Abstract
Natural ectoine, (+)-(4S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid, is an extremely important small biomolecule belonging to the class of osmolytic/osmoprotective compounds. It stabilizes biomacromolecules such as DNA and proteins and protects them from denaturation by heat, dehydration, and UV radiation. The rapidly growing interest in this compound resulted in currently exclusive biotechnological production, while a chemical process along with enantioseparation as an alternative has not yet been established. An improved chemical synthesis of racemic ectoine starting from γ-butyrolactone in very good yield is described. Regioselective monoacetylation is achieved by the complexation of a copper(II)-ion with two molecules of 2,4-diamonobutyric acid in the key synthetic step. The racemic ectoine was synthesized with the aim of being successfully enantioseparated for the first time by high-performance liquid chromatography (HPLC) using a teicoplanin-based Chiral-T column in different solvent systems. The presence of (+)-ectoine was determined and quantified using an HPLC protocol on the Synergy Polar-RP column in fermentation broths inoculated with different strains of Streptomyces sp. bacteria isolated from the Adriatic Sea and grown on different NaCl concentrations.
1. Introduction
Ectoine, (+)-(4S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid, belongs to a group of compounds called osmolytes or osmoprotectors. Galinski et al. [1] discovered ectoine in 1985 in the highly halophilic bacterium Halorhodospira halophila, which was isolated from the Wadi El-Natrun valley in Egypt. Ectoine is classified as either a heterocyclic amino acid or a partially hydrogenated pyrimidine derivative. Ectoine was originally thought to be an extremely rare osmolyte, but improved screening techniques using HPLC and 13C NMR spectroscopy showed that ectoine is very abundant in microorganisms in response to elevated salinity. The presence of ectoine has been observed in a number of bacteria [2,3,4], archaea [5], protists [6,7,8], and some microalgae [9], where it has an osmoprotective effect on the aforementioned microorganisms and also stabilizes biomacromolecules such that they are protected from denaturation, heat, drying, and UV radiation [2]. At physiological pH, ectoine contains a negatively charged carboxyl group attached to a heterocyclic ring with a delocalized positive charge between two nitrogen atoms. The resulting interaction between hydrophilic and hydrophobic forces affects the water–water and water–solute interactions, and therefore, has strong effects on the hydration of ectoine itself and ion binding, as well as having an influence on the local water structure. Molecular dynamics simulations have shown that ectoine is a strong water-binding agent and that it can accumulate seven water molecules around itself at a distance of less than 0.6 nm [10], which is due to the creation of a large number of hydrogen bonds. It was shown that the accumulation of water around the ectoine molecule was not affected even at a high salt concentration. These physicochemical properties of ectoine enable physiologically appropriate hydration of the cytoplasm after ectoine accumulation in response to osmotic stress, which has a protective effect on protein stability and macromolecules functionality [11,12]. Due to its positive effect on human, animal, and other cell cultures, ectoine is emphasized as a molecule of interest in both the scientific community and industrial production [13]. Recently, ectoine has been discovered as an effective treatment of eye surface irritations and inflammation [14]. Ectoine is currently produced exclusively by biotechnological processes [2,15,16,17,18], and only a few synthetic methods of ectoine synthesis are known [19,20].
S. Himdi-Kabbab et al. [19] proposed two synthetic routes for the preparation of ectoine, which are shown in Scheme 1. Both synthetic routes are based on the cyclization of 2,4-diaminobutyric acid derivatives. In one synthetic route, the thermal cyclization of 4-acetamido-2-aminobutyric acid or 2-acetamido-4-aminobutyric acid is carried out (Scheme 1(i)). The second synthetic route, involves the cyclization of 2-amino-4-(1-ethoxyethylidene)aminobutyric acid or 4-amino-2-(1-ethoxyethylidene) aminobutyric acid (Scheme 1(ii)).
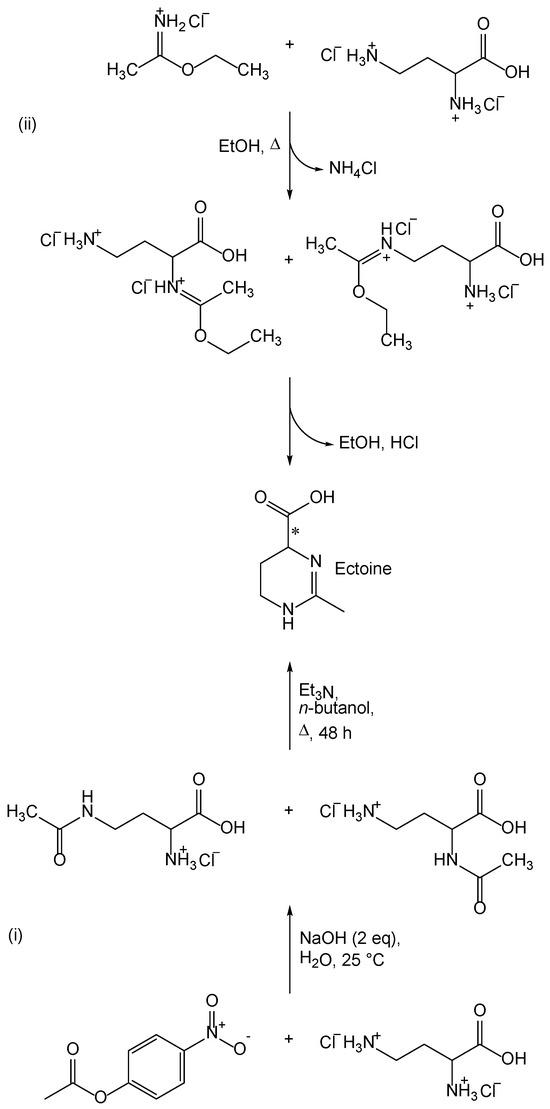
Scheme 1.
Two synthetic routes described by S. Himdi-Kabbab et al. [19]. (i) acetylation with 4-nitrophenyl acetate; (ii) reaction with ethyl ethanimidate.
The starting compound for both synthetic routes (Scheme 1) is the dihydrochloride of 2,4-diaminobutyric acid. The most favorable are the synthetic routes starting from dihydrofuran-2(3H)-one (γ-butyrolactone), which after reaction with potassium phthalimide finally gives the desired α-aminoxyline [21,22]. One of the synthetic ways to obtain dihydrochloride of 2,4-diaminobutyric acid was proposed by Talbot et al. [21], and involves three reactions. The first reaction starts from commercially available 3-bromodihydrofuran-2(3H)-one [23]. Another route for the preparation of 2,4-diaminobutanoic acid dihydrochloride was proposed by Zaoral in his article [22]. The next step in the synthesis is the acetylation of 2,4-diaminobutanoic acid (Scheme 1). Since 2,4-diaminobutanoic acid contains two free amino groups that have almost the same chemical properties, it is important to achieve selective acetylation of only one amino group. For obtaining ectoine, however, it is not important whether the amino group at position two or four is acetylated, because the thermal cyclization of both 2-acetamido-4-aminobutyric acid and 4-acetamido-2-aminobutyric acid yields ectoine as a product. The introduction of an acetyl group at the amino group of 2,4-diaminobutyric acid can be carried out with 4-nitrophenylacetate. Leclerc and Benoiton have described the dependence of selective acetylation on pH and on the equivalents of 4-nitrophenyl-acetate [24]. A more favorable method for the preparation of monoacetylated 2,4-diaminobutyric acid involves the complexation of 2,4-diaminobutyric acid with copper(II)-ions and the acetylation of the resulting complex with 4-nitrophenylacetate [25,26,27]. This method has not yet been used for the preparation of ectoine and its derivatives.
In this work, racemic ectoine was prepared in purpose to develop an efficient chromatographic method for its enantioseparation, which can be used for the determination of the optical purity of ectoine samples, but also to test individually their osmolytic/osmoprotective activities. Moreover, this approach can lead to an alternative method for ectoin enantiomers production based on preparative HPLC/SMB technology. The separation of enantiomers by liquid chromatography can be performed by direct and indirect methods [28]. Indirect methods are based on the addition of chiral additives to the mobile phase, whereupon the optical isomers react with the chiral additive, and consequently, diastereomers are formed, which can then be separated on an achiral stationary phase [29]. On the other hand, direct methods separate enantiomers on a chiral stationary phase by the formation of diastereomeric labile complexes of different stability, where enantiomer that forms a more stable diastereoisomer complex remains longer on the chiral stationary phase. Chiral stationary phases can be categorized according to basic structural features and the nature of the attractive forces acting between the stationary phase and the individual enantiomers [30,31].
The most commonly used chiral stationary phases built from synthetic polymers are polysaccharides, which consist mainly of derivatized celluloses and amyloses. These chiral stationary phases are most commonly used with non-polar mobile phases, but there are also polysaccharide-based chiral stationary phases intended for the reverse phase (Chiralpak AD-RH, Chiralpak AS-RH, Chiralcel OD-RH, Chiralcel OJ-RH, etc.). The next group of chiral stationary phases that form inclusion compounds with the analyte are cyclodextrins. Brush-type or Pirke-type chiral stationary phases contain chiral selectors with small molecular masses. Protein-based chiral stationary phases contain immobilized proteins on a silica gel support.
Chiral stationary phases that have proven to be most suitable for the separation of non-derivatized amino acids are chiral stationary phases containing macrocyclic glycopeptides, crown ethers, and chiral selectors for ion exchange. Macrocyclic glycopeptides (e.g., vancomycin, teicoplanin) covalently bound to a silica gel support represent a type of chiral stationary phase that allows the use of a wide range of mobile phases, non-polar and polar, ideal for the analytical and preparative separation of neutral, polar, and ionic compounds. These chiral stationary phases interact with the analyte through six different molecular interactions, which include ionic interactions, hydrogen bonding, π–π interactions, dipole–dipole interactions, and hydrophobic and steric interactions. In addition to the mentioned molecular interactions, macrocyclic glycopeptides contain several inclusion sites that promote selectivity with regard to the geometric shape of the analyte. The most important feature of these stationary phases is the ability to generate ionic interactions between the analyte and the chiral stationary phase, which enables the separation of non-derivatized amino acids, which are present as zwitter-ions at physiological pH [30]. Chiral stationary phases containing crown ethers covalently bonded to a silica gel support have also been shown to be a good choice for the separation of enantiomers of non-derivatized natural and non-natural amino acids. It is necessary to use acidic polar mobile phases in order to obtain the best separation of amino acids [30]. Ion-exchange chiral stationary phases contain anion- or cation-exchange functional groups, or zwitter-ion, which serve as chiral selectors and immobilized on a silica gel support. These chiral stationary phases can be used with all common HPLC solvents, and due to the ionic character of the chiral selector as well as the analyte, it is necessary to add an acid or a base to the mobile phase [32].
2. Results and Discussion
2.1. Synthesis of (±)-Ectoine
The (±)-ectoine, (±)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid 7, was synthesized by combining different synthetic approaches and improved the method. The overall conducted synthesis is shown in Scheme 2.
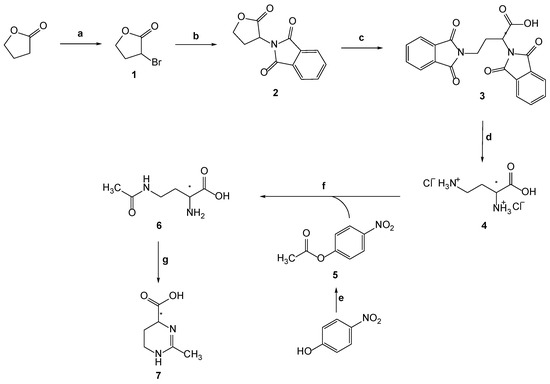
Scheme 2.
Synthesis of (±)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid (7): (a) PBr3, Br2; (b) potassium phthalimide, DMF; (c) (1) potassium phthalimide, DMF, (2) acetic acid (conc.); (d) HCl (aq) (1:1, v/v); (e) Ac2O; (f) (1) NaOH, Cu(CH3COO)2 × H2O, acetone, compound 5, (2) quinolin-8-ol, acetone, water; (g) Et3N, n-butanol.
Compound 1 was prepared from the γ-butyrolactone (dihydrofuran-2(3H)-one) by reaction with elemental bromine and with a catalytic amount of phosphorus tribromide. In this reaction 2,4-dibromobutyric acid was formed as intermediate with the release of HBr. In regular conditions only a small amount of 2,4-dibromobutyric acid changes to product, and some interventions in form of vacuum distillation or addition of base KOH are needed [18]. When the reaction was completed, the residual acid was neutralized with Na2CO3. Compound 1 was isolated after extraction with dichloromethane (DCM).
The synthesis of compound 4 includes three steps: (i) nucleophilic substitution of bromine with potassium phthalimide, (ii) lactone ring opening with the second molecule of potassium phthalimide, and (iii) hydrolysis. The first step involves the SN2 reaction of compound 1 with potassium phthalimide resulting in substitution of bromine with phthalimide and the formation of 2-(oxotetrahydrofuran-3-yl)isoindolin-1,3-dione (2) (Scheme 2) [16].
The second step involves the reaction of compound 2 with another molecule of potassium phthalimide and the formation of intermediate potassium salt, which with concentrated acetic acid gave 2,4-bis(1,3-dioxoisoindolin-2-yl)butyric acid (3) in 79% yield (Scheme 2). The third step was hydrolysis of compound 3 with HCl to remove the phthalate groups. Compound 4 was obtained in 71% yield (Scheme 2). The reaction of 4-nitrophenol and acetic anhydride, in which the oxygen from hydroxyl group of 4-nitrophenol nucleophilically attacks the carbonyl carbon in the acetic anhydride, gives compound 5 in 98% yield (Scheme 2). The synthesis of compound 6 was obtained in two different methods (A and B). Method A is the general procedure for the preparation of compound 6. We examined six modifications (A1-A6) of this method, which are described in the Experimental section. According to Bazureau et al. [19], the expected products are 4-acetamido-2-aminobutyric acid and 2-acetamido-4-aminobutyric acid in different ratios depending on the base added. We observed that in each of the six modifications of the general procedure, three products were observed, i.e., 4-acetamido-2-aminobutyric acid and 2-acetamido-4-aminobutyric acid, which were expected, but another compound not described in the literature was also found: 2,4-diacetamidobutyric acid, as well as a small amount of the starting compound 4.
Isolation of the desired product is difficult due to the similar nature of the compounds. For this reason, a different synthetic route was discussed, involving complexation with copper(II)-ions and then subsequent acetylation with 4-nitrophenyl acatate (5) (Scheme 3).
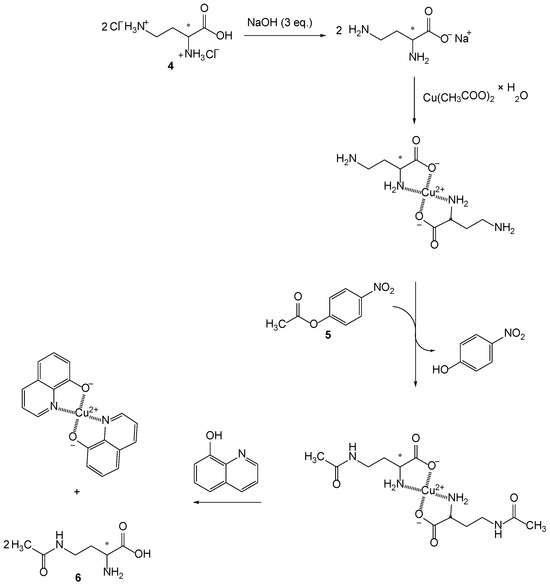
Scheme 3.
Synthesis of 4-acetamido-2-butyric acid (6): (i) complexation of 2,4-diaminobutyric acid dihydrochloride (4) in base conditions with copper(II)-ions; (ii) selective monoacetylation of 2,4-diaminobutyric acid copper(II)complex with 4-nitrophenylacetate (5) resulting in release of 4-nitrophenol and acetylation of amino groups of copper(II) complex; (iii) reaction with 8-hydroxyquinoline results in copper(II)-quinoline complex and two molecules of monoacetylated diaminobutyric acid (6).
Complexation of copper(II)-ion was obtained with two molecules of 2,4-diamonobutyric acid where copper(II)-ion coordinate hydroxyl and α-amino group from two 2,4-diaminobutyric acids. This pathway protects the α-amino group and only allows monoacetylation of the γ-amino group (Scheme 3). Since the 8-hydroxyaminoquinoline is a better ligand for copper(II)-ions, its addition to the suspension of copper(II)-4-acetamido-2-aminobuytic acid results with release of two molecules of 4-acetamido-2-aminobutyric acid (6) and the formation of the copper(II)-quinoline complex.
Since the compound 6 is soluble in water and formed copper(II) quinoline complex is not, the isolation of compound 6 was obtained simply by filtration. After evaporation of the solvent, the compound 6 was obtained in 82% yield.
Racemic mixture of ectoine 7 was obtained by thermal cyclization of compound 6 (Scheme 2). The pure compound 7 was obtained in 63% yield after trituration with cold ethanol in 63% yield. A better yield of 82% was obtained when a cation exchange resin (Amberlite IR 120 H*) was used for purification.
2.2. (±)-Ectoine Enantiomers Separation
To our best knowledge this is the first time that enantiomers separation of (±)-ectoine has been performed using chiral stationary phases. In general, there are several subgroups of chiral stationary phases, and some of these subgroups were examined: polysaccharide-based chiral stationary phases (cellulose, amylose), brush-type chiral stationary phases, and stationary phases with macrocyclic selector respectively macrocyclic glycopeptide (teicoplanine). Several factors must be considered when developing the method. The first is the solubility of (±)-ectoine, which is soluble in water and MeOH, less soluble in EtOH and ACN, and insoluble in non-polar organic solvents. This physical property restricted the choice of stationary phase to reverse phases only. Furthermore, the ectoine is small organic molecule present in zwitter-ion without chromophores. Ectoine does not possess an π–π system to enable π–π interactions with suitable chiral selectors. The interactions that ectoine can form include hydrogen bonding, electrostatic interactions, dipole–dipole interaction, and charge transfer interaction. The interactions can be attractive or repulsive, they can be single or multiple, but at least one must be stereochemically dependent.
Separations on brush-type ((R,R)-WhelkO1) and polysaccharide-type columns (Chiralpak AD-RH, Chiralcel OD-RH, Chiralcel OJ-RH and Chiralpak AS-H and Chirallica PST-4) gave no results (methods 1–15. Due to the nature of ectoine, there was no interactions with these stationary phases and the ectoine was eluted without retention. The exchange of solvents did not give the results. On the other hand, almost all tested methods on teicoplanin-based chiral stationary phase, the Chiral-T column (Table 1, methods 16–35) separate the enantiomers to baseline (except methods 17–19 method conditions shown in Table S1). This separation of ectoine enantiomers is the result of the ability to form an interaction between ectoine and the chiral stationary phase. Chromatographic parameters such as retention time of the first and the second eluting enantiomer (tR1, tR2), retention factor of the first and the second eluting enantiomer (k1, k2), separation factor (α), and resolution (Rs) are summarized in Table 1. During the development of enantioseparation method on the Chiral-T column, the following parameters were varied: mobile phase, temperature, and flow rate. Due to the solubility of ectoine, mostly water and MeOH in different ratios were used as the mobile phase. The influence of temperature was measured in range of 30–45 °C. The flow rate was varied from 0.5 to 1.2 mL/min. The addition of formic acid (0.1% and 0.01%), Et3N (0.01%), and ammonium formate (25 mM, pH = 3.0) was examined. The addition of formic acid in MeOH (methods 17 and 18) leads to a drastic shortening of the retention time, a deformation and broadening of the peaks and thus to a loss of resolution. On the other hand, the addition of Et3N (method 19) results in long retention time, which is caused by the formation of an ion-pair between ectoine and Et3N.

Table 1.
Chromatographic parameters for (±)-ectoine enantioseparation on the Chiral-T column for different methods.
Good results were obtained with ammonium formate (methods 33 and 34), but the best results were obtained with water and MeOH alone. Water and MeOH were also used in the scale-up method for the purpose of preparative isolation of (+)- and (−)-ectoine. The effect of flow rate (method 27, 28 and 31) was investigated after the determining the optimum ratio of H2O:MeOH as 1:1 (Figure 1). According to the chromatograms (Figure 1) and the separation and resolution values (Table 1), the method 27 with flow rate at 0.5 mL/min is proved to be the best choice for the enantioseparation of racemic ectoine.
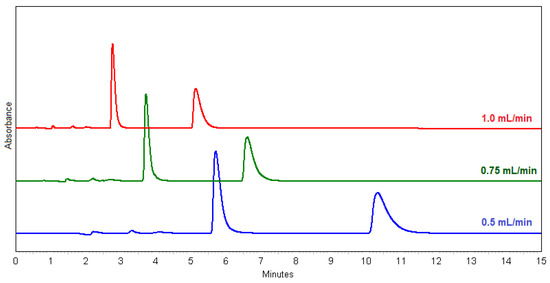
Figure 1.
Overlay chromatograms of (±)-ectoine at 0.5 mL/min (method 27, blue); at 0.75 mL/min (method 28, green); at 1.0 mL/min (method 31, red).
Temperature effect on separation and resolution (methods 29–32) are shown in the chromatograms in Figure 2.
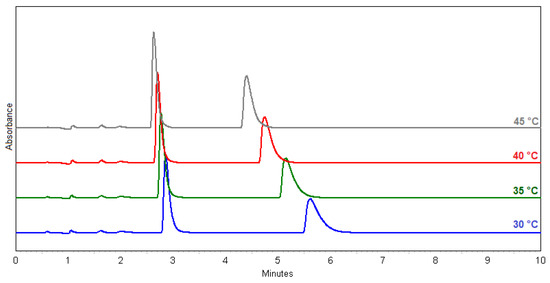
Figure 2.
Overlay chromatograms of (±)-ectoine: method 29 (blue); 30 (green); 31 (red); 32 (grey).
In terms of the resolution (Rs), the best result was obtained with method 23 (MeOH:H2O = 7.5:2.5; Rs = 10.3), but the method 30 (MeOH:H2O = 1:1; Rs = 8.3) was chosen because it uses less MeOH, the flow rate is higher, and the peak shapes are better. This method is optimal for the Chiral-T column in terms of its properties and characteristics. The chosen method is also suitable for a scale-up. Figure 3 shows the identification of (+)- and (−)-ectoine enantiomers compared to standard, natural (+)-ectoine using the selected method 30.
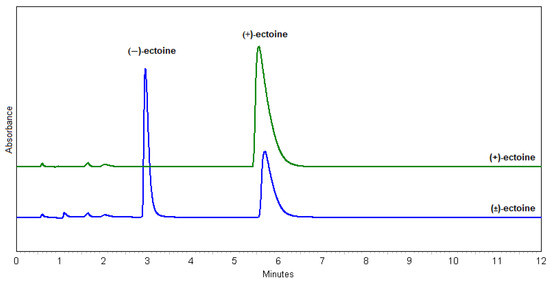
Figure 3.
Chromatograms of (+)-ectoine (green) and (±)-ectoine (blue) using method 30.
2.3. Detection of (+)-Ectoine in Fermentation Broths of Salt-Tolerant Marine Streptomyces
In our program of screening marine bacteria for bioactive compounds, numerous Streptomyces sp. have been isolated from marine sediments and various organisms from the Adriatic Sea. Their taxonomic affiliation to the genus was verified by 16S rRNA gene sequencing (Table S2). Here, we used HPLC for the rapid detection of (+)-ectoine directly in the fermentation broths of 11 salt-tolerant marine Streptomyces isolated from sponge (n = 1), tunicates (n = 3), mussel (n = 1), and sediments (n = 6). As previously reported, under osmotic stress, bacteria mainly retain the produced ectoine in cells and membranes to protect themselves from osmotic stress [33]. The presence of (+)-ectoine in samples was analyzed using Synergy Polar-RP HPLC column at 220 nm in gradient as described in Experimental. As shown in Table 2 the method used in this study successfully detected ectoine in the culture broth of all isolates and reference strain if grown in media supplemented with different concentrations of NaCl. The ectoine concentration in the samples was calculated from calibration curve.
Table 2.
HPLC detection of (+)-ectoine in the fermentation broth of salt-tolerant Streptomyces strains isolated from the Adriatic Sea.
As shown in Table 2 and Figure 4, the HPLC method used in this study successfully detects microgram quantities of ectoine directly in the fermentation medium and can be used for rapid screening of microorganisms with increased potential for the synthesis of this osmolyte. Among the tested strains, BC81 (sample 6) and BC167 (sample 16) showed the highest potential for ectoine production and secretion into the medium, which indicates that these strains have the ability for large-scale fermentation and ectoine production. However, this possibility should be further investigated.
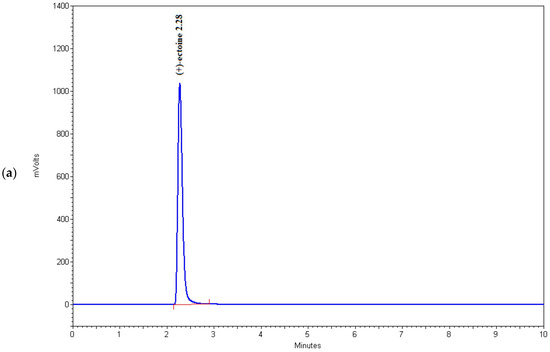

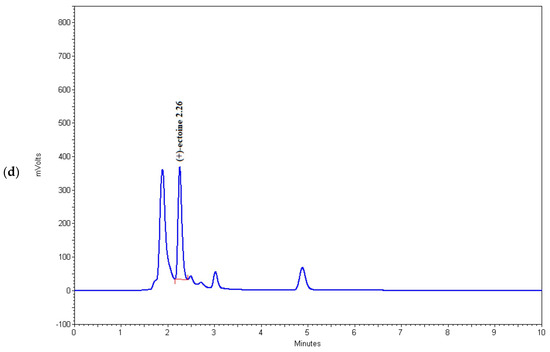
Figure 4.
Chromatograms of (a) (+)-ectoine, (b) sample 6, (c) sample 15, and (d) sample 16.
3. Materials and Methods
3.1. General
Commercial reagents and solvents were purchased as follows: γ-butyrolactone (dihydrofuran-2(3H)-one), (±)-ectoine (4S)-2-methyl-1,4,5,6-tetrahydropirimidine-4-carboxylic acid and N,N-diethylethanamine (Et3N) from Sigma Aldrich (Munich, Germany), bromine (Br2) and copper(II) acetate monohydrate [Cu(CH3COO)2 × H2O] from Fluka (Buchs, Switzerland), phosphorus tribromide (PhBr3) and potassium phthalimide from Merck (Darmstadt, Germany), quinolin-8-ol, sodium carbonate (Na2CO3), sodium hydroxide (NaOH), sodium chloride (NaCl), anhydrous sodium sulfate (Na2SO4), glacial acetic acid (AcOH), and hydrochloric acid (HCl) from Kemika (Zagreb, Croatia), and dimethylformamide (DMF), acetonitrile (ACN), n-butanol (n-BuOH), isopropanol (i-PrOH), methanol (MeOH), acetone, dichloromethane (DCM), and ethanol (EtOH) from Carlo Erba (Van de Reuil, France).
Thin-layer chromatography (TLC) was performed on silica-gel plates TLC silica gel 60 F254 (Merck, Darmstadt, Germany) in different solvent systems: A, EtOH:hexane 9:1; B, DCM:MeOH 25:1; C, i-PrOH:AcOH:H2O 7:3:2. Spots were detected with UV/254 nm, and/or KMnO4 or ninhydrin reagent, followed by heating. The preparative TLC was performed on Analtec Silica gel GF 254, 20,000 µM from Merck (Damstad, Germany). The compounds were characterized using 1H and 13C NMR spectra recorded on Bruker Technologies (Ettlingen and Leipzig, Germany) AV 300, 25 °C, 300 MHz (1H) and 75 MHz (13C) in deuterated chloroform (CDCl3), deuterated water (D2O), deuterated dimethyl sulfoxide (DMSO-d6) and deuterated methanol (CD3OD). The spectra were visualized with MestReNova. Chemical shifts (δ) were expressed as parts per million (ppm). The internal standard in the 1H spectra was tetramethylsilane (TMS), and in the 13C spectra dioxane-d8 or solvent signal. IR spectra were recorded on PerkinElmer UATR Two spectrometer Perkinelmer Inc., Waltman, MA, USA) using the ATR method in the range of 450 to 4000 cm−1, with a resolution of 4 cm−1 and 8 scans per spectrum.
Melting points were determined using an Olympus BX51 polarizing microscope equipped with a Linkam TH600 hot stage and PR600 temperature controller and the Olympus C 5050 ZOOM digital camera (Olympus, Tokyo, Japan). Speed of cycles of heating samples was 5 °C/min.
An Agilent 8860 (Waldbronn, Germany) GC system with FID detector and autosampler was used to monitor preparation of 3-bromodihydrofurane-2-(3H)-one. An Agilent Technologies HP-5 (30 m × 0.35 mm, 0.25 µm) column was used. Heating method: 0→2 min 100 °C; 3 min 110 °C; 4 min 120 °C; 5 min 130 °C; 6 min 140 °C; 7 min 150 °C. The samples were prepared by dissolving 1 mg in 1 mL DCM, and 2 µL of these solutions were injected.
High-resolution mass spectrometry (HRMS) was performed on an Agilent 1290/6550 LC/Q-TOF MS (Agilent Technologies, Santa Clara, CA, USA), using electrospray ionization (ESI) in a positive mode. The samples were dissolved in methanol and injected directly into the MS detector in methanol/water (95/5, v/v) containing 0.1% formic acid under isocratic elution program at a flow rate of 0.2 mL min−1. For the MS analysis, the mass scan ranged from m/z 50 to 400. For MS/MS analysis the product ion m/z 143 was performed with collision energy of 17 V. The fragmentor voltage was 110 V. The capillary voltage and atomizing gas pressure were 3.5 kV and 35 psi, respectively. The flow rate of drying gas was 12 mL min−1, the gas temperature was 200 °C, and the sheath gas temperature 250 °C. Recording and data processing were performed using the Agilent MassHunter Acquisition Software (V.10.1) and Agilent MassHunter Qualitative Software (V.10.0).
3.2. Synthesis of (±)-Ectoine
3.2.1. Synthesis of 3-Bromodihydrofuran-2-(3H)-one (1)
In a two-necked flask, dihydrofuran-2(3H)-one (4.00 mL, 0.052 mol) was added along with a catalytic amount of phosphorus tribromide (100 µL, 1.10 mmol). The reaction mixture was stirred and heated to 100 °C, and then bromine (2.68 mL, 0.052 mol) was added dropwise for 4 h. The reaction mixture was stirred for additional 4 h at 130 °C.
The remaining dihydrofuran-2(3H)-one was distilled in vacuo, and the saturated solution of Na2CO3 (20 mL) was added to the residue and extracted three times with DCM (20 mL). The organic layers were dried over anhydrous Na2SO4, and DCM was evaporated. Compound 1 was obtained as a yellow liquid and used without purification.
Compound 1 C4H5BrO2: Rf(A) = 0.87; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.46–2.55 (m, 1H), 2.74–2.87 (m, 1H), 4.40–4.47 (m, 2H), 4.49–4.57 (m, 1H). 13C NMR (CDCl3, 75 MHz) δ/ppm: 33.8, 37.6, 67.1, 173.1; IR ṽ/cm−1: 2916.75 (C-H stretching), 1770.10 (C=O, stretching), 1478.90 (C-H, twisting), 1200.75 (C-O, stretching), 677.42 (C-Br, stretching).
3.2.2. Synthesis of 2-(2-Dioxotetrahydrofuran-3-il)isoindolin-1,3-dione (2)
Compound 1 (20.00 g, 0.121 mol) and potassium phthalimide (22.42 g, 0.121 mol) were dissolved in DMF (60 mL) in a round bottom flask and stirred at 100 °C for 4 h. The reaction mixture was cooled in ice water, 300 mL of distilled water was added, and then filtered through the Büchner funnel. The precipitate was washed with cold distilled water (2 × 50 mL) and dried at 60 °C. Compound 2 (24.48 g, 87%) was obtained as a beige powder.
Compound 2 C6H10N2O2: Rf(B) = 0.68; mp = 162.7–164.1 °C; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.55–2.66 (m, 1H), 2.73–2.88 (m, 1H), 4.36–4.48 (m, 1H), 4.67 (td, J = 9.2, J = 2.2 Hz, 1H), 5.11 (dd, J = 11.0 Hz, J = 9.6 Hz, 1H), 7.73–7.92 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 26.7, 47.3, 66.0, 123.9, 131.8, 134.6, 167.1, 172.3; IR ṽ/cm−1: 2922.88 (C-H, stretching), 1760.09 (C=O, stretching, γ-lacone), 1706.37 (C=O, stretching, tertiary amide), 1188.32 (C-N, stretching).
3.2.3. Synthesis of 2,4-bis-(1,3-Dioxoisoindoline-2-il)butyric Acid (3)
A suspension of compound 2 (24.42 g, 0.105 mol) and potassium phthalimide (19.54 g, 0.105 mol) in DMF (60 mL) was refluxed for one hour. After completion of the reaction, water (300 mL) and glacial acetic acid (40 mL) were added. The reaction was cooled in an ice bath and the precipitate was filtered off through a Büchner funnel, washed twice with water (50 mL), and dried at 60 °C. Compound 3 (31.48 g, 79%) was obtained as a white powder.
Compound 3 C20H14N2O6: mp = 186.2–188.1 °C; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.61–2.89 (m, 2H), 3.70–3.87 (m, 2H), 4.93 (dd, J = 10.3 Hz, J = 5.4 Hz, 1H), 7.66–7.88 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 27.4, 35.2, 49.6, 123.5, 123.8, 131.9, 132.0, 134.2, 134.4, 167.7, 168.3, 173.7; IR ṽ/cm−1: 3061.20 (O-H, stretching), 1776.71 (C=O, stretching, carboxylic acid), 1711.12 (C=O, stretching, tertiary amide), 1612.36 (N-H, twisting), 1396.70 (O-H, twisting).
3.2.4. Synthesis of 2,4-Diaminobutyric Acid (4)
Compound 3 was suspended in diluted HCl (v/v 1:1.6 mL) and refluxed for 16 h. The reaction mixture was cooled and filtered through a Büchner funnel. The filtrate was evaporated and resuspended in cold absolute ethanol, and the precipitate was filtrated and dried. Compound 4 (8.51 g, 71%) was obtained as an off-white powder.
Compound 4 C4H12Cl2N2O2: Rf(C) = 0.16; mp = 180.0–181.4 °C; 1H NMR (D2O, 300 MHz) δ/ppm: 2.20–2.41 (m, 2H), 3.17–3.34 (m, 2H), 4.13 (dd, J = 9 Hz, J = 6 Hz, 1H); 13C NMR (D2O, 75 MHz) δ/ppm: 27.4, 36.1, 50.8, 171.2; IR ṽ/cm−1: 2876.59 (O-H, streching), 1732.95 (C=O, streching), 1580.19 (N-H, twisting).
3.2.5. Synthesis of 4-Nitrophenol Acetate (5)
4-Nitrophenol (15.00 g, 0.108 mol) was suspended in acetic anhydride (23.60 mL, 0.250 mol) and refluxed for 2 h. The reaction mixture was cooled, neutralized with 30% Na2CO3 and extracted three times with DCM (30 mL). The organic layers were collected, dried over anhydrous Na2SO4, and evaporated. Compound 5 (19.22 g, 98.4%) was obtained as a light yellow powder.
Compound 5 C8H7NO4: Rf(B) = 0.89; mp = 78.6–79.7 °C; 1H NMR (CDCl3, 75 MHz) δ/ppm: 2.35 (s, 3H), 7.27–7.31 (m, 2H), 8.24–8.30 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 21.2, 122.6, 125.3, 145.5, 155.5, 168.5; IR ṽ/cm−1: 3113.20 (C-H, streching), 1757.62 (C=O, streching), 1516.87 (N-O, streching).
3.2.6. Synthesis of N,N-Diacetyl-2,4-diaminobutyric Acid (6)
Method A Compound 4 in water (5 mL) was incubated with different amount of compound 5 and base. The reaction conditions are given in Table 3.
Table 3.
Reaction conditions for modified general method A for the synthesis of compound 6.
Method B Compound 4 was dissolved in water solution of NaOH (3 mol/44.3 mL) and solution of copper(II) acetate monohydrate in water (4.255 g, 0.0222 mol/22.2 mL) was added. The compound 5 (9.63 g, 0.0532 mol) in acetone (89.2 mL) was added dropwise and reaction mixture was stirred at 25 °C for 48 h. The reaction mixture was filtered and the precipitate washed with a mixture of acetone and water (2/1, v/v) and dried. The copper(II) complex of 4-acetamido-2-aminobutyric acid (7.07 g, 83%) was obtained as a light blue colored powder.
IR ṽ/cm−1: 3310.81 (N-H stretching), 3228.18 (O-H, stretching), 2911.63 (C-H, stretching), 1614.16 (C=O, stretching).
The prepared copper(II) complex (7.07 g, 0.0184 mol) was suspended in acetone (36.8 mL) and stirred for 15 min at 25 °C. Distilled water (36.8 mL) was then added and stirred for additional 10 min. Quinolin-8-ol (6.94 g, 0.0478 mol) and distilled water (220.8 mL) were then added to the reaction mixture and stirred for 4 h. The precipitate of copper(II) quinolin-8-ol was filtered, and the mother liquor containing the product was evaporated. Compound 6 (5.84 g, 99%) was obtained as an off-white powder.
Compound 6 C6H12N2O3: Rf(C) = 0.37; 1H NMR (D2O, 300 MHz) δ/ppm: 2.01 (s, 3H), 2.04–2.18 (m, 2H), 3.25–3.44 (m, 2H), 3.72 (dd, J = 7.6 Hz, J = 5.6 Hz, 1H); 13C NMR (D2O, 75 MHz) δ/ppm: 21.9, 30.3, 35.5, 52.5, 174.3, 174.8; IR ṽ/cm−1: 3251.80 (N-H stretching), 2958.90 (O-H stretching), 2891.83 (C-H, stretching), 1632.81 (C=O, twisting), 1412.2 (C-H, twisting).
3.2.7. Synthesis of (±)-Ectoine (7)
Compound 6 (5.84 g, 0.0365 mol) was suspended in n-butanol, and Et3N (10.7 mL, 0.078 mol) was added and the reaction mixture was refluxed for 48 h. After completion of the reaction the solvent was evaporated and the product was purified with cold absolute ethanol and filtered-off. The pure compound 7 (3.26 g, 63%) was obtained as a white powder.
Compound 7 [1] C6H10N2O2: Rf(C) = 0.41; 1H NMR (D2O, 300 MHz) δ/ppm: 2.04–2.24 (m, 2H), 3.02 (s, 3H), 3.25–3.50 (m, 2H), 4.08 (t, J = 5.5 Hz, 1H); 13C NMR (D2O, 75 MHz) δ/ppm: 18.2, 21.5, 37.3, 53.2, 160.5, 176.7; IR ṽ/cm−1: 3191.94 (N-H, stretching), 2851.12 (C-H, stretching), 2663.07 (O-H, stretching), 1655.22 (C=N, stretching); HRMS Calcd. for C6H10N2O2 (H+) 143.08205, Found 143.0817.
3.3. Separation of (±)-Ectoine Enantiomeres
Instruments HPLC analyses were performed using an Agilent 1200 Series system (Agilent Technologies, Waldbronn, Germany)) consisting of a vacuum degasser G1322A, a quaternary pump G1311A, a thermostated column compartment G1330B, an autosampler G1329A and a variable wavelength detector G1311D. Agilent EZChrom Elite software version 3.1.7 (Agilent Technologies, Waldbronn, Germany) was used for data analysis.
(R,R)-WhelkO1 was purchased from Regis Technologies, Inc. (Morton Grove, IL, USA). Polysaccharide-type chiral columns: Chiralpak AD (based on amylose tris(3,5-dimethylphenylcarbamate)), Chiralcel OD-RH (based on cellulose tris(3,5-dimethylphenylcarbamate)), Chiralcel OJ-RH (based on cellulose tris(4-methylbenzoate)), and Chiralpak AS-H (based on amylose tris[(S)-α-methylbenzylcarbamate] were purchased from Daicel Chiral Technologies Co., Ltd. (Tokyo, Japan), and Chirallica PTS-4 (based on amylose tris(3,5-dimethylphenylcarbamate) was prepared at Institute Ruđer Bošković (Zagreb, Croatia). (R,R)-WhelkO1 and polysaccharide-type chiral columns with identical dimensions (250 mm × 4.6 mm I.D., 5 μm particle size). Chiral-T column (150 × 4.6 mm I.D., 2.7 µm) was purchased from Agilent Technologies (Waldbronn, Germany).
The mobile phase systems used are listed in Table S1.
The retention factor (k) was calculated from the equation:
where tR and t0 are the retention times of analyte and the unretained solute, respectively. The enantioseparation factor (α) was calculated using the equation:
where k1 and k2 are the retention factors of the first and second eluted enantiomers, respectively. The resolution (Rs) was calculated using the following equation:
where tR1 and tR2 are the retention times of the first and second eluted enantiomers, respectively, and wb1 and wb2 are the baseline peak widths of the first and second eluted enantiomers, respectively. The dead time value (t0) was determined by solvent peak.
k = (tR − t0)/t0,
α = k2/k1,
Rs = 2 × (tR2 − tR1)/(wb1 + wb2),
3.4. Detection of (+)-Ectoine in Fermentation Broths
Sample collection and bacteria isolation. Marine-derived Streptomyces were isolated from three different invertebrates and sediments collected at several locations in the Adriatic Sea, Croatia (Table S2). Briefly, samples were collected in sterile bags which were kept on ice and processed immediately upon arrival to the laboratory. Marine invertebrates were washed several times in sterile seawater and approximately two grams of sponge tissue and tunicate or mussel digestive system were macerated and incubated in 10 volumes of sterile seawater. Sea sediments (5 g) were transferred into 5 mL of sterile seawater and vigorously shaken for about 10 min. The supernatants were 10-fold serially diluted (10−2, 10−3, 10−4, 10−5) and 100 μL of each dilution was plated on ISP2 plates supplemented with cycloheximide (100 μg/mL) and nalidixic acid (25 μg/mL). Plates were incubated at 12 °C for two to three weeks, and the appearance and growth of colonies was monitored regularly.
Selection and molecular identification of salt-tolerant Streptomyces. Colonies that morphologically resemble streptomycetes (velvety surface, ingrowth into agar medium, production of diffuse pigment) were selected, and a pure culture was obtained from a mixed population using the streak-plate method. The obtained isolates were further tested for salt tolerance by inoculation on ISP2 plates containing 3, 6 or 9% (w/v) NaCl. Plates were incubated at 30 °C for five days and inspected for growth. Strains tolerant to 9% (w/v) NaCl were inoculated in nutrient broth (Biolab Diagnostics Laboratory Inc., Budapest, Hungary) and incubated in a heated shaker (200 rpm) at 30 °C for three days. Cells were harvested by centrifugation (4 °C, 6000× g, 10 min) and total genomic DNA was extracted with DNeasy Blood and Tissue Kit (Qiagen, Venlo, The Netherlands). PCR amplification of the 16S rDNA gene sequence was performed using the universal bacterial primers 27F (5′-GAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) [34]. Amplified PCR products were visualized by agarose gel electrophoresis, purified with QIAquick PCR Purification Kit (Qiagen, The Netherlands) and sequenced using 27F primer at Macrogen Europe, Amsterdam, The Netherlands. The obtained sequences were analyzed using NCBI BLAST tool (http://www.ncbi.nlm.nih.gov/BLAST, accessed on 31 August 2024).
Growth conditions for bacterial isolates and the HPLC analysis of (+)-ectoine. Salt-tolerant marine Streptomyces strains (n = 11) and reference strain Streptomyces coelicolor A3(2) were inoculated in 10 mL of minimal liquid medium [35] supplemented with the indicated NaCl concentrations. The bacteria were first kept at room temperature for 24 h without shaking, then transferred to a shaker (30 °C, 200 rpm) and grown for 48 h. The bacterial cells were pelleted by centrifugation (4 °C, 6000× g, 10 min), and the supernatant (fermentation broth) was stored at −20 °C until analysis.
A quantitative analysis of ectoine in samples was performed on an HPLC Shimadzu LC.2010HTC (Kyoto, Japan) with DAD detector (wavelengths 210 and 220 nm) and a Synergi Polar-RP 80 Å column (150 mm × 4.6 mm i.d., 4 µm, Phenomenex, Torrance, CA, USA). The supernatants were filtered through PTFE filters (0.20 µm, Ø13 mm) without dilution and further processing. A solvent gradient was used for HPLC analysis: 0→7 min 100% H2O; 7→20 min 100%H2O→100%ACN; 20→25 min 100%ACN; 25→25.01 min 100%ACN→100%H2O; 25.01→30 min 100% H2O. The flow rate was 1 mL/min, the column temperature 30 °C, and the run time 30 min. The samples were injected in portions of 20 µL. The ectoine concentration in the samples was calculated using the calibration curve generated from the known (+)-ectoine concentrations. The data for the calibration curve were recorded in two replicates.
4. Conclusions
The chemical synthesis of (±)-ectoine was greatly improved by the use of copper(II)-complexes as selective protection for the α-amino group of 2,4-diaminobutyric acid in the cyclization step. The obtained racemic ectoine was successfully separated by chiral HPLC. For this reason, different chiral columns and methods were investigated. The chiral column Chiral-T with the glycopeptide teicoplanin as chiral stationary phase showed very good results in the base-line separation of racemic ectoine while brush-type and polysaccharide-type chiral stationary phases were not suitable. RP-HPLC was successfully used for the rapid detection of (+)-ectoine directly in the fermentation broth of marine streptomycetes isolated from different habitats. These strains showed a greater tolerance to increased salt concentrations than the reference strain, S. coelicolor. In addition, the HPLC approach enabled the rapid identification of a strain with a high potential for ectoine production, and is, therefore, interesting for further research with regard to biotechnological applications. The prepared (±)-ectoine will serve as a starting material for the method optimization in preparative chiral HPLC/SMB enantioseparation. After separation of the enantiomers, their individual osmolytic/osmoprotective activities will be compared. Further investigations in this direction are currently underway and will be reported in due course.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app14188353/s1. Figure S1.1.–S1.24.: NMR spectra Figure S2.1.–S2.8.: IR spectra; Figure S4.2.1.–S4.2.18.: HPLC chromatograms of (+)-ectoine in fermentation broths of salt-tolerant marine Streptomyces; Figure S5.1.–S5.4.: MS spectra of compound 7 and standard (+)-ectoine; Table S1: Separation of (±)-ectoine enantiomers; Table S2: Streptomyces strains.
Author Contributions
M.Š.—formal synthesis and analysis; M.J.—analysis, methodology and writing original draft; D.V.—investigation and writing original draft; A.Š.—investigation and writing original draft; A.J.—analysis and writing original draft; M.R.—conceptualization, supervision, data analysis, revision, and final approval. All authors have read and agreed to the published version of the manuscript.
Funding
European Regional Development Fund—The Competitiveness and Cohesion Operational Programme KK.01.1.1.02 granted to the Scientific Centre of Excellence for Marine Bioprospecting-BioProCro.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Acknowledgments
We would like to thank Luka Bočkor from the Centre for Applied Bioanthropology, Institute for Anthropological Research, Zagreb for the MS analysis.
Conflicts of Interest
Authors declare no conflicts of interest.
References
- Galinski, E.A.; Pfeiffer, H.-P.; Truper, H.G. 1,4,5,6-Tetrahydro-2-Methyl-4-Pyrimidinecarboxylic Acid. A Novel Cyclic Amino Acid from Halophilic Phototrophic Bacteria of the Genus Ectothiorhodospira. Eur. J. Biochem. 1985, 149, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Pastor, J.M.; Salvador, M.; Argandoña, M.; Bernal, V.; Reina-Bueno, M.; Csonka, L.N.; Iborra, J.L.; Vargas, C.; Nieto, J.J.; Cánovas, M. Ectoines in Cell Stress Protection: Uses and Biotechnological Production. Biotechnol. Adv. 2010, 28, 782–801. [Google Scholar] [CrossRef] [PubMed]
- Widderich, N.; Höppner, A.; Pittelkow, M.; Heider, J.; Smits, S.H.J.; Bremer, E. Biochemical Properties of Ectoine Hydroxylases from Extremophiles and Their Wider Taxonomic Distribution among Microorganisms. PLoS ONE 2014, 9, e93809. [Google Scholar] [CrossRef]
- Reshetnikov, A.S.; Khmelenina, V.N.; Mustakhimov, I.I.; Kalyuzhnaya, M.; Lidstrom, M.; Trotsenko, Y.A. Diversity and Phylogeny of the Ectoine Biosynthesis Genes in Aerobic, Moderately Halophilic Methylotrophic Bacteria. Extremophiles 2011, 15, 653–663. [Google Scholar] [CrossRef]
- Widderich, N.; Czech, L.; Elling, F.J.; Könneke, M.; Stöveken, N.; Pittelkow, M.; Riclea, R.; Dickschat, J.S.; Heider, J.; Bremer, E. Strangers in the Archaeal World: Osmostress-responsive Biosynthesis of Ectoine and Hydroxyectoine by the Marine Thaumarchaeon Nitrosopumilus maritimus. Environ. Microbiol. 2016, 18, 1227–1248. [Google Scholar] [CrossRef]
- Weinisch, L.; Kühner, S.; Roth, R.; Grimm, M.; Roth, T.; Netz, D.J.A.; Pierik, A.J.; Filker, S. Identification of Osmoadaptive Strategies in the Halophile, Heterotrophic Ciliate Schmidingerothrix salinarum. PLoS Biol. 2018, 16, e2003892. [Google Scholar] [CrossRef]
- Moreira, D.; López-García, P. Protist Evolution: Stealing Genes to Gut It Out. Curr. Biol. 2017, 27, R223–R225. [Google Scholar] [CrossRef][Green Version]
- Harding, T.; Roger, A.J.; Simpson, A.G.B. Adaptations to High Salt in a Halophilic Protist: Differential Expression and Gene Acquisitions through Duplications and Gene Transfers. Front. Microbiol. 2017, 8, 944. [Google Scholar] [CrossRef]
- Fenizia, S.; Thume, K.; Wirgenings, M.; Pohnert, G. Ectoine from Bacterial and Algal Origin Is a Compatible Solute in Microalgae. Mar. Drugs 2020, 18, 42. [Google Scholar] [CrossRef]
- Schwibbert, K.; Marin-Sanguino, A.; Bagyan, I.; Heidrich, G.; Lentzen, G.; Seitz, H.; Rampp, M.; Schuster, S.C.; Klenk, H.; Pfeiffer, F.; et al. A Blueprint of Ectoine Metabolism from the Genome of the Industrial Producer Halomonas elongata DSM 2581T. Environ. Microbiol. 2011, 13, 1973–1994. [Google Scholar] [CrossRef]
- Richter, A.A.; Kobus, S.; Czech, L.; Hoeppner, A.; Zarzycki, J.; Erb, T.J.; Lauterbach, L.; Dickschat, J.S.; Bremer, E.; Smits, S.H.J. The Architecture of the Diaminobutyrate Acetyltransferase Active Site Provides Mechanistic Insight into the Biosynthesis of the Chemical Chaperone Ectoine. J. Biol. Chem. 2020, 295, 2822–2838. [Google Scholar] [CrossRef] [PubMed]
- Czech, L.; Hermann, L.; Stöveken, N.; Richter, A.; Höppner, A.; Smits, S.; Heider, J.; Bremer, E. Role of the Extremolytes Ectoine and Hydroxyectoine as Stress Protectants and Nutrients: Genetics, Phylogenomics, Biochemistry, and Structural Analysis. Genes 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Kadam, P.; Khisti, M.; Ravishankar, V.; Barvkar, V.; Dhotre, D.; Sharma, A.; Shouche, Y.; Zinjarde, S. Recent Advances in Production and Applications of Ectoine, a Compatible Solute of Industrial Relevance. Bioresour. Technol. 2024, 393, 130016. [Google Scholar] [CrossRef]
- Bilstein, A.; Heinrich, A.; Rybachuk, A.; Mösges, R. Ectoine in the Treatment of Irritations and Inflammations of the Eye Surface. BioMed Res. Int. 2021, 2021, 8885032. [Google Scholar] [CrossRef]
- Kunte, H.; Lentzen, G.; Galinski, E. Industrial Production of the Cell Protectant Ectoine: Protection Mechanisms, Processes, and Products. Curr. Biol. 2014, 3, 10–25. [Google Scholar] [CrossRef]
- Sauer, T.; Galinski, E.A. Bacterial Milking: A Novel Bioprocess for Production of Compatible Solutes. Biotechnol. Bioeng. 1998, 57, 306–313. [Google Scholar] [CrossRef]
- Cantera, S.; Di Benedetto, F.; Tumulero, B.F.; Sousa, D.Z. Microbial Conversion of Carbon Dioxide and Hydrogen into the Fine Chemicals Hydroxyectoine and Ectoine. Bioresour. Technol. 2023, 374, 128753. [Google Scholar] [CrossRef]
- Czech, L.; Höppner, A.; Kobus, S.; Seubert, A.; Riclea, R.; Dickschat, J.S.; Heider, J.; Smits, S.H.J.; Bremer, E. Illuminating the Catalytic Core of Ectoine Synthase through Structural and Biochemical Analysis. Sci. Rep. 2019, 9, 364. [Google Scholar] [CrossRef]
- Himdi-Kabbab, S.; Lavrador, K.; Bazureau, J.P.; Hamelin, J. Synthesis of 1, 4, 5, 6-Tetrahydro 2-Methyl 4-Pyrimidine Carboxylic Acid: Osmoprotector Amino Acid. Synth. Commun. 1995, 25, 2223–2227. [Google Scholar] [CrossRef]
- Motitschke, L.; Driller, H.; Galinski, E.A. Ectoin and Ectoin Derivatives as Moisturizers in Cosmetics. U.S. Patent US6060071, 9 May 2000. [Google Scholar]
- Talbot, G.; Gaudry, R.; Berlinguet, L. Synthesis of 4-Aminobutyric Acid and 2,4-Diaminobutyric Acid from Butyrolactone. Can. J. Chem. 1958, 36, 593–596. [Google Scholar] [CrossRef]
- Zaoral, M. Amino Acids and Peptides. XXIV. Preparation of DL-α,γ-Diaminobutyric Acid and Its Derivatives from γ-Butyrolactone. Collect. Czech. Chem. Commun. 1959, 24, 1314–1319. [Google Scholar] [CrossRef]
- Livak, J.E.; Britton, E.C.; VanderWeele, J.C.; Murray, M.F. Synthesis of D/L-Methionine. J. Am. Chem. Soc. 1945, 67, 2218–2220. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Benoiton, L. On the Selectivity of Acylation of Unprotected Diamino Acids. Can. J. Chem. 1968, 46, 1047–1051. [Google Scholar] [CrossRef]
- Wiejak, S.; Masiukiewicz, E.; Rzeszotarska, B. Improved Scalable Syntheses of Mono- and Bis-Urethane Derivatives of Ornithine. Chem. Pharm. Bull. 2001, 49, 1189–1191. [Google Scholar] [CrossRef]
- Kurtz, A.C. Use of Copper (II) Ion in Masking α-Amino Group of Amino Acids. J. Biol. Chem. 1949, 180, 1253–1267. [Google Scholar] [CrossRef]
- Pícha, J.; Buděšínský, M.; Macháčková, K.; Collinsová, M.; Jiráček, J. Optimized Syntheses of Fmoc Azido Amino Acids for the Preparation of Azidopeptides. J. Pep. Sci. 2017, 23, 202–214. [Google Scholar] [CrossRef]
- Brichac, J.; Honzatko, A.; Picklo, M.J. Direct and Indirect High-Performance Liquid Chromatography Enantioseparation of Trans-4-Hydroxy-2-Nonenoic Acid. J. Chromatogr. A 2007, 1149, 305–311. [Google Scholar] [CrossRef][Green Version]
- Yu, L.; Wang, S.; Zeng, S. Chiral Separations: Methods and Protocols, 2nd ed.; Scriba, G.K.E., Ed.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2013; Volume 970, ISBN 978-1-62703-262-9. [Google Scholar]
- Okamoto, Y.; Ikai, T. Chiral HPLC for Efficient Resolution of Enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef]
- Francote, E. Chirality in Drug Research; Francotte, E., Lindner, W., Eds.; Methods and Principles in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2006; ISBN 978-3-527-31076-0. [Google Scholar]
- Hyun, M.H. Liquid Chromatographic Ligand-Exchange Chiral Stationary Phases Based on Amino Alcohols. J. Chromatogr. A 2018, 1557, 28–42. [Google Scholar] [CrossRef]
- Ng, H.S.; Wan, P.-K.; Kondo, A.; Chang, J.-S.; Lan, J.C.-W. Production and Recovery of Ectoine: A Review of Current State and Future Prospects. Processes 2023, 11, 339. [Google Scholar] [CrossRef]
- Duran, R.; Bielen, A.; Paradžik, T.; Gassie, C.; Pustijanac, E.; Cagnon, C.; Hamer, B.; Vujaklija, D. Exploring Actinobacteria Assemblages in Coastal Marine Sediments under Contrasted Human Influences in the West Istria Sea, Croatia. Environ. Sci. Pollut. Res. 2015, 22, 15215–15229. [Google Scholar] [CrossRef]
- Kieser, T. Practical Streptomyces Genetics; Kieser, T., Ed.; Innes: Norwich, UK, 2000; ISBN 978-0-7084-0623-6. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).