
Stability of Paracetamol Amorphized by Co-Melting with Various Cellulose Derivatives




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Differential Scanning Calorimetry
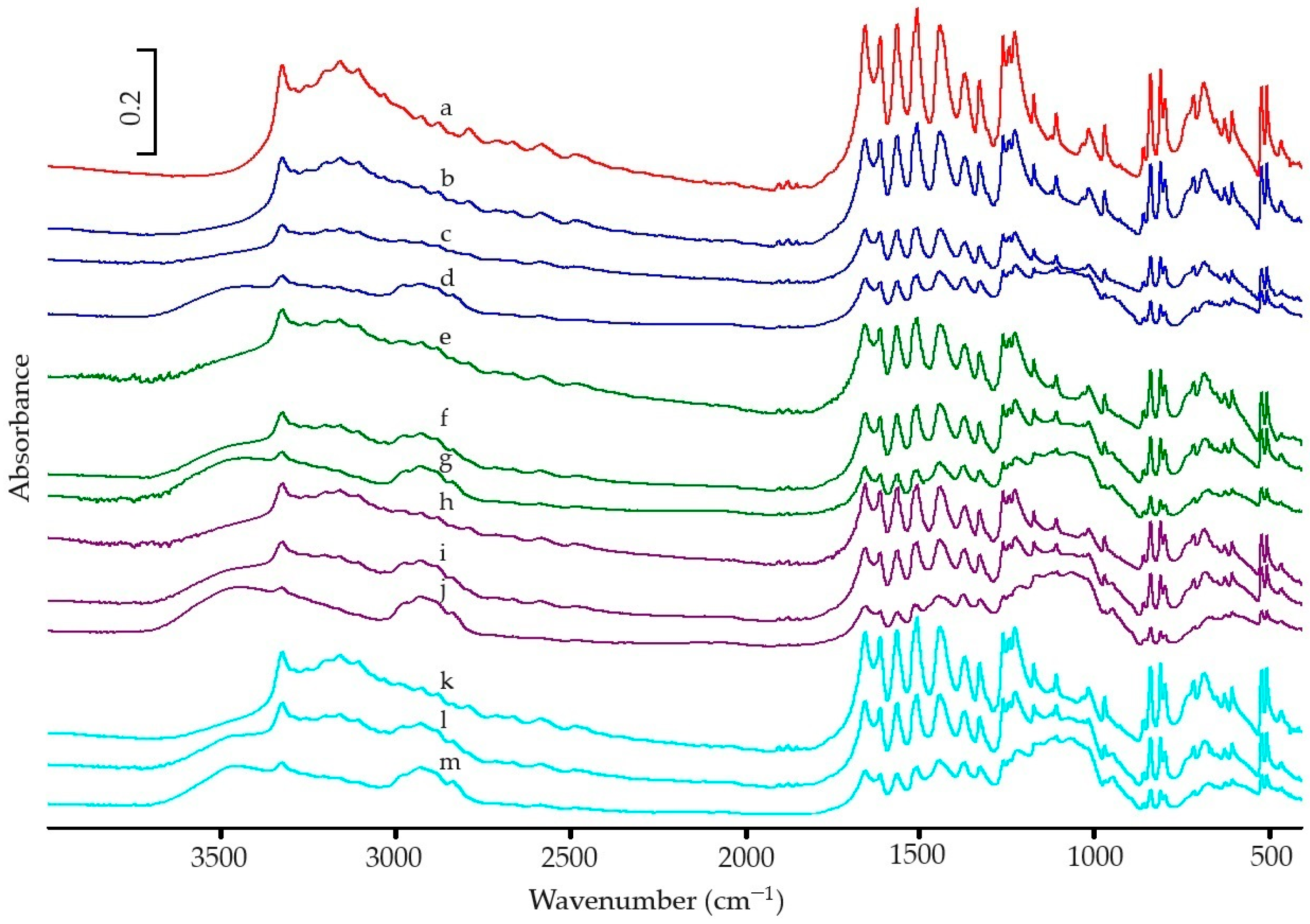
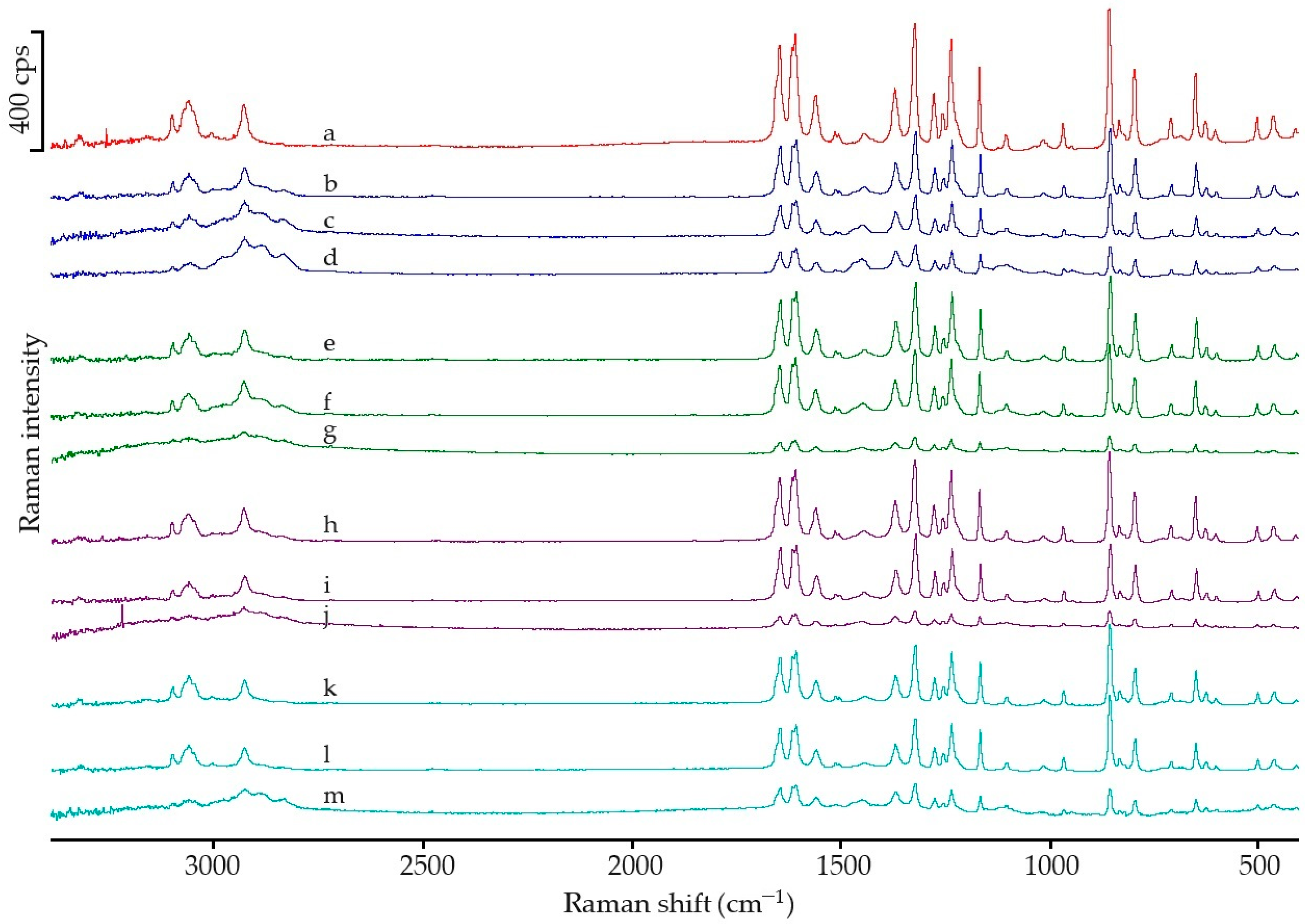
2.2.2. Spectroscopic Analysis
2.2.3. Chromatographic Analysis
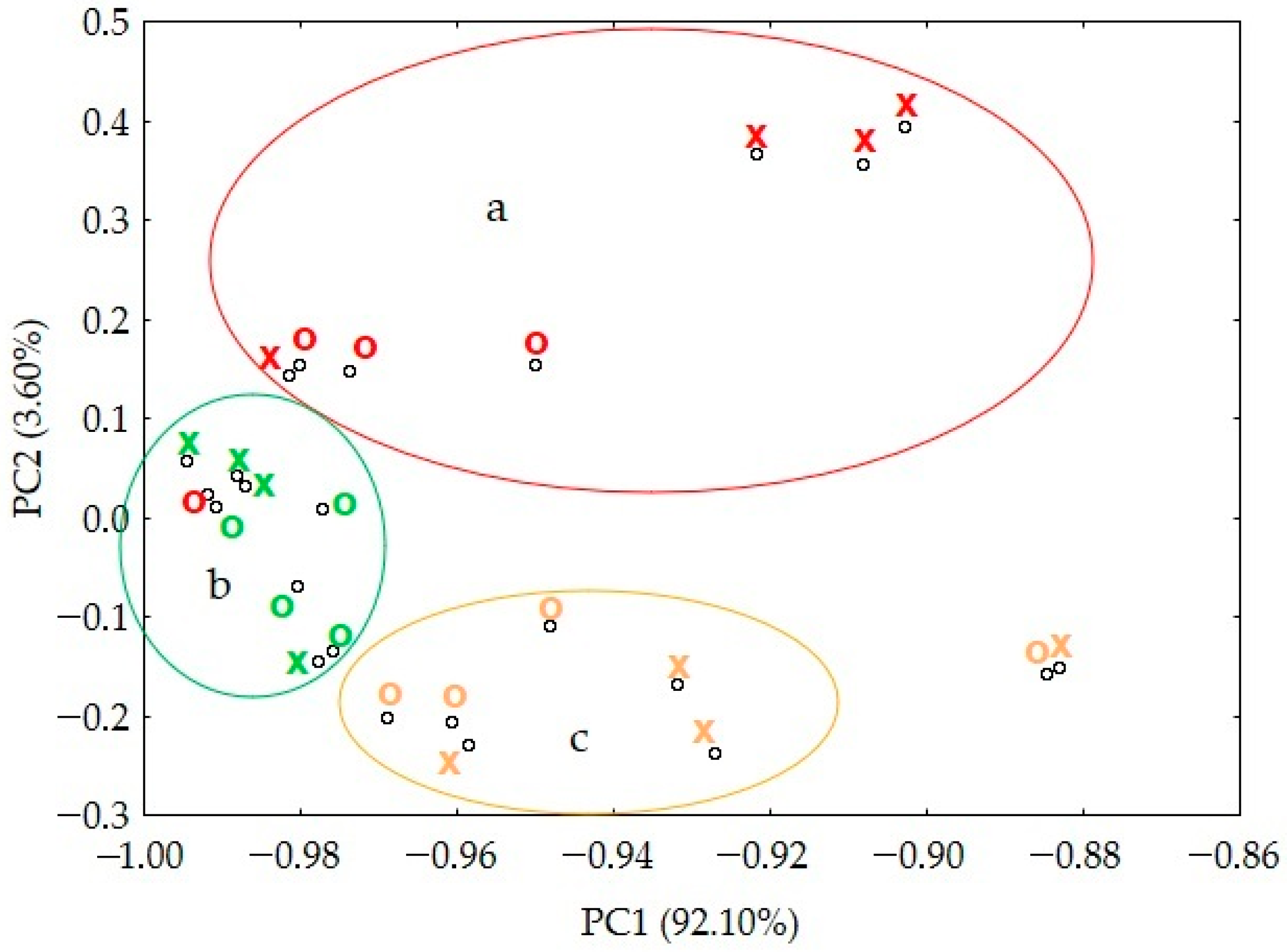
2.2.4. Chemometric Analysis
3. Results
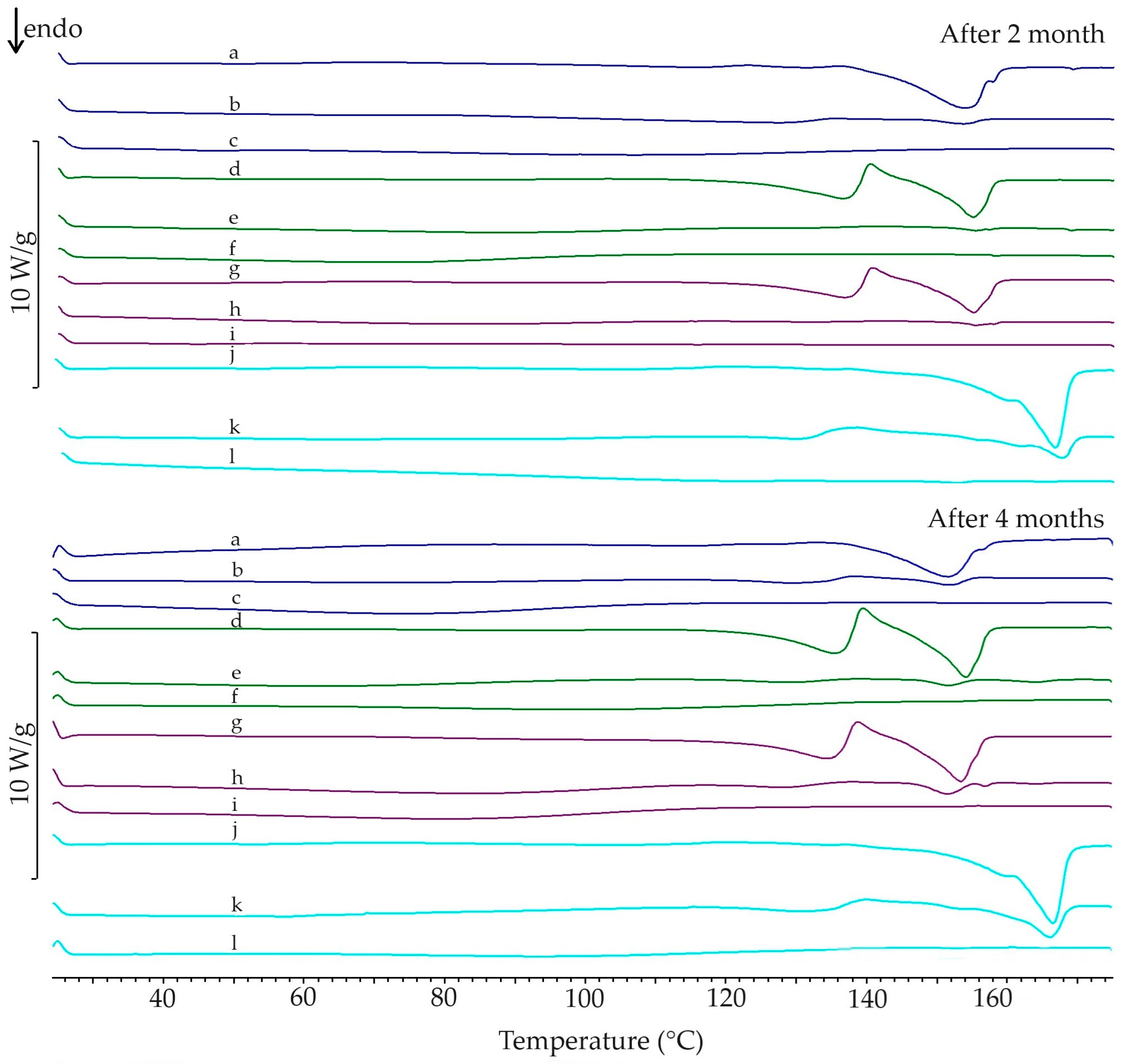
3.1. DSC Analysic
3.1.1. Paracetamol Amorphization
3.1.2. Stability of Amorphous Form
3.2. Spectroscopic Analysic
3.3. Stability of Paracetamol after Melting
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Singhal, D.; Curatolo, W. Drug polymorphism and dosage form design: A practical perspective. Adv. Drug Deliv. Rev. 2004, 56, 335–347. [Google Scholar] [CrossRef]
- Dong, W.; Su, X.; Xu, M.; Hu, M.; Sun, Y.; Zhang, P. Preparation, characterization, and in vitro/vivo evaluation of polymer-assisting formulation of atorvastatin calcium based on solid dispersion technique. Asian J. Pharm. Sci. 2018, 13, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.M.; Shakeel, F.; Ibrahim, M.A.; Elzayat, E.M.; Altamimi, M.; Mohsin, K.; Almeanazel, O.T.; Alkholief, M.; Alshetaili, A.; Alsulays, B.; et al. Dissolution and bioavailability improvement of bioactive apigenin using solid dispersions prepared by different techniques. Saudi Pharm. J. 2019, 27, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.D.; Sung, J.H.; Kim, K.K.; Kim, D.W.; Kim, J.O.; Lee, B.J.; Yong, C.S.; Choi, H.G. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int. J. Pharm. 2012, 422, 202–210. [Google Scholar] [CrossRef]
- Rashid, R.; Kim, D.W.; Din, F.; Mustapha, O.; Yousaf, A.M.; Park, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.G. Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr. Polym. 2015, 130, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ema.europa.eu/en/glossary/bioavailability (accessed on 17 June 2024).
- Atkinson, A.J. Chapter 4—Drug Absorption and Bioavailability. In Principles of Clinical Pharmacology, 3rd ed.; Atkinson, A.J., Huang, S.-M., Lertora, J.J.L., Markey, S.P., Eds.; Academic Press: Cambridge, MA, USA, 2012; pp. 41–53. [Google Scholar] [CrossRef]
- Chow, S. Bioavailability and bioequivalence in drug development. WIREs Comput. Stat. 2014, 6, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.R.; Lee, P.I.; Pang, K.S. Finding Tmax and Cmax for Multicompartmental Models. Drug Metab. Dispos. 2018, 46, 1796–1804. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, L.; Wu, T.; Cai, T.; Gunn, E.M.; Yu, L. Stability of amorphous pharmaceutical solids: Crystal growth mechanisms and effect of polymer additives. AAPS J. 2012, 14, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Li, F.; Yeh, S.; Wang, Y.; Xin, J. Physical stability of amorphous pharmaceutical solids: Nucleation, crystal growth, phase separation and effects of the polymers. Int. J. Pharm. 2020, 590, 19925. [Google Scholar] [CrossRef] [PubMed]
- Kerlin, R.L.; Li, X. Chapter 24—Pathology in Non-Clinical Drug Safety Assessment. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology, 3rd ed.; Haschek, W.M., Rousseaux, C.G., Wallig, M.A., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 725–750. [Google Scholar] [CrossRef]
- van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef]
- Veith, H.; Wiechert, F.; Luebbert, C.; Sadowski, G. Combining crystalline and polymeric excipients in API solid dispersions—Opportunity or risk? Eur. J. Pharm. Biopharm. 2021, 158, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Dattachowdhury, B.; Vangara, K.; Patel, N.; Alexander, K.; Boddu, S. Excipients That Facilitate Amorphous Drug Stabilization. In Excipient Applications in Formulation Design and Drug Delivery; Narang, A., Boddu, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 463–495. [Google Scholar] [CrossRef]
- Ayoub, S.S. Paracetamol (acetaminophen): A familiar drug with an unexplained mechanism of action. Temperature 2021, 8, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Rassat, M.; Ouandji, I.; Guiblin, N.; Espeau, P. Obtaining two polymorphic forms of paracetamol within the phase diagram with PEG 1500. Eur. J. Pharm. Biopharm. 2024, 195, 114173. [Google Scholar] [CrossRef] [PubMed]
- Shtukenberg, A.G.; Tan, M.; Vogt-Maranto, L.; Chan, E.J.; Xu, W.; Yang, J.; Tuckerman, M.E.; Hu, C.T.; Kahr, B. Melt Crystallization for Paracetamol Polymorphism. Cryst. Growth Des. 2019, 19, 4070–4080. [Google Scholar] [CrossRef]
- Yeh, K.L.; Lee, H.L.; Lee, T. Crystallization of Form II Paracetamol with the Assistance of Carboxylic Acids toward Batch and Continuous Processes. Pharmaceutics 2022, 14, 1099. [Google Scholar] [CrossRef]
- Leyk, E.; Wesolowski, M. Interactions Between Paracetamol and Hypromellose in the Solid State. Front. Pharmacol. 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Leyk, E.; Wesolowski, M. The effect of cellulose derivatives on paracetamol crystallinity reduction. J. Therm. Anal. Calorim. 2022, 147, 10037–10048. [Google Scholar] [CrossRef]
- Burgina, E.B.; Baltakhinov, V.P.; Boldyreva, E.V.; Shakhtschneider, T.P. IR Spectra of Paracetamol and Phenacetin. 1. Theoretical and Experimental Studies. J. Struct. Chem. 2004, 45, 64–73. [Google Scholar] [CrossRef]
- Zapata, F.; López-Fernández, A.; Ortega-Ojeda, F.; Quintanilla, G.; García-Ruiz, C.; Montalvo, G. Introducing ATR-FTIR Spectroscopy through Analysis of Acetaminophen Drugs: Practical Lessons for Interdisciplinary and Progressive Learning for Undergraduate Students. J. Chem. Educ. 2021, 98, 2675–2686. [Google Scholar] [CrossRef]
- Elbagerma, M.A.; Edwards, H.G.M.; Munshi, T.; Scowen, I.J. Identification of a new cocrystal of citric acid and paracetamol of pharmaceutical relevance. CrystEngComm 2011, 13, 1877–1884. [Google Scholar] [CrossRef]
- Sibik, J.; Sargent, M.J.; Franklin, M.; Zeitler, J.A. Crystallization and phase changes in paracetamol from the amorphous solid to the liquid phase. Mol. Pharm. 2014, 11, 1326–1334. [Google Scholar] [CrossRef]
- Cruz, P.C.; Rocha, F.A.; Ferreira, A.M. Application of selective crystallization methods to isolate the metastable polymorphs of paracetamol: A review. Org. Process Res. Dev. 2019, 23, 2592–2607. [Google Scholar] [CrossRef]
- Giordano, F.; Rossi, A.; Bettini, R.; Savioli, A.; Gazzaniga, A.; Nováket, C. Thermal Behavior of Paracetamol-Polymeric Excipients Mixtures. J. Therm. Anal. Calorim. 2002, 68, 575–590. [Google Scholar] [CrossRef]
- Ritters, L.; Tian, Y.; Reichl, S. Spray-Dried Paracetamol/Polyvinylpyrrolidone Amorphous Solid Dispersions: Part I-Stability of Powders and Tablets. Pharmaceutics 2021, 13, 1938. [Google Scholar] [CrossRef]
- Balamurugan, S.D.; Venkatesan, A.; Radhakrishnan, A.; Kuppusamy, G.; Singh, S.K. Preliminary Investigation for Preparing Amorphous Paracetamol. Res. J. Pharm. Technol. 2021, 14, 1487–1492. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAR | Mixture with HPMC 86 | Mixture with HPMC 90 | Mixture with HPMC 120 | Mixture with MC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PAR Content in Mixture | 75% | 50% | 25% | 75% | 50% | 25% | 75% | 50% | 25% | 75% | 50% | 25% | ||
| 1st heating | Enthalpy of melting (J/g) | 221.8 | 142.8 | 72.8 | 29.7 | 135.4 | 57.3 | 26.5 | 131.2 | 64.1 | 38.7 | 141.8 | 81.3 | 34.5 |
| Calculated amount of I form (%) | 64.4 | 32.8 | 13.4 | 61.0 | 25.8 | 12.0 | 59.1 | 28.9 | 17.4 | 63.9 | 36.7 | 15.6 | ||
| Ton (°C) | 170.1 | 169.3 | 168.0 | 168.1 | 168.1 | 166.9 | 168.1 | 167.8 | 167.8 | 167.6 | 168.8 | 166.9 | 166.3 | |
| Tp (°C) | 171.3 | 171.3 | 172.2 | 171.7 | 171.2 | 170.8 | 171.2 | 171.5 | 171.3 | 171.2 | 171.9 | 171.1 | 171.5 | |
| 2nd heating | Enthalpy of melting (J/g) | 202.7 | 36.8 | - | - | 5.0 | - | - | 5.0 | - | - | 70.7 | - | - |
| Ton (°C) | 158.0 | 153.3 | - | - | 153.2 | - | - | 153.9 | - | - | 163.9 | - | - | |
| Tp (°C) | 159.9 | 156.7 | - | - | 157.4 | - | - | 157.8 | - | - | 168.9 | - | - | |
| after 2 months | Enthalpy of melting (J/g) | 46.8 | 6.4 | - | 49.2 | 3.0 | - | 43.8 | 4.0 | - | 95.2 | 27.3 | 1.5 | |
| Ton (°C) | 142.3 | 140.1 | - | 149.6 | 152.2 | - | 149.4 | 152.7 | - | 158.4 | 162.6 | 158.9 | ||
| Tp (°C) | 153.5 | 153.7 | - | 154.9 | 155.4 | - | 155.1 | 155.6 | - | 162.8 | 167.6 | 152.5 | ||
| after 4 months | Enthalpy of melting (J/g) | 54.5 | 7.2 | - | 47.9 | 3.8 | - | 43.6 | 6.5 | - | 101.1 | 36.0 | 0.9 | |
| Ton (°C) | 140.9 | 145.8 | - | 148.0 | 147.9 | - | 146.7 | 147.9 | - | 158.8 | 160.4 | 160.6 | ||
| Tp (°C) | 151.8 | 151.9 | - | 154.3 | 152.0 | - | 153.6 | 151.7 | - | 166.2 | 166.2 | 165.5 | ||
| FTIR | Raman Spectroscopy | ||||||
|---|---|---|---|---|---|---|---|
| References [25,26] | Measured Values for PAR | References [25,27] | Measured Values for PAR | ||||
| Wave Number (cm−1) | Molecular Vibration | Peak (cm−1) | Normalized Peak Intensity | Raman Shift (cm−1) | Molecular Vibration | Peak (cm−1) | Normalized Peak Intensity |
| 3327 | OH st | 3325 | 1.060 | 3110 | as CH st | 3102 | 1.210 |
| 3161 | PhH st | 3160 | 1.084 | 3058 | s CH3 st | 3062 | 1.826 |
| 2926 | CH3 st | 2929 | 0.649 | 2937 | s CH3 st | 2930 | 1.619 |
| 1653 | C=O st, CNH def | 1654 | 1.314 | 1649 | C=O st | 1646 | 4.076 |
| 1610 | Ph st | 1611 | 1.247 | 1611 | NH def | 1609 | 4.525 |
| 1565 | CNH def, Ph st | 1563 | 1.324 | 1559 | NH st, C=O st | 1560 | 2.017 |
| 1506 | CNH def, Ph st | 1506 | 1.425 | 1371 | CH3 def (umbrellas) | 1370 | 2.314 |
| 1442 | CH3 def, Ph st | 1440 | 1.311 | 1324 | CH b″ | 1322 | 4.956 |
| 1371 | CH3 def, Ph st | 1369 | 1.007 | 1278 | OH with CO | 1276 | 2.130 |
| 1327 | CH3 def | 1327 | 0.960 | 1237 | CN st | 1235 | 4.290 |
| 1260 | PhH def, CN st | 1259 | 1.244 | 1169 | CH of Ph b′ | 1167 | 3.288 |
| 1227 | PhH def, CC st | 1225 | 1.278 | 856 | CH of Ph st | 856 | 5.698 |
| 1172 | PhH def, COH def | 1171 | 0.880 | 839 | CH b″ | 832 | 1.000 |
| 1107 | PhH def, CH3 def | 1107 | 0.749 | 796 | Ph st | 795 | 3.061 |
| 1015 | PhH def, COH def | 1014 | 0.656 | 649 | Ph b′ | 649 | 2.932 |
| 968 | PhH def, CN st | 968 | 0.672 | 501 | Ph b″ | 502 | 1.131 |
| 856 | PhH b″ | 857 | 0.532 | 463 | Ph b″ | 463 | 1.193 |
| 837 | PhH b″ | 837 | 1.000 | ||||
| 810 | Ph def, amide def | 808 | 1.033 | ||||
| 795 | Ph def, amide def | 796 | 0.849 | ||||
| 715 | CNH b″, PhH b″ | 713 | 0.866 | ||||
| 668 | CNH b″, PhH b″ | 684 | 0.943 | ||||
| 624 | Ph b″, amide b″ | 625 | 0.719 | ||||
| 604 | Ph b″, amide b″ | 603 | 0.766 | ||||
| 520 | Ph-amide c | 518 | 0.930 | ||||
| 504 | Ph-amide c | 503 | 0.930 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leyk, E.; Plenis, A.; Kasprowicz, J.; Dziurkowska, E. Stability of Paracetamol Amorphized by Co-Melting with Various Cellulose Derivatives. Appl. Sci. 2024, 14, 6709. https://doi.org/10.3390/app14156709
Leyk E, Plenis A, Kasprowicz J, Dziurkowska E. Stability of Paracetamol Amorphized by Co-Melting with Various Cellulose Derivatives. Applied Sciences. 2024; 14(15):6709. https://doi.org/10.3390/app14156709
Chicago/Turabian StyleLeyk, Edyta, Alina Plenis, Julia Kasprowicz, and Ewelina Dziurkowska. 2024. "Stability of Paracetamol Amorphized by Co-Melting with Various Cellulose Derivatives" Applied Sciences 14, no. 15: 6709. https://doi.org/10.3390/app14156709
APA StyleLeyk, E., Plenis, A., Kasprowicz, J., & Dziurkowska, E. (2024). Stability of Paracetamol Amorphized by Co-Melting with Various Cellulose Derivatives. Applied Sciences, 14(15), 6709. https://doi.org/10.3390/app14156709