoring path is selected as the optimal alignment. The scoring system considers mismatches, gaps, and the overall coherence of the alignment within the graph structure;
- Handling Complexity:
- GraphAligner is specifically adept at handling complex graph structures with varying paths, such as those introduced by repeats or structural variants, which are often challenging for linear alignment methods.
GraphAligner is particularly useful in genomic studies where the reference genome includes significant alternative paths, such as in cancer genomics, where somatic mutations, insertions, deletions, and rearrangements are common, or in research involving highly polymorphic species.
The complexity of GraphAligner depends on the size of the reference graph and the length of the read sequences. It typically has a worst-case time complexity of O(k × n × m), where k is the number of nodes in the graph, n is the length of the reference sequence, and m is the length of the read sequence.
2.4. Hashing Techniques in Genome Alignment
Hashing is a cornerstone computational technique used extensively across various domains, including bioinformatics and genomics. In the context of genome alignment, hashing techniques facilitate the rapid mapping of sequencing reads to a reference genome by efficiently indexing sequence data. This method leverages hash functions to convert variable-length sequences into fixed-length hash values, which serve as unique identifiers for subsequences or “k-mers” within the genomic data. Through creating hash tables that map these hash values to their corresponding locations in the reference genome, alignment tools can quickly identify potential matching regions, significantly speeding up the alignment process.
2.4.1. Tools Utilizing Hashing Techniques
Several alignment tools employ hashing techniques to enhance their performance and accuracy, each optimized for specific types of analyses:
- SOAP: SOAP (short oligonucleotide alignment program) uses a straightforward hashing strategy to align short reads against a reference genome [51]. It is particularly efficient in handling throughput sequencing data, making it a popular choice for SNP discovery and genotyping analyses (Figure 10). The complexity of SOAP depends on the size and complexity of the input sequences. It typically has a worst-case time complexity of O(n2), where n is the length of the input sequences;
 Figure 10. SOAP algorithm pseudo-code.
Figure 10. SOAP algorithm pseudo-code.
- Stampy: While primarily relying on the Burrows–Wheeler transform for alignment, Stampy incorporates hashing to improve alignment accuracy in regions with high sequence variability [52]. This makes it exceptionally useful for aligning reads from organisms with significant genetic diversity or in studies focusing on evolutionary differences;
- BLAST: The aasic local alignment search tool (BLAST) is a widely-used algorithm for comparing an inquiry sequence against a database or reference sequence [34]. BLAST employs hashing to rapidly find regions of local similarity, facilitating a broad range of analyses from gene identification to comparative genomics;
- GMAP: Optimized for spliced alignments, such as those needed in RNA-seq data analysis, GMAP uses hashing to index the genome [53], allowing efficient identification of splice junctions and exons (Figure 11). Its capability to handle long reads makes it suitable for transcriptome studies where reads span multiple exons. GMAP’s complexity is O(n), where n is the length of the reference sequence.
 Figure 11. GMAP algorithm pseudo-code.
Figure 11. GMAP algorithm pseudo-code.
2.4.2. Efficiency and Applications
Hashing techniques contribute to the efficiency of genome alignment tools by reducing the computational complexity of searching for sequence matches. This efficiency is critical for analyzing the vast datasets generated through NGS technologies, enabling real-time data processing and analysis.
- Efficiency in SNP discovery: Tools like SOAP leverage hashing for rapid identification of potential SNP locations via quickly mapping reads to the reference genome [54], a process integral to variant calling and genotyping studies;
- Handling genetic diversity: Stampy’s use of hashing to accommodate high sequence variability allows researchers to study populations with significant genetic differences, making it invaluable for evolutionary biology and conservation genetics [55];
- Comparative genomics with BLAST: BLAST’s hashing-based search mechanism is efficient in identifying homologous sequences across species, aiding in the annotation of novel genomes and the discovery of evolutionary conserved regions [56].
In conclusion, hashing techniques enhance the capability of genome alignment tools to manage the complexities and demands of modern genomic analyses. Through facilitating rapid and accurate alignment of sequencing reads to reference genomes, tools employing hashing are indispensable for a wide range of genomic studies, from variant discovery and comparative genomics to transcriptomics and beyond. Their continued development and integration into bioinformatics software will remain a key factor in the advancement of genetic and genomic research.
3. Algorithmic Aspects of WGA Algorithms
3.1. Performance Characteristics
The performance of whole-genome alignment (WGA) algorithms is often evaluated based on their speed, memory usage, scalability, and accuracy [44]. The speed at which an algorithm can process and align genomes is crucial in handling the ever-increasing size of genomic data. Memory usage determines the size of genomes or datasets that the algorithm can handle at once [45].
Scalability in the context of WGA algorithms refers to the ability of an algorithm to handle increasing volumes of data without a proportional increase in computational resources. This is a critical characteristic, especially when dealing with larger and more complex genomes [46].
The accuracy of WGA algorithms is usually assessed via comparing the generated alignment with a reference alignment or evaluating the biological relevance of the alignment. Accuracy is influenced by the algorithm’s ability to correctly identify homologous regions, handle genomic rearrangements, and align divergent sequences [39,47].
3.2. Methodological Underpinnings
The methodological underpinnings of WGA algorithms can be broadly categorized into heuristic and exact methods. Heuristic methods, such as MUMmer and BLASTZ, generate approximate alignments rapidly but may not always produce the optimal alignment [57,58]. These methods are often used for the initial stage of alignment to identify regions of similarity, which are then refined using more accurate methods [59].
Exact methods, on the other hand, guarantee finding the optimal alignment but are typically slower and require more computational resources. Such methods, including those based on dynamic programming, are often used for refining alignments or for aligning smaller regions of the genome [60,61].
Recent advancements in WGA algorithms have seen the development of methods that strike a balance between speed and accuracy. For instance, tools like Cactus and Progressive Cactus utilize a combination of heuristic and exact methods to generate accurate alignments quickly [62]. These tools use heuristic methods for the initial alignment and then apply exact methods to refine the alignment, resulting in a balance of speed and accuracy.
WGA algorithms also vary in their ability to handle genomic rearrangements, such as inversions, duplications, and translocations. Some algorithms, like Mauve and Progressive Cactus, have built-in features for handling these rearrangements [27]. Other algorithms may require additional steps or tools to correctly align regions with genomic rearrangements [27].
Finally, the choice of a suitable WGA algorithm often depends on the specific requirements of the research question. For instance, studies focusing on closely related species may benefit from algorithms that excel at aligning highly similar sequences, while studies on divergent species may require algorithms that can handle high levels of sequence divergence [63,64].
In summary, the algorithmic aspects of WGA algorithms, from their performance characteristics to their methodological underpinnings, are key factors influencing their suitability for different research applications. Continued advancements in these aspects will undoubtedly contribute to the ongoing evolution of the field of whole-genome alignment [65,66].
4. Recent Advancements in WGA Algorithms
The field of whole-genome alignment has seen significant progress over the years. With the rapid advancements in sequencing technologies and the ever-increasing amount of genomic data, the development of efficient and accurate WGA algorithms has become more critical than ever [67].
One of the major advancements in WGA algorithms has been the development of methods that can handle large-scale genomic rearrangements. While traditional alignment algorithms often struggle with these rearrangements, newer tools like Cactus and Progressive Cactus have incorporated mechanisms to accurately align regions with inversions, duplications, and translocations [62]. These tools use graph-based approaches, allowing them to model and align complex genomic structures [68] (Table 1).
Another significant advancement is the development of algorithms that can align divergent genomes. Traditionally, aligning divergent genomes has been challenging due to the high levels of sequence divergence and the presence of unique genomic elements. However, tools like LASTZ and MULTIZ have been designed to tackle these challenges, enabling the alignment of divergent genomes [25,69].
The advent of cloud computing and parallel processing has also influenced the development of WGA algorithms. Tools like Cloud Aligner (alpha version) and ParaGraph have leveraged these technologies to perform alignments on a massive scale, handling large volumes of data and multiple genomes simultaneously [70,71]. This has significantly improved the speed and scalability of WGA algorithms, allowing researchers to undertake more ambitious projects.
Incorporating machine learning techniques into WGA algorithms is another promising direction. Through learning from existing alignments, these algorithms can potentially improve their accuracy and efficiency. For instance, tools like Deep Align have demonstrated the potential of machine learning to improve the accuracy of sequence alignments [72].
Furthermore, the integration of WGA algorithms with other bioinformatics tools has also seen notable progress. For example, pipelines like EAGER2 integrate WGA tools with other bioinformatics software, providing a comprehensive solution for genomic analysis [73]. This not only simplifies the analysis process but also enhances the utility of WGA algorithms in research.
In conclusion, the recent advancements in WGA algorithms have significantly improved their performance and versatility, catering to a wider range of research applications. As the field continues to progress, it will be exciting to see how these advancements will shape the future of whole-genome alignment [74].
5. Comprehensive Analysis of Genomic Comparison Tools: Human and Diverse Genomes
To comprehensively evaluate the effectiveness of various whole-genome alignment tools across diverse evolutionary contexts, we conducted a rigorous study utilizing multiple alignment methodologies. Our analysis encompassed alignments between human genomes, human vs. mouse genomes, and genomes of Caenorhabditis elegans (C. elegans) and Saccharomyces cerevisiae (baker’s yeast), representing species with considerable evolutionary divergence.
This study included the utilization of well-established alignment tools such as MUMmer4 (suffix tree-based), SibeliaZ (graph-based), Dialign2 (anchor-based), and Minimap2 (anchor-based). Additionally, we incorporated Progressive Cactus 2.8 and GraphAlign tools into our evaluation framework to assess their performance alongside the established tools.
These analyses were conducted on a server equipped with dual E5-2699 v4 processors and 512 GB of RAM, operating on CentOS 7. Through including alignments across different species and evolutionary distances, our study aimed to provide researchers with a comprehensive understanding of alignment tool performance under varying biological contexts.
5.1. Analyses of Human Genomes
The initial phase of our study used MUMmer4, Sibeliaz, Dialign2, and Minimap2 for analyzing two human genomes. The contrasting results from these tools provided insights into their specific functionalities and limitations:
- MUMmer4 demonstrated a remarkable ability to detect near-identical sequences in human genomes, reporting a similarity of almost 100%. This high similarity index indicates that MUMmer4 is particularly efficient at aligning genomes with minimal genetic variations, making it an ideal tool for studies where the genomes are closely related;
- Sibeliaz presented a different perspective, reporting a much lower similarity percentage. Sibeliaz’s methodology, which focuses on k-mer pattern analysis, allows it to uncover subtle variations that direct sequence comparison methods might miss. This attribute is particularly beneficial for research that delves into genomic diversity, mutation analysis, and evolutionary biology;
- Dialign2 encountered limitations with the human genomes, primarily due to their size. This outcome underscores the necessity of considering genomic data scale when selecting analytical tools, particularly for large and complex genomes;
- Mirroring MUMmer4 in effectiveness, Minimap2 showed a 100% mapping rate, aligning the human genomes completely. It demonstrated additional capabilities in managing complex mapping situations, indicated by the presence of secondary and supplementary alignments, thus offering a broader scope in genomic analysis;
- The alignment process using GraphAligner failed, resulting in an empty output GAM file.
- Progressive Cactus encountered challenges in achieving the anticipated alignments across whole human genomes, demonstrating successful alignment only for chromosomes. This outcome underscores the necessity for refinement in accommodating the diversity and complexity of genomic datasets. The alignment between human Chromosome 1 and Chromosome 2 was effectively visualized using Circos, based on the output generated by Progressive Cactus. This visualization highlighted the comparative genomic architecture and facilitated the identification of conserved and divergent regions between these two chromosomes (Figure 12).
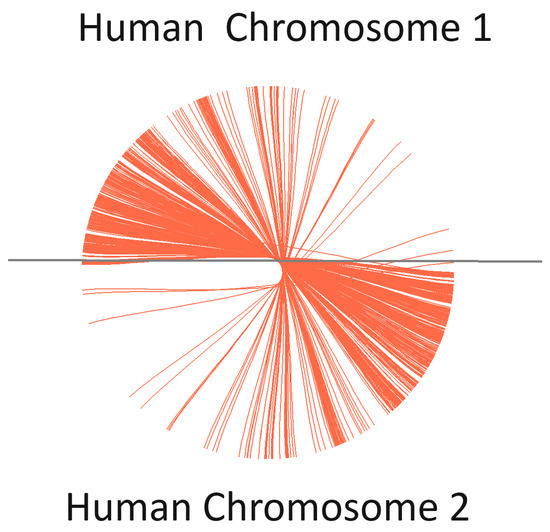 Figure 12. Comparative genomic visualization of human Chromosomes 1 and 2, using Circos based on Progressive Cactus output.
Figure 12. Comparative genomic visualization of human Chromosomes 1 and 2, using Circos based on Progressive Cactus output.
5.2. Analysis of Human and Mouse Genomes
Performing whole-genome alignment between human and mouse genomes offers valuable insights into evolutionary relationships, genetic conservation, and functional similarities between these species. Through comparing their entire genomic sequences, researchers can elucidate conserved regions, identify genetic variations, and unravel the molecular mechanisms underlying biological processes shared across mammalian species.
- Mummer4: During the execution of the “nucmer” command, we encountered an error related to the construction of the suffix tree, indicating that the input sequence length exceeded the maximum allowable limit. As a result, the alignment process failed to proceed, preventing the comparison between the human and mouse genomes using MUMmer;
- SibeliaZ: The analysis indicated that the human and mouse genomes shared a substantial number of junctions, highlighting common genomic features and evolutionary conservation between the two species. The identification of 1,784,620 blocks suggested regions of significant sequence similarity, potentially corresponding to conserved genes, regulatory elements, or functional domains across the genomes. The coverage level of 15% suggested that a considerable portion of the genomes aligned with each other, indicating a substantial level of evolutionary conservation and shared ancestry between humans and mice.
- Progressive Cactus: This method faced obstacles in achieving the expected alignments between entire human and mouse genomes, mirroring its performance limitations observed in human-to-human genome comparisons. This suggests that the tool may be better suited for shorter read comparisons, such as chromosome-level alignments, rather than whole-genome analyses spanning across species.
- Dialign2: When applied to the alignment of human versus mouse genomes, Dialign2 faced significant challenges, attributable in particular to the substantial size of these genomes. In the same way, difficulties were encountered in the human vs. human genome alignment, resulting in a failure to achieve the alignment. This outcome mirrored the difficulties encountered in aligning large genomes and underscored the importance of refining alignment methodologies to effectively handle such complex datasets.
- GraphAligner: the alignment process using GraphAligner failed, resulting in an empty output GAM file;
- Minimap2: Out of the total reads processed (2589), 98.45% reads were successfully mapped. The high mapping rate of 98.45% indicated a close evolutionary relationship between the human and mouse genomes, facilitating robust alignment of sequencing reads between these species.
5.3. Analysis of C. elegans and Baker’s Yeast Genomes
The study was then extended to a comparison of C. elegans and baker’s yeast genomes to evaluate the tools under significantly different genomic conditions:
- In stark contrast to its performance with human genomes, MUMmer4 yielded no output for the C. elegans and baker’s yeast genomes. This indicated MUMmer4′s limitations in aligning genomes that are not closely related, thereby suggesting its niche application in genomic studies;
- Sibeliaz revealed only 12% coverage in conserved regions between C. elegans and baker’s yeast, identifying 21,816 distinct blocks. This low coverage indicated a substantial evolutionary distance between these species. Sibeliaz’s strength lies in its ability to analyze and compare genomes with significant structural and evolutionary variations, making it a versatile tool for comparative genomics across diverse species;
- Like its performance with human genomes, Dialign2 was unsuccessful in processing the data from C. elegans and baker’s yeast, further highlighting its limitations in handling diverse genomic data.
- Unlike its effective mapping of human genomes, Minimap2 failed to map the C. elegans and baker’s yeast genomes, indicating challenges in aligning significantly divergent genomes.
5.4. Execution Times and Tool Complexity
In our comparative analysis of genomic alignment tools applied to human genomes, we observed significant variations in execution times, reflecting each tool’s computational approach and efficiency. MUMmer4 took approximately 15 h and 34 min, indicating its intensive computational process, especially effective for aligning highly similar sequences. Sibeliaz completed its analysis of the human genomes in about 6 h and 19 min, demonstrating its efficiency in handling complex k-mer pattern analysis despite the large data size. For the comparison between human and mouse genomes, the analysis took 22 h, indicating a longer processing time due to the increased complexity of comparative genomic analysis. Dialign2 faced challenges with the human genome size, leading to a crash and underscoring the need for more scalable solutions in genomic analysis tools. Minimap2 stood out for its speed, completing the mapping in just 44 min, demonstrating its capability for rapid and efficient genomic alignment. These varying execution times are indicative of the underlying algorithms and computational strategies employed by each tool, highlighting the importance of considering both accuracy and efficiency when selecting genomic analysis tools for large-scale studies (Figure 13). GraphAlign consistently failed during the analyses, probably due to computational constraints, especially when aligning large genomes used for analyses (human vs. human and human vs. mouse).
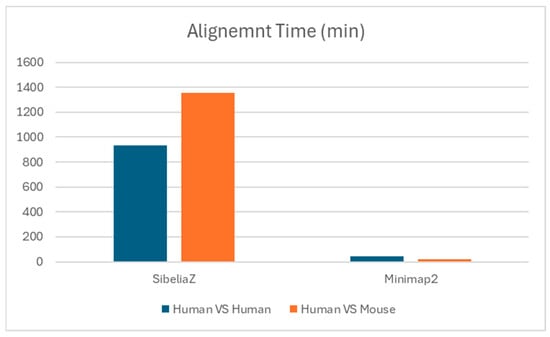
Figure 13.
Human vs. human and human vs. mouse whole-genome alignment (WGA) execution time for Minimap2 and SibeliaZ software.
5.5. Methodological Insights
Our study revealed distinct strengths and challenges for each tool. MUMmer4 and Minimap2, effective for similar genomes, struggled with significant genomic divergence. Sibeliaz excelled in analyzing complex and varied genomic data. Dialign2’s limitations underscore the necessity for robust, scalable tools in modern genomic research.
This comparative analysis emphasizes the importance of tool selection based on genomic data characteristics. The choice of tools, influenced by their methodologies—suffix trees, k-mer analysis, or hashing and chaining algorithms—significantly impacts genomic data interpretation. Furthermore, computational time, a critical factor in genomic research, ranged from 44 min (Minimap2) to over 15 h (MUMmer4), underscoring the need to balance accuracy and efficiency in tool selection.
On the other hand, GraphAligner, Progressive Cactus, and Dialign2 failed to achieve human vs. human whole-genome alignment. This outcome highlights the challenges faced by these tools, potentially attributed to computational limitations or the complexity of the task at hand. (Figure 14).
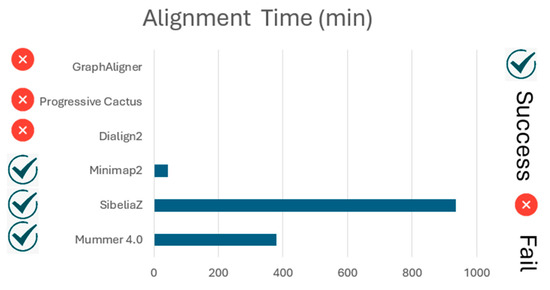
Figure 14.
Human vs. human whole-genome alignment (WGA) execution time per software.
6. Challenges in Whole-Genome Alignment
Whole-genome alignment (WGA) has been a tremendous asset to the scientific community, enabling insights into evolutionary biology, comparative genomics, and disease pathology, among other fields. However, the complex nature of genomic data and the increasing demands for accurate and efficient analysis present several challenges that need to be addressed. Additionally, the dynamic nature of this field also opens up new directions for future research and development [75].
6.1. Computational Challenges
Computational challenges in WGA span from the handling of large and complex datasets to the efficiency of alignment algorithms. The exponential increase in the volume of genomic data due to advancements in sequencing technologies necessitates WGA algorithms that can handle large datasets efficiently and accurately [76].
Temporal and spatial complexities are significant challenges. As the size of the genomes increases, the time taken for alignment and the memory required for processing also increase exponentially. Even with advancements in algorithmic design and computational power, dealing simultaneously with large genomes or multiple alignments remains a daunting task [77].
The implementation of parallel and distributed computing strategies can help mitigate these challenges to some extent. However, these approaches also require specialized knowledge and resources and can be associated with significant costs [78].
6.2. Biological Relevance
Ensuring biological relevance in the results produced with WGA algorithms is paramount. The complexity of genomes, including structural variations and non-coding regions, presents a significant challenge for alignment [79]. Misinterpretation or oversights can lead to inaccurate biological conclusions, highlighting the importance of precision in WGA algorithms [80].
Understanding the biological significance of alignment results is another challenge. This often involves complex bioinformatics analyses, requiring a wide array of skills and expertise [81].
6.3. Future Directions
There are several promising directions for future research in WGA. The development of more efficient and scalable algorithms remains a top priority. This includes methods that can handle large genomes and multiple alignments more efficiently, as well as algorithms that can better account for structural variations and non-coding regions [82].
Furthermore, there is a need for user-friendly and accessible WGA tools. These would make WGA more accessible to researchers from various backgrounds, promoting interdisciplinary research and broadening the applications of WGA [70].
The integration of machine learning and artificial intelligence techniques in WGA also presents a promising direction. These techniques could potentially enhance the accuracy, efficiency, and scalability of WGA algorithms and offer new ways to interpret and visualize alignment results [83].
In summary, the field of WGA continues to evolve, with numerous challenges to address and exciting avenues for future research. Collaboration between bioinformaticians, computer scientists, biologists, and other stakeholders will be critical in pushing the boundaries of what can be achieved with WGA [84].
7. Discussion
The advancements in whole-genome alignment (WGA) over the past few decades have been remarkable, providing critical insights into the genetic underpinnings of life. However, as we have discussed in the preceding sections, there are still significant challenges and opportunities that lie ahead [75].
The computational challenges in WGA, particularly those associated with handling large and complex genomic datasets, remain a key obstacle. Despite the significant strides made in improving the computational efficiency of WGA algorithms, the exponential growth of genomic data continues to pose challenges [82]. This necessitates the development of even more scalable and efficient algorithms that can cope with the volume and complexity of modern genomic datasets. Also, the potential of parallel and distributed computing in enhancing the efficiency of WGA has been recognized, but the cost, resources, and expertise required by these approaches limit their adoption [85].
Ensuring the biological relevance of WGA results is another critical challenge. The complexity of genomes, including the presence of structural variations and non-coding regions, increases the difficulty of producing biologically accurate alignments. It also amplifies the need for precise and sophisticated WGA algorithms. Even with accurate alignments, understanding the biological significance of the results requires complex bioinformatic analyses, highlighting the need for interdisciplinary collaboration in the field [86].
The integration of machine learning and artificial intelligence techniques into WGA could potentially enhance the accuracy, efficiency, and scalability of WGA algorithms and offer innovative ways to interpret and visualize alignment results [87]. Additionally, developing user-friendly and accessible WGA tools would not only make WGA more accessible to a broader range of researchers but also promote interdisciplinary research, ultimately expanding the applications of WGA [72].
Therefore, although there are significant challenges to overcome, the future of WGA is undoubtedly promising. Through continued research, collaboration, and innovation, we can look forward to new breakthroughs that will further our understanding of genomes and fuel discoveries in various fields of biology [84].
8. Conclusions
Whole-genome alignment (WGA) stands as a cornerstone in comparative genomics, illuminating insights into evolutionary processes, species relationships, and functional genomics. As we have reviewed, the field has seen substantial advancement in terms of algorithmic development, with diverse methodologies introduced and continually refined to tackle the inherent complexity of genomic data.
However, we have also highlighted that this field continues to face significant computational and biological challenges. The sheer scale of genomic data, coupled with its complexity, calls for increasingly efficient and scalable algorithms. The need for biological relevance further underscores the necessity for sophisticated and precise WGA methodologies. Moreover, the effective interpretation of WGA results requires deep bioinformatic analyses, thus emphasizing the need for interdisciplinary collaboration.
There is considerable potential for this field. The incorporation of machine learning and artificial intelligence techniques into WGA methodologies offers exciting prospects for enhancing accuracy and scalability. Efforts towards making WGA tools more user-friendly and accessible will democratize the use of these tools, promoting interdisciplinary research and expanding the applications of WGA.
In conclusion, the challenges faced in the field of WGA are significant, but they are outweighed by the potential for growth and discovery. As we continue to refine and innovate WGA methodologies, we can anticipate new insights into genomic organization and function, further driving the fields of evolutionary biology, genomics, and beyond.
Author Contributions
B.S. wrote the initial draft of the review. E.S. updated various sections and revised the references. J.Z., M.M.M.M. and T.Z. thoroughly revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by: (1) 2020–2022 National Natural Science Foundation of China under Grand No. 52001039; (2) 2020–2022 Shandong Natural Science Foundation in China No. ZR2019LZH005; (3) 2022–2025 National Natural Science Foundation of China under Grand, No.52171310; (4) Science and Technology on Underwater Vehicle Technology Laboratory under Grant 2021JCJQ-SYSJJ-LB06903.
Data Availability Statement
The genomic datasets used in our analysis are available in the NCBI database. For the human genome, we accessed the primary human reference genome (GRCh38) and the mitochondrial genome (GRCh38 mitochondrial assembly). For the comparative analysis involving different species, the genome of Caenorhabditis elegans (accession GCF_000002985.6) was used, and for the baker’s yeast, Saccharomyces cerevisiae, we utilized the genome with the accession GCA_000146045.2. For Mus musculus (house mouse), we used the reference genome (GRCm39). The source code used for these analyses can be downloaded from the Github repository: https://github.com/BacemDataScience/WholeGenomeAlignment (accessed on 12 March 2024).
Acknowledgments
We would like to thank all the organizers of the 2017 Bioinformatics and Genome Analyses course which was held at the Pasteur Institute in Tunisia. The participation of Bacem Saada in this training inspired him to write this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guerfali, F.; Laouini, D.; Boudabous, A.; Tekaia, F. Designing and running an advanced Bioinformatics and genome analyses course in Tunisia. PLoS Comput. Biol. 2019, 15, e1006373. [Google Scholar] [CrossRef]
- Goldfeder, R.L.; Wall, D.P.; Khoury, M.J.; Ioannidis, J.P.A.; Ashley, E.A. Human Genome Sequencing at the Population Scale: A Primer on High-Throughput DNA Sequencing and Analysis. Am. J. Epidemiol. 2017, 186, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef] [PubMed]
- Medina-Medina, N.; Broka, A.; Lacey, S.; Lin, H.; Klings, E.; Baldwin, C.; Steinberg, M.; Sebastiani, P. Comparing Bowtie and BWA to align short reads from a RNA-Seq experiment. In Proceedings of the 6th International Conference on Practical Applications of Computational Biology & Bioinformatics, Salamanca, Spain, 28–30 March 2012; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Pinese, M.; Lacaze, P.; Rath, E.M.; Stone, A.; Brion, M.-J.; Ameur, A.; Nagpal, S.; Puttick, C.; Husson, S.; Degrave, D.; et al. The Medical Genome Reference Bank contains whole genome and phenotype data of 2570 healthy elderly. Nat. Commun. 2020, 11, 435. [Google Scholar] [CrossRef]
- Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.; Calvo, F.; Eerola, I.; Gerhard, D.S.; Guttmacher, A. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Blake, J.A.; Baldarelli, R.; Kadin, J.A.; Richardson, J.E.; Smith, C.L.; Bult, C.J.; Mouse Genome Database Group. Mouse Genome Database (MGD): Knowledgebase for mouse–human comparative biology. Nucleic Acids Res. 2021, 49, D981–D987. [Google Scholar] [CrossRef]
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Ai, R.; Aken, B.; Akiyama, J.A.; Jammal, O.A.; Amrhein, H.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar]
- Morgenstern, B. DIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 1999, 15, 211–218. [Google Scholar] [CrossRef]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar] [CrossRef]
- Gusfield, D. Algorithms on stings, trees, and sequences: Computer science and computational biology. ACM Sigact News 1997, 28, 41–60. [Google Scholar] [CrossRef]
- Farruggia, A.; Gagie, T.; Navarro, G.; Puglisi, S.J.; Siren, J. Relative Suffix Trees. Comput. J. 2018, 61, 773–788. [Google Scholar] [CrossRef]
- Tian, Y.; Tata, S.; Hankins, R.A.; Patel, J.M. Practical methods for constructing suffix trees. VLDB J. 2005, 14, 281–299. [Google Scholar] [CrossRef]
- Delcher, A.L.; Kasif, S.; Fleischmann, R.D.; Peterson, J.; White, O.; Salzberg, S.L. Alignment of whole genomes. Nucleic Acids Res. 1999, 27, 2369–2376. [Google Scholar] [CrossRef]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Yang, T.; Liu, R.; Luo, Y.; Hu, S.; Wang, D.; Wang, C.; Pandey, M.K.; Ge, S.; Xu, Q.; Li, N.; et al. Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat. Genet. 2022, 54, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Soares, I.; Goios, A.; Amorim, A. Sequence comparison alignment-free approach based on suffix tree and L-words frequency. Sci. World J. 2012, 2012, 450124. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Mäkinen, V. Compressed full-text indexes. ACM Comput. Surv. (CSUR) 2007, 39, 2-es. [Google Scholar] [CrossRef]
- Su, W.; Liao, X.; Lu, Y.; Zou, Q.; Peng, S. Multiple sequence alignment based on a suffix tree and center-star strategy: A linear method for multiple nucleotide sequence alignment on spark parallel framework. J. Comput. Biol. 2017, 24, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Guo, M.Z.; Wang, X.K.; Zhang, T.T. An Algorithm for DNA Multiple Sequence Alignment Based on Center Star Method and Keyword Tree. Acta Electonica Sin. 2009, 37, 1746–1750. [Google Scholar]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2016, 17, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Linard, B.; Lecompte, O.; Poch, O. A comprehensive benchmark study of multiple sequence alignment methods: Current challenges and future perspectives. PLoS ONE 2011, 6, e18093. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S.; Program, N.C.S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]
- Wan, X.; Karniadakis, G.E. An adaptive multi-element generalized polynomial chaos method for stochastic differential equations. J. Comput. Phys. 2005, 209, 617–642. [Google Scholar] [CrossRef]
- Eppstein, D.; Galil, Z.; Giancarlo, R.; Italiano, G.F. Sparse dynamic programming I: Linear cost functions. J. ACM 1992, 39, 519–545. [Google Scholar] [CrossRef]
- Popendorf, K.; Tsuyoshi, H.; Osana, Y.; Sakakibara, Y. Murasaki: A fast, parallelizable algorithm to find anchors from multiple genomes. PLoS ONE 2010, 5, e12651. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.A.; Madden, T.L. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999, 174, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Tromp, J.; Li, M. PatternHunter: Faster and more sensitive homology search. Bioinformatics 2002, 18, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Kent, W.J.; Smit, A.; Zhang, Z.; Baertsch, R.; Hardison, R.C.; Haussler, D.; Miller, W. Human–mouse alignments with BLASTZ. Genome Res. 2003, 13, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Bu, L.; Wang, Q.; Gu, W.; Yang, R.; Zhu, D.; Song, Z.; Liu, X.; Zhao, Y. Improving read alignment through the generation of alternative reference via iterative strategy. Sci. Rep. 2020, 10, 18712. [Google Scholar] [CrossRef] [PubMed]
- Minkin, I.; Medvedev, P. Scalable multiple whole-genome alignment and locally collinear block construction with SibeliaZ. Nat. Commun. 2020, 11, 6327. [Google Scholar] [CrossRef]
- Al Ait, L.; Yamak, Z.; Morgenstern, B. DIALIGN at GOBICS—Multiple sequence alignment using various sources of external information. Nucleic Acids Res. 2013, 41, W3–W7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.R.; Kaufmann, M.; Morgenstern, B. DIALIGN-TX: Greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol. Biol. 2008, 3, 6. [Google Scholar] [CrossRef]
- Song, B.; Marco-Sola, S.; Moreto, M.; Johnson, L.; Buckler, E.S.; Stitzer, M.C. AnchorWave: Sensitive alignment of genomes with high sequence diversity, extensive structural polymorphism, and whole-genome duplication. Proc. Natl. Acad. Sci. USA 2022, 119, e2113075119. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Dewey, C.N. Aligning multiple whole genomes with Mercator and MAVID. Comp. Genom. 2008, 221–235. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2010, 27, 334–342. [Google Scholar] [CrossRef]
- Minkin, I.; Medvedev, P. Scalable pairwise whole-genome homology mapping of long genomes with BubbZ. IScience 2020, 23, 101224. [Google Scholar] [CrossRef]
- Dabbaghie, F.; Ebler, J.; Marschall, T. BubbleGun: Enumerating bubbles and superbubbles in genome graphs. Bioinformatics 2022, 38, 4217–4219. [Google Scholar] [CrossRef]
- Armstrong, J.; Hickey, G.; Diekhans, M.; Fiddes, I.T.; Novak, A.M.; Deran, A.; Fang, Q.; Xie, D.; Feng, S.; Stiller, J.; et al. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 2020, 587, 246–251. [Google Scholar] [CrossRef]
- Rautiainen, M.; Marschall, T. GraphAligner: Rapid and versatile sequence-to-graph alignment. Genome Biol. 2020, 21, 253. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Cui, Y.; Liao, X.; Peng, S.; Lu, Y.; Yang, C.; Wang, B.; Wu, C. Large-scale neo-heterogeneous programming and optimization of SNP detection on Tianhe-2. In Proceedings of the High Performance Computing: 30th International Conference, ISC High Performance 2015, Frankfurt, Germany, 12–16 July 2015; Proceedings 30. Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Capblancq, T.; Butnor, J.R.; Deyoung, S.; Thibault, E.; Munson, H.; Nelson, D.M.; Fitzpatrick, M.C.; Keller, S.R. Whole-exome sequencing reveals a long-term decline in effective population size of red spruce (Picea rubens). Evol. Appl. 2020, 13, 2190–2205. [Google Scholar] [CrossRef]
- Kuznetsov, A.; Bollin, C.J. NCBI genome workbench: Desktop software for comparative genomics, visualization, and GenBank data submission. Mult. Seq. Alignment Methods Protoc. 2021, 261–295. [Google Scholar] [CrossRef]
- Saada, B.; Zhang, J. DNA sequences compression algorithm based on extended-ASCII representation. In Proceedings of the World Congress on Engineering and Computer Science, San Francisco, CA, USA, 21–23 October 2015. [Google Scholar]
- Silva, M.; Pratas, D.; Pinho, A.J. Efficient DNA sequence compression with neural networks. GigaScience 2020, 9, giaa119. [Google Scholar] [CrossRef]
- Corbett, R.D.; Eveleigh, R.; Whitney, J.; Barai, N.; Bourgey, M.; Chuah, E.; Johnson, J.; Moore, R.A.; Moradin, N.; Mungall, K.L. A distributed whole genome sequencing benchmark study. Front. Genet. 2020, 11, 612515. [Google Scholar] [CrossRef]
- Marco-Sola, S.; Eizenga, J.M.; Guarracino, A.; Paten, B.; Garrison, E.; Moreto, M. Optimal gap-affine alignment in O(s) space. Bioinformatics 2023, 39, btad074. [Google Scholar] [CrossRef]
- Alser, M.; Rotman, J.; Deshpande, D.; Taraszka, K.; Shi, H.; Baykal, P.I.; Yang, H.T.; Xue, V.; Knyazev, S.; Singer, B.D.; et al. Technology dictates algorithms: Recent developments in read alignment. Genome Biol. 2021, 22, 249. [Google Scholar] [CrossRef]
- Rhie, A.; Nurk, S.; Cechova, M.; Hoyt, S.J.; Taylor, D.J.; Altemose, N.; Hook, P.W.; Koren, S.; Rautiainen, M.; Alexandrov, I.A.; et al. The complete sequence of a human Y chromosome. Nature 2023, 621, 344–354. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, J.; Wu, Y.; Zhang, W.; Jin, J. A completeness-independent method for pre-selection of closely related genomes for species delineation in prokaryotes. BMC Genom. 2020, 21, 183. [Google Scholar] [CrossRef]
- Gardner, S.N.; Hiddessen, A.L.; Williams, P.L.; Hara, C.; Wagner, M.C.; Colston, B.W., Jr. Multiplex primer prediction software for divergent targets. Nucleic Acids Res. 2009, 37, 6291–6304. [Google Scholar] [CrossRef]
- Dewey, C.N. Whole-Genome Alignment. In Evolutionary Genomics: Statistical and Computational Methods, Volume 1; Anisimova, M., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 237–257. [Google Scholar]
- Löytynoja, A. Alignment methods: Strategies, challenges, benchmarking, and comparative overview. In Volutionary Genomics: Statistical and Computational Methods, Volume 1; Springer: Berlin/Heidelberg, Germany, 2012; pp. 203–235. [Google Scholar]
- Couronne, O.; Poliakov, A.; Bray, N.; Ishkhanov, T.; Ryaboy, D.; Rubin, E.; Pachter, L.; Dubchak, I. Strategies and tools for whole-genome alignments. Genome Res. 2003, 13, 73–80. [Google Scholar] [CrossRef]
- Govek, K.W.; Yamajala, V.S.; Camara, P.G. Clustering-independent analysis of genomic data using spectral simplicial theory. PLoS Comput. Biol. 2019, 15, e1007509. [Google Scholar] [CrossRef]
- Wu, Y.; Johnson, L.; Song, B.; Romay, C.; Stitzer, M.; Siepel, A.; Buckler, E.; Scheben, A. A multiple alignment workflow shows the effect of repeat masking and parameter tuning on alignment in plants. Plant Genome 2022, 15, e20204. [Google Scholar] [CrossRef]
- Kille, B.; Balaji, A.; Sedlazeck, F.J.; Nute, M.; Treangen, T.J. Multiple genome alignment in the telomere-to-telomere assembly era. Genome Biol. 2022, 23, 182. [Google Scholar] [CrossRef]
- Huang, C.; Li, R.; Li, A. Parallel Implementation of Key Algorithms for Intelligent Processing of Graphic Signal Data of Consumer Digital Equipment. Mob. Netw. Appl. 2023. [Google Scholar] [CrossRef]
- Nolle, T.; Seeliger, A.; Thoma, N.; Mühlhäuser, M. DeepAlign: Alignment-based process anomaly correction using recurrent neural networks. In Proceedings of the International Conference on Advanced Information Systems Engineering, Grenoble, France, 8–12 June 2020; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Peltzer, A.; Jäger, G.; Herbig, A.; Seitz, A.; Kniep, C.; Krause, J.; Nieselt, K. EAGER: Efficient ancient genome reconstruction. Genome Biol. 2016, 17, 60. [Google Scholar] [CrossRef]
- Song, B.; Buckler, E.S.; Stitzer, M.C. New whole-genome alignment tools are needed for tapping into plant diversity. Trends Plant Sci. 2024, 29, 355–369. [Google Scholar] [CrossRef]
- Earl, D.; Nguyen, N.; Hickey, G.; Harris, R.S.; Fitzgerald, S.; Beal, K.; Seledtsov, I.; Molodtsov, V.; Raney, B.J.; Clawson, H. Alignathon: A competitive assessment of whole-genome alignment methods. Genome Res. 2014, 24, 2077–2089. [Google Scholar] [CrossRef]
- Schadt, E.E.; Linderman, M.D.; Sorenson, J.; Lee, L.; Nolan, G.P. Computational solutions to large-scale data management and analysis. Nat. Rev. Genet. 2010, 11, 647–657. [Google Scholar] [CrossRef]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef]
- Kshemkalyani, A.D.; Singhal, M. Distributed Computing: Principles, Algorithms, and Systems; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Volozonoka, L.; Miskova, A.; Gailite, L. Whole genome amplification in preimplantation genetic testing in the era of massively parallel sequencing. Int. J. Mol. Sci. 2022, 23, 4819. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Girisha, M.N.; Badiger, V.P.; Pattar, S. A comprehensive review of global alignment of multiple biological networks: Background, applications and open issues. Netw. Model. Anal. Health Inform. Bioinform. 2022, 11, 9. [Google Scholar] [CrossRef]
- Hennig, A.; Nieselt, K. Efficient merging of genome profile alignments. Bioinformatics 2019, 35, i71–i80. [Google Scholar] [CrossRef]
- Armstrong, J.; Fiddes, I.T.; Diekhans, M.; Paten, B. Whole-genome alignment and comparative annotation. Annu. Rev. Anim. Biosci. 2019, 7, 41–64. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Voet, T. Single cell genomics: Advances and future perspectives. PLoS Genet. 2014, 10, e1004126. [Google Scholar] [CrossRef]
- Shi, L.; Wang, Z. Computational strategies for scalable genomics analysis. Genes 2019, 10, 1017. [Google Scholar] [CrossRef]
- Ryva, B.; Zhang, K.; Asthana, A.; Wong, D.; Vicioso, Y.; Parameswaran, R. Wheat germ agglutinin as a potential therapeutic agent for leukemia. Front. Oncol. 2019, 9, 100. [Google Scholar] [CrossRef]
- Taylor, J.; Yudkowsky, E.; LaVictoire, P.; Critch, A. Alignment for advanced machine learning systems. Ethics Artif. Intell. 2016, 342–382. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).