Abstract
Aristolochic acids, compounds derived from Aristolochiaceae plant species, are associated with significant renal nephrotoxicity and carcinogenicity. Aristolochic acid I (AAI), the most predominant and potent of these compounds, is a primary etiological agent in acute and chronic kidney diseases such as Aristolochic Acid Nephropathy (AAN) and Balkan Endemic Nephropathy (BEN). Due to the kidneys’ critical role in xenobiotic excretion, they are the primary organs affected by AAI toxicity. Recent in vitro and in vivo studies have highlighted mitochondrial dysfunction as a crucial factor in the pathogenesis of these kidney diseases. This review provides an update on the recent advances in understanding the causes of acquired mitochondrial dysfunction within the context of AAN and BEN. Key findings include the identification of mitochondrial DNA depletion, loss of mitochondrial membrane potential, and decreased ATP production as significant contributors to kidney damage. Additionally, oxidative stress markers and inflammatory mediators have been implicated in disease progression. Potential therapeutic approaches, such as the use of antioxidants like vitamin C and catalpol, have shown promise in mitigating AAI-induced cytotoxicity. Furthermore, future predictive approaches like pharmacogenomics could pave the way for novel mitochondria-targeted treatments. A comprehensive characterization of mitochondrial function, its underlying molecular mechanisms, and specific biomarkers could offer valuable insights and potential therapeutic options, significantly impacting the current management of AAN and BEN.
1. Introduction
During the last decades, several peculiar kidney diseases of unknown origin have been described in various countries, their etiology being subsequently linked to the environmental phytotoxins aristolochic acids (AAs). The most studied compound among AAs and their metabolites is aristolochic acid I (AAI), this compound being the most abundant in Aristolochia plants [1]. AAI is one of the most nephrotoxic and carcinogenic compounds found in Aristolochiaceae family plants, which is used in traditional medicine for several ailments affecting various organs [2,3,4]. There are two well-described environmentally induced kidney diseases, Balkan Endemic Nephropathy (BEN) and Aristolochic Acid Nephropathy (AAN) [5,6]; although these kidney diseases were discovered 40 years apart, the common environmental factor causing them is AAI [7,8]. For over 60 years, BEN, a slowly progressing loss of kidney function, has been affecting residents of rural villages in Romania, Bulgaria, Serbia, Croatia, and Bosnia-Herzegovina. Over 25,000 individuals have suffered from this disease, with variations in the age of onset from 40 to 70 years [9]. Many possible causing factors were suggested in BEN causation since its original description in the 1950s, and as early as 1969, research from Serbia suggested that the BEN outbreak in the Balkan Peninsula could be due to iatrogenic exposure to AAs through the food chain, proposing the ingestion of flour contaminated with Aristolochia seeds [10]. Further studies demonstrated that AAs could indeed contaminate crop plants through absorption from the soil and uptake by neighboring food crops through their roots, being deposited into the grains and other cultivated vegetables [11]. Despite intensive research to find other potential causative agents, AAs were shown to be one of the main culprits of this disease [12]. Evidence from BEN studies provides an example of toxic renal injury induced by phytotoxins found in the local environment. Those affected by BEN present tubular proteinuria, impaired renal concentrating capacity, reduced glomerular filtration rate (GFR), and interstitial fibrosis with tubular atrophy [13]. In the early 1990s, a new epidemic of rapidly progressive tubulointerstitial nephritis was reported in a cohort of young female patients in Belgium. This acute kidney disease was rapidly associated with the intake of slimming pills containing Chinese herbs, which incidentally contained AAs, and the disease was named AAN [14]. Aristolochia plant species have been used since ancient times in traditional Chinese medicine to treat a wide range of diseases and symptoms. Even if AAs-containing herbs have been banned by the Chinese government since 2003, new cases of AAN keep emerging [15,16,17,18]. A high dose of AAs-containing medication induced irreversible acute tubular necrosis and resulted in progressive chronic renal failure. Long-term intake of AAs-containing medications resulted in chronic tubulointerstitial nephropathy [19,20,21]. The accumulating feature of AAI is a significant factor in the rapidly progressive renal dysfunction in AAN patients [22]. Investigating methods that facilitate the bio-elimination of these chemicals from the body could be valuable for protecting the kidney [23]. Since the discovery of these diseases, many experimental in vitro and in vivo studies have been performed to understand the cellular and molecular mechanisms of pathogenesis. Recent findings emphasize the involvement of mitochondria in the progression of chronic kidney damage, particularly due to a reduction in mitochondrial DNA (mtDNA) copy number, loss of mitochondrial membrane potential (Δψm), and a drop in ATP production [24,25]. Mitochondria are involved in apoptosis and the epithelial-to-mesenchymal transition of renal tubular epithelial cells, contributing to the fibrogenic process [26]. Understanding mitochondrial dysfunction in kidney diseases is crucial because mitochondria are the powerhouses of cells, supplying the energy required for cellular functions. Dysfunctional mitochondria can lead to energy deficits, increased oxidative stress, and apoptosis, exacerbating kidney damage [27]. Mitochondria also play a vital role in cellular signaling and homeostasis, and their impairment can have far-reaching consequences on renal function and overall health [28]. A comprehensive characterization of mitochondrial function, its underlying molecular mechanisms, and specific markers could offer insights and possible therapeutic options that could significantly impact the current management of AAN and BEN [24,25]. Emerging evidence suggests that dysfunctional mitochondria play a primary role in the development of chronic kidney diseases (CKD) and the associated comorbidities, highlighting their potential as therapeutic targets [29]. A variety of agents, such as food-derived antioxidants and natural plant compounds combined with conventional therapies, could help clinicians improve the outcomes of these kidney diseases and identify proper targets for interventions [30]. For instance, vitamin C has been shown to suppress AAI-induced cytotoxicity in kidney cells by attenuating the increased levels of H2O2 and caspase-3 activation [22,23]. These findings suggest that supplementation with vitamin C or catalpol may be beneficial for reducing AAI-induced renal damage [23]. Future predictive approaches like pharmacogenomics could pave the way for novel mitochondria-targeted treatments for AAN, BEN, and other types of CKDs [31]. This review will first discuss the background of AAI-induced nephropathy, followed by a detailed analysis of mitochondrial dysfunction and potential therapeutic strategies.
2. Mitochondria and Kidney Diseases
Mitochondria are the primary suppliers of cellular energy, producing large amounts of ATP through the electron transport chain (ETC) located in the inner mitochondrial membrane. Mitochondria have their own genetic material encoded by mitochondrial DNA (mtDNA), which consists of 37 genes: 22 encode transfer RNAs, two encode ribosomal RNA, and the remaining ones encode polypeptides that are part of the OXPHOS enzyme complexes I–V (7 units of complex I, cytochrome b of complex III, three subunits of complex IV, and two subunits of complex V) [12,13]. Mitochondria contain channels and carrier proteins that facilitate the accumulation of xenobiotics in the matrix and/or the inner mitochondrial membrane through selective trapping mechanisms [14,15].
The kidneys are the second most energy-demanding organs in the body, with the second highest mitochondrial content and oxygen consumption after the heart [13]. Mitochondria, as a dynamic network of organelles, produces ATP to supply the energy required for basal cellular functions in the kidneys. They modulate metabolic reactions, generate reactive oxygen species (ROS), maintain intracellular calcium homeostasis, thermogenesis, and regulate proliferation and intrinsic apoptotic pathways. Evidence shows that both acute and chronic kidney injuries can lead to mitochondrial respiratory chain-derived oxidative stress, ultrastructural defects, abnormal activation of the mitochondrial pathway of apoptosis, unstable mtDNA, and defective clearance of damaged mitochondria. Mitochondrial dysfunction increases the risk of developing tubulointerstitial diseases, cystic kidney disease, and nephrotic syndromes. A deeper understanding of the cellular and molecular mechanisms governing mitochondrial dysfunction in kidney pathology could facilitate the development of improved therapeutic strategies [16].
In the past decade, both clinical and experimental studies have suggested a causal link between acute kidney injury (AKI) and chronic kidney disease (CKD), a process termed AKI-to-CKD transition. This transition is often due to incomplete or maladaptive repair after AKI. AKI has been identified as a significant risk factor for the onset and progression of CKD, with symptoms including hypoxia, endothelial dysfunction, nephron loss, alterations in renal cell functions, cell cycle arrest, inflammation, mitochondrial fragmentation, and epigenetic modifications [17]. Mitochondria are particularly sensitive to environmental changes that can cause mitochondrial dysfunction, leading to decreased ATP generation, increased ROS levels, and the induction of apoptosis (Figure 1). These factors collectively contribute to the development and progression of AKI, CKD, and the AKI-to-CKD transition. CKD is one of the fastest-growing causes of death worldwide and is projected to become the fifth leading global cause of death by 2040 [18].
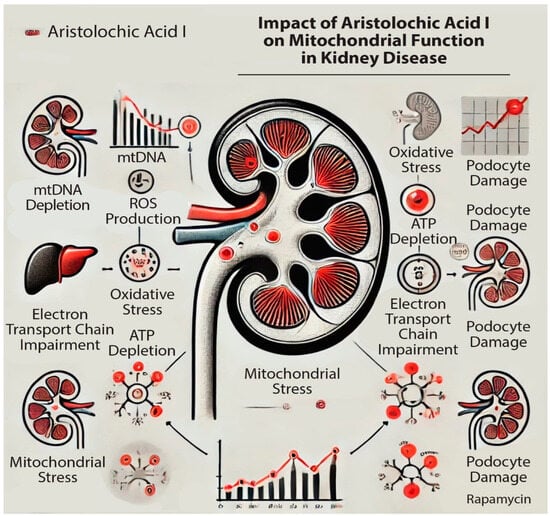
Figure 1.
Impact of aristolochic acid I (AAI) on mitochondrial activity in the kidney: impairing the mitochondrial DNA (mtDNA) production and Electron Transfer Chain (ETC); increasing the Reactive Oxygen Species (ROS) production.
3. Mitochondrial Dysfunction in AAI-Induced Kidney Diseases
We have found a total of 22 articles in the scientific literature, dated from 1998 to 2022, which describe the effects of AAI on mitochondrial activity and dysfunction. Because AAI is the most concentrated compound found in Aristolochiaceae family plants, and the most potent carcinogenic and nephrotoxic molecule, this was the most frequently involved in both acute and chronic kidney diseases, like AAN and BEN.
3.1. Methods of Investigations and Biomarker Detection in AAI-Induce Mitochondrial Dysfunction
To comprehensively understand the effects of aristolochic acid I (AAI) on mitochondrial function, both in vivo and in vitro studies are essential. These studies typically involve the treatment of biological samples, such as kidney tissues or kidney cell lines, with AAI to assess mitochondrial function. Although the literature contains numerous studies on the impact of AAI on mitochondrial functions, most focus on the compound’s acute nephrotoxic effects. This section outlines the in vitro and in vivo investigation methods, as well as the biological markers used to evaluate mitochondrial activity affected by AAI.
3.2. Investigation Methods of Mitochondrial Function
Studies on AAI nephrotoxicity consistently show mitochondrial dysfunction. Evaluations of AAI’s effects on the ETC and cellular respiration have involved isolating mitochondria from kidney cells of both animals and humans. The isolation process involves several steps: mechanical disruption of tissues or cells through homogenization, differential centrifugation to remove debris and large cellular organelles, and subsequent isolation and collection of mitochondria. To proceed with further investigations, the Biuret method is used to measure protein concentration. Isolated mitochondria are then analyzed to assess the functionality of the ETC (Figure 2) [16,17].
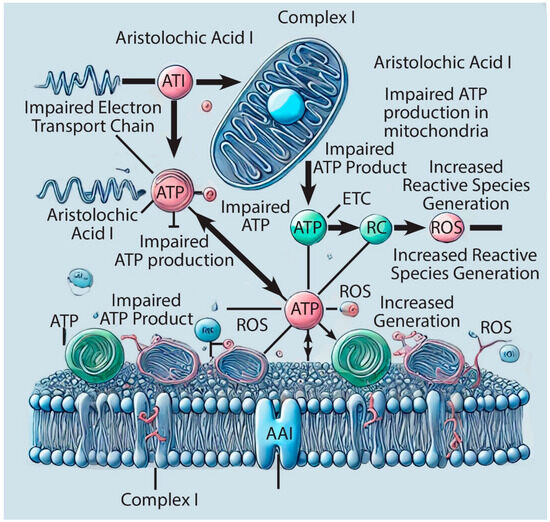
Figure 2.
Schematic representation of the aristolochic acid I (AAI) impact on complex I of the Electron Transfer Chain (ETC).
The following assays measure the effects of AAI on respiratory chain complexes in mitochondria: measurement of oxygen consumption, respiratory chain complexes, membrane potential of mitochondria (MMP), and adenine nucleotide translocator (ANT) activity.
There are also other assays performed on isolated mitochondria which assess the functionality and the impact of AAI on mitochondrial DNA, such as mitochondrial DNA (mtDNA).
3.2.1. Assay of Oxygen Consumption Measurement
Mitochondrial respiration rates are evaluated by measuring oxygen consumption in the presence of various substrates. Glutamate and malate, which selectively fuel electron transport at Complex I, or succinate combined with an NADH dehydrogenase inhibitor (like rotenone) to target Complex II, are commonly used. ADP-stimulated respiration (State 3) is assessed with ADP present, while ADP-independent respiration (State 4) is measured after ADP depletion. The respiratory control ratio (RCR), calculated as the ratio of State 3 to State 4 oxygen consumption, indicates the efficiency of ATP synthesis coupled to respiration and reflects mitochondrial integrity [12]. Oxygen consumption rate, determined by the decline in dissolved oxygen within the cell culture, serves as a direct measure of mitochondrial respiratory function. Studies have shown that AAI treatment leads to a reduction in respiratory function after 12 h [19]. Moreover, a significant decrease in mtDNA copy number is observed after 24 h of exposure to 50 µM AAI [2], further highlighting the detrimental impact of AAI on mitochondrial function.
3.2.2. Respiratory Chain Complexes
The specific activities of the oxidative phosphorylation system (OXPHOS) enzyme complex I and II were assessed in isolated mitochondria. The effects of AAI on mitochondrial function were investigated in rat kidneys by the two mitochondrial energy metabolism indicators: ATP content and the respiratory control ratio (RCR). AAI treatment resulted in a dose-dependent reduction in ATP content and a decrease in RCR [11]. In the presence of substrates for complex I, characterized by NADH oxidoreductase activity, AAI reduced the RCR in a dose-dependent manner. The substrate specificity indicates that the inhibition of complex I was more pronounced than that of respiratory complex II, which is represented by succinate oxidoreductase-specific activity. These results suggest that the activity of respiratory complex I, which is partially encoded by mtDNA, was significantly more reduced than the activity of respiratory complex II, which is entirely encoded by nuclear DNA [12].
3.2.3. Mitochondrial Membrane Potential (MMP)
The mitochondrial bioenergetic state is evaluated in living cells by the mitochondrial membrane potential (Δψ) (MMP). These assays are used for semi-quantitative Δψ analysis of fluorescent lipophilic cations (e.g., tetramethylrhodamine methyl ester (TMRM), rhodamine 123 (Rhod123)), which enter the cells and accumulate in the mitochondrial matrix under normal conditions. Mitochondrial permeability transition pore (MPT), a protein complex formed in the inner mitochondrial membrane, appears under pathological conditions. An MPT pore opens when mitochondrial membranes become permeable, allowing molecules below 1500 Daltons molecular weight to enter, therefore AAI, which has 341 Da. In the literature, MMP was studied by staining the cells with a lipophilic cationic probe, 5,6,6-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl-carbocyanine iodide (JC-1). A decrease was observed in MMP in cells cultured with AAI for 12 h [2,19,20]. Another experiment monitored MMP with Rhod123 in isolated mitochondria preincubated with succinate, followed by AAI and Ca2+. Therefore, an accumulation of Rhod123 was observed in HK-2 cells when these were treated with the immunosuppressant cyclosporin A (CsA) and bongkrekic acid (BA) as MPT inhibitors [20]. When TMRM is used as a reagent, active mitochondria sequester it giving a fluorescent signal [22]. In cell cultures treated with AAI, an increased production of nitric oxide (NO) can be identified, leading to the production of reactive oxygen species (ROS), which cause mitochondrial dysfunction. In this assay, FCCP (ionophore uncoupler of oxidative phosphorylation) is used as a positive control because it eliminates MMP [22].
3.2.4. Assay of Adenine Nucleotide Translocator (ANT) Activity
AAI-induced mitochondrial permeability transition (MPT) in isolated rat kidney mitochondria and human renal tubular epithelial cells is likely related to the inhibition of adenine nucleotide translocator (ANT). MPT contributes to HK-2 cell injury induced by AAI. ANT activity decreased with AAI treatment of isolated mitochondria [20].
3.2.5. Measurement of Calcium
The cytotoxic effects of aristolochic acid I (AAI) are potentiated by increased concentrations of Ca2+ [2]. Exposure of kidney homogenates to AAI (50 μM) along with Ca2+ for 24 h resulted in mitochondrial swelling, Ca2+ efflux, membrane depolarization, cytochrome C release, decreased ATP production, and high caspase 3 activity [5,6]. In another study, a higher concentration of AAI (100 μM) was used on a canine kidney cell line (MDCK), which induced cellular stress, increased intracellular Ca2+ concentration, and altered mitochondrial function [23]. Ca2+ efflux was measured after isolated mitochondria from rat kidneys were energized with succinate in the presence of AAI, which induced significant swelling that was prevented by the addition of cyclosporin A (CsA) and ADP. The release of Ca2+ in response to AAI occurred in a dose-dependent manner, indicating the induction of mitochondrial permeability transition (MPT). AAI caused damage to HK-2 cells through a mitochondria-dependent mechanism, evidenced by mitochondrial swelling, release of Ca2+ from kidney mitochondria, and membrane depolarization in both mitochondria and HK-2 cells. A low concentration of Ca2+ and AAI alone cannot induce MPT in isolated mitochondria. However, Ca2+ levels are enhanced in tubular cells treated with AAI, suggesting tissue-specific toxicity of AAI that may account for its MPT-induced effects on renal tubular cells [20].
In a study performed on MH1C1-A23187 treated cells, a delay in the increase in cytosolic Ca2+ concentration ([Ca2+]c) was observed, and this delay was prevented by the addition of AAI [32]. AAI increased intracellular Ca2+ concentration ([Ca2+]i) in MDCK cells in the presence of extracellular Ca2+; even in the absence of extracellular Ca2+, AAI could still induce an increase in [Ca2+]i. The AAI-induced increase in [Ca2+]i led to the release of endoplasmic reticulum (ER) stores and the influx of extracellular Ca2+ [23].
3.2.6. Mitochondrial DNA (mtDNA)
Aristolochic Acid I (AAI) has been shown to reduce complex I activities of the respiratory chain, encoded by the mitochondrial DNA (mtDNA), in a dose-dependent manner without affecting complex II activities, encoded by the nuclear DNA (nDNA). The low mtDNA content in the kidneys of the AAI-treated animals suggests that AAI impairs its replication and suppresses oxidative phosphorylation inducing nephrotoxicity (Figure 1). This effect explains the sensitivity of the proximal tubule to AAI toxicity, as this segment exhibits the highest densities of mitochondria in the kidney. mtDNA may be more susceptible than nDNA to AAI-induced damage due to the absence of DNA repair mechanisms in mitochondria. mtDNA damage and respiratory chain defects occur in the kidney following acute AAI exposure, and these are considered possible mechanisms for AAI-induced acute nephrotoxicity [12]. AAI-induced renal toxicity in rats was linked to mtDNA depletion and respiratory chain defects. AAI decreases the respiratory control ratio (RCR), indicating respiratory and mitochondrial dysfunction, by suppressing ATP synthesis in a dose-dependent manner after treatment with AAI in HK-2 cells [20].
3.3. Other Types of Investigation Methods and Markers of Mitochondrial Dysfunction
In addition to methods specific to mitochondrial activity evaluation, various other techniques are employed to assess markers involved in AAI-induced cell and tissue toxicity. These methods are summarized in Table 1.

Table 1.
Assay methods used in AAI-induced in vitro and in vivo toxicity.
The aforementioned methods have proven valuable in both research and in preclinical and clinical diagnosis of AAI-induced kidney pathologies, particularly in identifying specific biomarkers. Fluorescent techniques, for instance, are highly useful in detecting AAI-induced mitochondrial dysfunction. These methods can be employed by departments such as pathological anatomy or immunology to evaluate biological samples—such as cells or biopsies—collected from affected patients. In addition to fluorescent techniques, common methods used in preclinical diagnosis include enzyme-linked immunosorbent assays (ELISA) for the detection of caspase activity, as well as biomarkers associated with oxidative stress pathways. Urinary biomarkers, like creatinine, are also frequently assessed to evaluate kidney function. A more specific, though costly, method involves the detection of mtDNA methylation. This technique is particularly valuable in the early clinical stages of kidney disease progression, offering insights into the molecular mechanisms underlying the disease.
The primary advantage of these methods lies in their specificity for detecting AAI, which enables the potential development of personalized treatment strategies. However, a significant drawback is their high cost, making them more suitable for research purposes [33,36] rather than routine diagnostic use in hospitals. Furthermore, these methods are typically applied only to patients suffering from CKDs or AAN [38], and not to those with (BEN), as these techniques have not yet been established as standard preclinical diagnostic tools in Balkan countries.
4. Discussions
4.1. Diagnosis and Prognosis of Mitochondrial Dysfunction in AAI-Induced Kidney Diseases
Understanding the role of mitochondrial dysfunction in the etiology and pathogenesis of Aristolochic Acid Nephropathy (AAN) and Balkan Endemic Nephropathy (BEN), as well as diagnosing and treating these conditions, remains limited. Specific diagnostic tools for detecting mitochondrial impairment in AAN and BEN are lacking. Mitochondrial functional assays are typically performed on fresh renal tissue, posing a significant limitation for clinical diagnosis due to the invasive nature of obtaining these samples. Despite this, mitochondria represent a promising pharmacological target for patients with renal impairment, especially if novel and noninvasive diagnostic approaches are developed. Combining mitochondrial-targeted therapies with conventional treatments and appropriate lifestyle modifications could enhance patient outcomes.
The scientific literature describes in vivo models of AAI-induced acute nephropathy in rats or mice and in vitro models where AAI cytotoxicity is tested on various cell lines and isolated mitochondria. These models reliably mimic the AAN condition observed in patients exposed to AAI through traditional medicines or contaminated foods. More than 90% of AAN patients consumed small amounts of AAI-containing medication for a median duration of 60 months. The nephrotoxic effects of AAI may manifest years after cessation, resulting in delayed diagnosis and treatment [10].
A comprehensive set of investigations is required to confirm tubulointerstitial nephropathy. These include renal biopsy, evaluation of clinical parameters such as hemoglobin (Hb), serum creatinine (Scr), and urinalysis. Additionally, risk factors for renal dysfunction should be documented, including cumulative AAI intake, high blood pressure, diabetes, cardiovascular diseases, smoking, obesity, and family history of kidney diseases. Some patients develop acute kidney injury (AKI) within three months of stopping AAI-containing herbs, leading to a diagnosis of acute AAN, with 50% progressing to chronic kidney disease (CKD) within four years of follow-up [10].
Pathological features of different AAN subtypes are diagnosed through immunofluorescence staining of renal biopsy specimens from patients with acute, chronic, and incipient tubular dysfunction. In acute AAN cases, light microscopy reveals tubular epithelial cell vacuolization or granulation. Electron microscopy can reveal disruption of the tubular brush border, mitochondrial and endoplasmic reticulum swelling, rupture of mitochondrial cristae, and an increased number of lysosomes. Acute AAN involves less tubular necrosis compared to chronic AAN, which shows patchy or diffuse tubular epithelial cell atrophy. Acute AAN cases, characterized by a large cumulative AAI intake (1.04 g) over a short period (14 days), exhibit different features compared to chronic AAN. In chronic AAN, cumulative AAI intake and the duration from cessation of AAI-containing medication to follow-up onset are independent factors correlated with the rate of eGFR decline, depending on the length of the period the patient had stopped taking AAI-containing medication [10].
4.2. Treatment of Mitochondrial Dysfunction in AAI-Induced Kidney Diseases
Future treatments for renal diseases caused by mitochondrial dysfunction will focus on protecting mitochondrial integrity using blockers of mitochondrial membrane depolarization (such as cyclosporin A), inhibitors of apoptosis, and reactive oxygen species (ROS) scavengers. Targeting mitochondria-derived oxidative stress could prevent or slow the progression of AAN, BEN, and potentially other AA-induced renal diseases. While the antioxidant properties of many of these agents are known, their use in clinical nephrology has only been partially investigated.
Patients with acute kidney injury (AKI) were treated with prednisone for one month, with the dose gradually reduced over the following two weeks. Chronic kidney disease (CKD) patients received standard treatments, including a low-protein diet and regular monitoring. During a one-year follow-up, 97% of AAN patients achieved normal blood pressure levels and hemoglobin concentrations, and metabolic acidosis and electrolyte disturbances were also normalized [10]. An in vitro study revealed that antioxidants containing a thiol group, such as glutathione and N-acetylcysteine, were effective in lowering AAI concentrations in kidney cells (HEK293) and preventing the formation of DNA adducts [39].
4.3. AAI-Induced Mitochondrial Dysfunction in Other Organs
It is worth noting that AAI can also target other organs, causing damage through mitochondrial stress and subsequent cellular modifications. For example, a study described impairment in oocyte development following AAI exposure. In AAI-exposed oocytes, mitochondria lost their normal localization entirely or partially. Additional findings revealed an increased level of ROS in AAI-exposed oocytes, leading to DNA damage, apoptosis, and defects in oocyte quality and fertilization ability. The reduced number of mitochondria and their aberrant distribution pattern suggest that AAI exposure causes mitochondrial dysfunction, compromising both nuclear and cytoplasmic maturation in oocytes. This impairment is likely induced by excessive oxidative stress. Exposure to AAI is harmful to both somatic and germ cells, potentially leading to various cancers, subfertility, or infertility in humans and animals [40].
Another organ affected by AAI is the liver, although studies on this topic are somewhat limited. Animal experiments have shown that in vivo exposure to different doses of AAI can induce deleterious effects on the liver, such as mitochondrial dysfunction, reduced numbers of organelles (e.g., endoplasmic reticulum, ribosomes), inflammatory cell infiltration, and fibrosis. Liver damage is mediated through the mitochondrial pathway of apoptosis and oxidative stress [41]. The concentration-dependent reduction in ATP levels due to AAI exposure was similar in both kidney cell lines and liver cell lines (L02) [37]. The toxic effects of AAI on liver cells (L02) are less severe compared to those on kidney cells (HEK293). Furthermore, exposure of human liver cells (L02) to AAI resulted in decreased glutathione levels and increased formation of DNA adducts [42].
4.4. The Cytoprotective Effects of AAI
Aristolochic acid I (AAI) is a phospholipase A2 (PLA2) inhibitor, and its inhibition of PLA2 activity is attributed to its cytotoxic effects [43]. Although AAI is cytotoxic to renal epithelial cells, it has been shown to be cytoprotective to certain non-renal cells under specific conditions [3,24,26,32,44]. By suppressing PLA2 activity, AAI may reduce arachidonic acid production and prevent mitochondrial permeability transition (MPT) [20,24,26].
In a model of apoptosis using Jurkat cell line cultures, a combination of an MPT inhibitor with AAI demonstrated potential in preventing cell death, mitochondrial membrane potential (MMP) loss, and cytochrome c release. Additionally, the apoptosis-inducing effects through Bax gene expression, followed by caspase 3 activation, DNA fragmentation, and PARP cleavage—consequences of MPT—were prevented by treatment with cyclosporin A (CsA) plus AAI [26].
AAI has been shown to induce apoptosis in pig kidney cells with epithelial morphology [45], associated with an increased intracellular calcium concentration ([Ca2+]i). Mitochondrial stress can be induced by an increase in [Ca2+]i upon AAI exposure, which activates caspase release and apoptosis. The anti-apoptotic protein Bcl-2, residing in mitochondrial membranes, may exert a cytoprotective effect against AAI-induced cytotoxicity by preventing MPT.
Other studies [32] have described the effects of a divalent cation ionophore (A23187) on apoptotic signaling in MH1C1 cells, where its addition caused a rapid rise in cytosolic Ca2+ ([Ca2+]c). The increase in [Ca2+]c led to phospholipid hydrolysis, which could be inhibited by AAI. These effects were followed by MPT pore opening and apoptosis in approximately 30% of the cell population. There is a cause–effect relationship between A23187 addition, cPLA2 activation, MPT pore opening, and cell death activation, but the [Ca2+]c rise was inhibited by AAI. The release of cytochrome c and cleavage of caspases 9 and 3 precede cell death, all of which were prevented by the addition of AAI and CsA.
cPLA2 is involved in triggering the inhibitory effects of AAI [46,47,48] and Ca2+ [49]. Additionally, the mitochondrial anion carrier protein UCP2 has a cytoprotective effect on HK-2 cells treated with AAI (40 µM) and AAI plus genipin. UCP2 also exhibits antioxidant and antiapoptotic properties in AAI-treated HK-2 cells by decreasing ROS production [33].
5. Conclusions and Future Perspectives
Aristolochic acid I (AAI) is a potent nephrotoxin and carcinogen, leading to kidney injury through exposure to contaminated medicinal plants, foods, or water. AAI is a well-known primary cause of Aristolochic Acid Nephropathy (AAN) and Balkan Endemic Nephropathy (BEN). To study mitochondrial functions at the renal level, various methods were employed using kidney cell lines or mitochondrial kidney homogenates. These methods included the measurement of oxygen consumption, respiratory chain complexes, mitochondrial membrane potential, adenine nucleotide translocator activity, mitochondrial DNA, imaging investigations, immunological methods, molecular biology, and biochemical assays.
In vitro models where kidney cells were exposed to AAI were used to investigate AAN and BEN. AAI cytotoxicity manifested in a dose-dependent manner, causing cell death mediated via caspase-dependent and independent pathways. Further studies revealed the inhibitory effects on AAI-induced toxicity of compounds such as catalpol, vitamin C, and probenecid. AAI induces mitochondrial stress, characterized by increased intracellular Ca2+, mtDNA depletion, and impaired respiratory chain function, leading to elevated ROS levels, decreased MMP, and ATP depletion. AAI-induced mitochondrial injury showed mitochondrial fragmentation, down-regulation of mitochondrial respiratory chain proteins, down-regulation of mitochondrial biogenesis-related proteins, and reduced mtDNA copy number.
Markers of AAI exposure were identified and analyzed, with abnormal levels contributing to kidney pathogenesis, including glutathione, intracellular Ca2+, DNA-adducts, ATP levels, cytochrome c, and caspase 3, which could be effectively used in the diagnosis and prognosis of AAN and BEN.
5.1. Future Directions
Emerging evidence suggests that dysfunctional mitochondria play a crucial role in the development of chronic kidney diseases (CKDs) and the associated comorbidities, highlighting their potential as therapeutic targets. A combination of food-derived antioxidants, natural plant compounds, and conventional therapies could help clinicians improve the outcomes of these kidney diseases and identify effective intervention targets.
Studies have shown that vitamin C can suppress AAI-induced cytotoxicity in kidney cells by reducing the increased levels of H2O2 and caspase-3 activation. These findings suggest that supplementation with vitamin C or catalpol may be beneficial in reducing AAI-induced renal damage [20,22]. Future predictive approaches, such as pharmacogenomics, should be pursued to develop novel mitochondria-targeted antioxidant treatments for AAN, BEN, and other types of CKDs.
Developing noninvasive diagnostic tools for detecting mitochondrial impairment in AAN and BEN should be prioritized in future research. Exploring the therapeutic potential of targeting mitochondrial-derived oxidative stress could prevent or slow the progression of AAN, BEN, and other AA-induced renal diseases.
5.2. Potential Therapeutic Approaches
Therapeutic approaches should focus on protecting mitochondrial integrity using blockers of mitochondrial membrane depolarization (such as cyclosporin A), inhibitors of apoptosis, and ROS scavengers. Combining these treatments with conventional therapies and appropriate lifestyle modifications could enhance patient outcomes. Predictive approaches, such as pharmacogenomics, should be undertaken to develop novel mitochondria-targeted antioxidant treatments for AAN, BEN, and other CKDs.
Recent advances in omics research, including genomics, proteomics, and metabolomics, have provided deeper insights into the molecular mechanisms underlying AAI-induced mitochondrial dysfunction in kidney disease [50]. Omics approaches have enabled the identification of novel biomarkers associated with mitochondrial damage, such as specific mitochondrial DNA mutations, alterations in the expression of mitochondrial proteins, and changes in metabolic pathways linked to energy production and oxidative stress [28]. These biomarkers not only improve our understanding of disease pathogenesis but also hold potential for the development of targeted therapies. For instance, mitochondrial proteomics has revealed a decrease in the expression of respiratory chain complexes, which correlates with reduced ATP production and increased susceptibility to oxidative stress in AAI-induced nephropathy [51,52]. Integrating these findings with current therapeutic strategies could lead to more effective interventions aimed at preserving mitochondrial function and mitigating renal damage.
5.3. Importance and Potential Impact
Understanding the mechanisms of AAI-induced mitochondrial dysfunction and identifying specific biomarkers is crucial for the early diagnosis and effective treatment of AAN and BEN. These findings highlight the potential for novel therapeutic strategies targeting mitochondrial health, which could significantly improve patient outcomes and prevent the progression of kidney diseases related to AAI exposure.
Recent research has shed light on the impact of Aristolochic Acid I (AAI) on mitochondrial function in kidney disease, particularly in the context of aristolochic acid nephropathy (AAN) [2]. AAI specifically targets proximal tubular epithelial cells (PTECs), which are known for their high mitochondrial content, making them particularly susceptible to mitochondrial dysfunction. Mitochondrial dysfunction is increasingly recognized as a critical factor in the pathogenesis of various kidney diseases, including AAN. However, despite this recognition, the precise status of mitochondrial function in PTECs following AAI exposure remains not fully elucidated.
5.4. Effects of AAI on Mitochondrial Function
AAI has been shown to reduce cell viability and induce apoptosis in a manner that is both dose- and time-dependent. In studies using NRK-52E cells, a model system for kidney epithelial cells, AAI treatment has been associated with several markers of mitochondrial dysfunction. These include elevated levels of reactive oxygen species (ROS), a reduction in mitochondrial membrane potential (MMP), a decrease in mtDNA copy number, and diminished ATP production. Importantly, these findings underscore the role of mitochondrial impairment in AAI-induced nephrotoxicity. The potential for therapeutic intervention has been explored with mitochondria-targeted antioxidants. For instance, the peptide Szeto-Schiller 31 (SS-31) has been demonstrated to mitigate AAI-induced mitochondrial dysfunction and reduce apoptosis, suggesting a promising avenue for treatment.
5.5. Mutational Signature and Carcinogenesis
Beyond its impact on mitochondrial function, AAI exposure has been linked to mutational changes that contribute to carcinogenesis. Specifically, DNA adducts derived from AAI serve as biomarkers for exposure, and a characteristic mutational signature, marked by A→T transversions, has been identified in urothelial carcinoma tumor tissues from patients exposed to AA. This signature provides critical insights into the carcinogenic mechanisms of AAI [7].
5.6. Clinical Implications
The clinical consequences of AAN are severe, often leading to irreversible kidney damage and end-stage renal failure. Despite the grave nature of this condition, no effective therapeutic regimen has been established to date. Alarmingly, AA-related adverse events continue to be reported, particularly in regions such as Asia and the Balkans, where exposure to AA-containing herbal products remains a concern.
Author Contributions
Conceptualization, A.T.L.-G., C.L.C., N.M.P. and C.A.T.; methodology, A.T.L.-G., C.L.C. and N.M.P.; formal analysis, I.-M.C., A.-G.S. and M.-A.P.; investigation, A.T.L.-G., C.L.C., F.A.E.S., I.-M.C. and M.-A.P.; data curation, A.T.L.-G., N.M.P. and C.A.T.; writing—original draft preparation, A.T.L.-G., N.M.P. and C.A.T.; writing—review and editing, A.T.L.-G., N.M.P. and C.A.T.; visualization, A.T.L.-G., N.M.P. and C.A.T.; supervision, V.L.O., N.M.P., V.P. and C.A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gifford, F.J.; Gifford, R.M.; Eddleston, M.; Dhaun, N. Endemic Nephropathy Around the World. Kidney Int. Rep. 2017, 2, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, J.; Wang, J.; Feng, X.; Wu, H.; Huang, R.; Fan, J.; Yu, X.; Yang, X. Mitochondrial Dysfunction Is Involved in Aristolochic Acid I-Induced Apoptosis in Renal Proximal Tubular Epithelial Cells. Hum. Exp. Toxicol. 2020, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, P.; Wei, F.; Lin, R.-C.; Khan, I.A.; Pasco, D.S. Structure Activity Relationships of Aristolochic Acid Analogues: Toxicity in Cultured Renal Epithelial Cells. Kidney Int. 2005, 67, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M. Aristolochic Acid as a Probable Human Cancer Hazard in Herbal Remedies: A Review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Debelle, F.D.; Vanherweghem, J.-L.; Nortier, J.L. Aristolochic Acid Nephropathy: A Worldwide Problem. Kidney Int. 2008, 74, 158–169. [Google Scholar] [CrossRef]
- Tatu, C.A.; Orem, W.H.; Finkelman, R.B.; Feder, G.L. The Etiology of Balkan Endemic Nephropathy: Still More Questions than Answers. Environ. Health Perspect. 1998, 106, 689. [Google Scholar] [CrossRef]
- Han, J.; Xian, Z.; Zhang, Y.; Liu, J.; Liang, A. Systematic Overview of Aristolochic Acids: Nephrotoxicity, Carcinogenicity, and Underlying Mechanisms. Front. Pharmacol. 2019, 10, 648. [Google Scholar] [CrossRef]
- Chan, C.-K.; Liu, Y.; Pavlović, N.M.; Chan, W. Etiology of Balkan Endemic Nephropathy: An Update on Aristolochic Acids Exposure Mechanisms. Chem. Res. Toxicol. 2018, 31, 1109–1110. [Google Scholar] [CrossRef]
- Jadot, I.; Declèves, A.-E.; Nortier, J.; Caron, N. An Integrated View of Aristolochic Acid Nephropathy: Update of the Literature. IJMS 2017, 18, 297. [Google Scholar] [CrossRef]
- Yang, L.; Su, T.; Li, X.-M.; Wang, X.; Cai, S.-Q.; Meng, L.-Q.; Zou, W.-Z.; Wang, H.-Y. Aristolochic Acid Nephropathy: Variation in Presentation and Prognosis. Nephrol. Dial. Transplant. 2012, 27, 292–298. [Google Scholar] [CrossRef]
- Braga, P.C.; Alves, M.G.; Rodrigues, A.S.; Oliveira, P.F. Mitochondrial Pathophysiology on Chronic Kidney Disease. IJMS 2022, 23, 1776. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Bao, Q.; Sun, L.; Huang, X.; Wang, T.; Zhang, S.; Li, H.; Zhang, L. Possible Role of mtDNA Depletion and Respiratory Chain Defects in Aristolochic Acid I-Induced Acute Nephrotoxicity. Toxicol. Appl. Pharmacol. 2013, 266, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Ham, Y.-H. Probing the Hidden Role of Mitochondrial DNA Damage and Dysfunction in the Etiology of Aristolochic Acid Nephropathy. Chem. Res. Toxicol. 2021, 34, 1903–1909. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; Benavides, G.A.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G.; et al. A Novel Approach to Measure Mitochondrial Respiration in Frozen Biological Samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A.J. Drug Delivery to Mitochondria: The Key to Mitochondrial Medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. IJMS 2021, 22, 11253. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Persistent Disruption of Mitochondrial Homeostasis after Acute Kidney Injury. Am. J. Physiol.-Ren. Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Zhou, Y.; Bian, X.; Fang, L.; He, W.; Dai, C.; Yang, J. Aristolochic Acid Causes Albuminuria by Promoting Mitochondrial DNA Damage and Dysfunction in Podocyte. PLoS ONE 2013, 8, e83408. [Google Scholar] [CrossRef]
- Qi, X.; Cai, Y.; Gong, L.; Liu, L.; Chen, F.; Xiao, Y.; Wu, X.; Li, Y.; Xue, X.; Ren, J. Role of Mitochondrial Permeability Transition in Human Renal Tubular Epithelial Cell Death Induced by Aristolochic Acid. Toxicol. Appl. Pharmacol. 2007, 222, 105–110. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, P.; Chen, J.; Yang, C.; Xia, F.; Zhang, J.; Tang, H.; Liu, D.; Gu, L.; Shi, Q.; et al. Dissection of Targeting Molecular Mechanisms of Aristolochic Acid-Induced Nephrotoxicity via a Combined Deconvolution Strategy of Chemoproteomics and Metabolomics. Int. J. Biol. Sci. 2022, 18, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.; Batuman, V. Aristolochic Acid I Induces Proximal Tubule Injury through ROS/HMGB1/Mt DNA Mediated Activation of TLRs. J. Cell. Mol. Med. 2022, 26, 4277–4291. [Google Scholar] [CrossRef] [PubMed]
- Hsin, Y.-H.; Cheng, C.-H.; Tzen, J.T.C.; Wu, M.-J.; Shu, K.-H.; Chen, H.-C. Effect of Aristolochic Acid on Intracellular Calcium Concentration and Its Links with Apoptosis in Renal Tubular Cells. Apoptosis 2006, 11, 2167–2177. [Google Scholar] [CrossRef]
- Tafani, M.; Schneider, T.G.; Pastorino, J.G.; Farber, J.L. Cytochrome C-Dependent Activation of Caspase-3 by Tumor Necrosis Factor Requires Induction of the Mitochondrial Permeability Transition. Am. J. Pathol. 2000, 156, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-K.; Wei, C.-W.; Pan, Y.-R.; Cherng, S.-H.; Chang, W.-J.; Wang, H.-F.; Yu, Y.-L. Vitamin C Attenuates the Toxic Effect of Aristolochic Acid on Renal Tubular Cells via Decreasing Oxidative Stress-Mediated Cell Death Pathways. Mol. Med. Rep. 2015, 12, 6086–6092. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Chen, S.-T.; Tafani, M.; Snyder, J.W.; Farber, J.L. The Overexpression of Bax Produces Cell Death upon Induction of the Mitochondrial Permeability Transition. J. Biol. Chem. 1998, 273, 7770–7775. [Google Scholar] [CrossRef]
- Jiménez-Uribe, A.P.; Pedraza-Chaverri, J. Promising Therapeutic Strategies Targeting Mitochondria in Kidney Diseases: From Small Molecules to Whole Mitochondria. Future Pharmacol. 2022, 2, 256–275. [Google Scholar] [CrossRef]
- Ho, H.-J.; Shirakawa, H. Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, 88. [Google Scholar] [CrossRef]
- Galvan, D.L.; Mise, K.; Danesh, F.R. Mitochondrial Regulation of Diabetic Kidney Disease. Front. Med. 2021, 8, 745279. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Wang, J.; Fan, J.; Feng, X.; Yu, X.; Yang, X. Possible Role of Mitochondrial Injury in Caulis Aristolochia Manshuriensis-Induced Chronic Aristolochic Acid Nephropathy. Drug Chem. Toxicol. 2017, 40, 115–124. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Schnellmann, R.G. Pharmacological Targeting of Mitochondria in Diabetic Kidney Disease. Pharmacol. Rev. 2023, 75, 250–262. [Google Scholar] [CrossRef]
- Penzo, D.; Petronilli, V.; Angelin, A.; Cusan, C.; Colonna, R.; Scorrano, L.; Pagano, F.; Prato, M.; Di Lisa, F.; Bernardi, P. Arachidonic Acid Released by Phospholipase A2 Activation Triggers Ca2+-Dependent Apoptosis through the Mitochondrial Pathway. J. Biol. Chem. 2004, 279, 25219–25225. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Anger, E.E.; Zhang, X.; Su, S.; Su, C.; Zhao, S.; Yu, F.; Li, J. Protective Effects of Mitochondrial Uncoupling Protein 2 against Aristolochic Acid I-Induced Toxicity in HK-2 Cells. IJMS 2022, 23, 3674. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Zhou, C.; Xu, Q.; Gao, H.; Huo, M.; Jiang, X.; Yu, W. Catalpol Attenuates Renal Injury by Regulating Oxidative Stress and Inflammation Response. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, S.; Wu, J.; Chen, W.; Sun, H.; Peng, W.; Yu, X.; Yang, X. Autophagy Inhibitors Promoted Aristolochic Acid I Induced Renal Tubular Epithelial Cell Apoptosis via Mitochondrial Pathway but Alleviated Nonapoptotic Cell Death in Mouse Acute Aritolochic Acid Nephropathy Model. Apoptosis 2014, 19, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, T.E.R.; Pozdzik, A.A.; Arlt, V.M.; De Prez, E.G.; Antoine, M.-H.; Quellard, N.; Goujon, J.-M.; Nortier, J.L. Probenecid Prevents Acute Tubular Necrosis in a Mouse Model of Aristolochic Acid Nephropathy. Kidney Int. 2012, 82, 1105–1113. [Google Scholar] [CrossRef]
- Yang, C.-C.; Wu, C.-T.; Chen, L.-P.; Hung, K.-Y.; Liu, S.-H.; Chiang, C.-K. Autophagy Induction Promotes Aristolochic Acid-I-Induced Renal Injury in Vivo and in Vitro. Toxicology 2013, 312, 63–73. [Google Scholar] [CrossRef]
- Zhou, Q.; Jiang, L.; Su, T.; Liu, G.; Yang, L. Overview of Aristolochic Acid Nephropathy: An Update. Kidney Res. Clin. Pract. 2023, 42, 579–590. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, C.-K.; Pavlović, N.M.; Chan, W. Effects of Diet on Aristolochic Acid-DNA Adduct Formation: Implications for Balkan Endemic Nephropathy Etiology. Chem. Res. Toxicol. 2023, 36, 438–445. [Google Scholar] [CrossRef]
- Zhang, Y.; ShiYang, X.; Zhang, Y.; Li, Y.; Shi, X.; Xiong, B. Exposure to Aristolochic Acid I Compromises the Maturational Competency of Porcine Oocytes via Oxidative Stress-Induced DNA Damage. Aging 2019, 11, 2241–2252. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.; Zhou, C.; Jia, Y.; Liu, S.; Xiong, Z.; Guo, X.; Fei, X.; Jiang, X.; Yu, W. Aristolochic Acid Induces Mitochondrial Apoptosis through Oxidative Stress in Rats, Leading to Liver Damage. Toxicol. Mech. Methods 2021, 31, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Au, C.-K.; Ham, Y.-H.; Chan, W. Bioaccumulation and DNA Adduct Formation of Aristolactam I: Unmasking a Toxicological Mechanism in the Pathophysiology of Aristolochic Acid Nephropathy. Chem. Res. Toxicol. 2023, 36, 322–329. [Google Scholar] [CrossRef]
- Fernandes, C.A.H.; Cardoso, F.F.; Cavalcante, W.G.L.; Soares, A.M.; Dal-Pai, M.; Gallacci, M.; Fontes, M.R.M. Structural Basis for the Inhibition of a Phospholipase A2-Like Toxin by Caffeic and Aristolochic Acids. PLoS ONE 2015, 10, e0133370. [Google Scholar] [CrossRef]
- Okada, H.; Watanabe, Y.; Inoue, T.; Kobayashi, T.; Kanno, Y.; Shiota, G.; Nakamura, T.; Sugaya, T.; Fukamizu, A.; Suzuki, H. Transgene-Derived Hepatocyte Growth Factor Attenuates Reactive Renal Fibrosis in Aristolochic Acid Nephrotoxicity. Nephrol. Dial. Transplant. 2003, 18, 2515–2523. [Google Scholar] [CrossRef]
- Gao, R.; Zheng, F.; Liu, Y.; Zheng, D.; Li, X.; Bo, Y.; Liu, Y. Aristolochic Acid I-Induced Apoptosis in LLC-PK1 Cells and Amelioration of the Apoptotic Damage by Calcium Antagonist. Chin. Med. J. 2000, 113, 418–424. [Google Scholar] [PubMed]
- Rosenthal, M.D.; Vishwanath, B.S.; Franson, R.C. Effects of Aristolochic Acid on Phospholipase A2 Activity and Arachidonate Metabolism of Human Neutrophils. Biochim. Biophys. Acta 1989, 1001, 1–8. [Google Scholar] [CrossRef]
- Chandra, V.; Jasti, J.; Kaur, P.; Srinivasan, A.; Betzel, C.; Singh, T.P. Structural Basis of Phospholipase A2 Inhibition for the Synthesis of Prostaglandins by the Plant Alkaloid Aristolochic Acid from a 1.7 Å Crystal Structure. Biochemistry 2002, 41, 10914–10919. [Google Scholar] [CrossRef]
- Vishwanath, B.S.; Fawzy, A.A.; Franson, R.C. Edema-Inducing Activity of Phospholipase A2 Purified from Human Synovial Fluid and Inhibition by Aristolochic Acid. Inflammation 1988, 12, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Gijón, M.A.; Leslie, C.C. Regulation of Arachidonic Acid Release and Cytosolic Phospholipase A2 Activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar] [CrossRef]
- Liu, X.; Shi, J.; Jiao, Y.; An, J.; Tian, J.; Yang, Y.; Zhuo, L. Integrated Multi-Omics with Machine Learning to Uncover the Intricacies of Kidney Disease. Brief. Bioinform. 2024, 25, bbae364. [Google Scholar] [CrossRef]
- Li, J.J.; Liu, J.; Lupino, K.; Liu, X.; Zhang, L.; Pei, L. Growth Differentiation Factor 15 Maturation Requires Proteolytic Cleavage by PCSK3, -5, and -6. Mol. Cell. Biol. 2018, 38, e00249-18. [Google Scholar] [CrossRef] [PubMed]
- Verissimo, T.; de Seigneux, S. New Evidence of the Impact of Mitochondria on Kidney Health and Disease. Nat. Rev. Nephrol. 2024, 20, 81–82. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).