



Virtual Screening and Molecular Dynamics Simulation to Identify Inhibitors of the m6A-RNA Reader Protein YTHDC1



Abstract
:1. Introduction
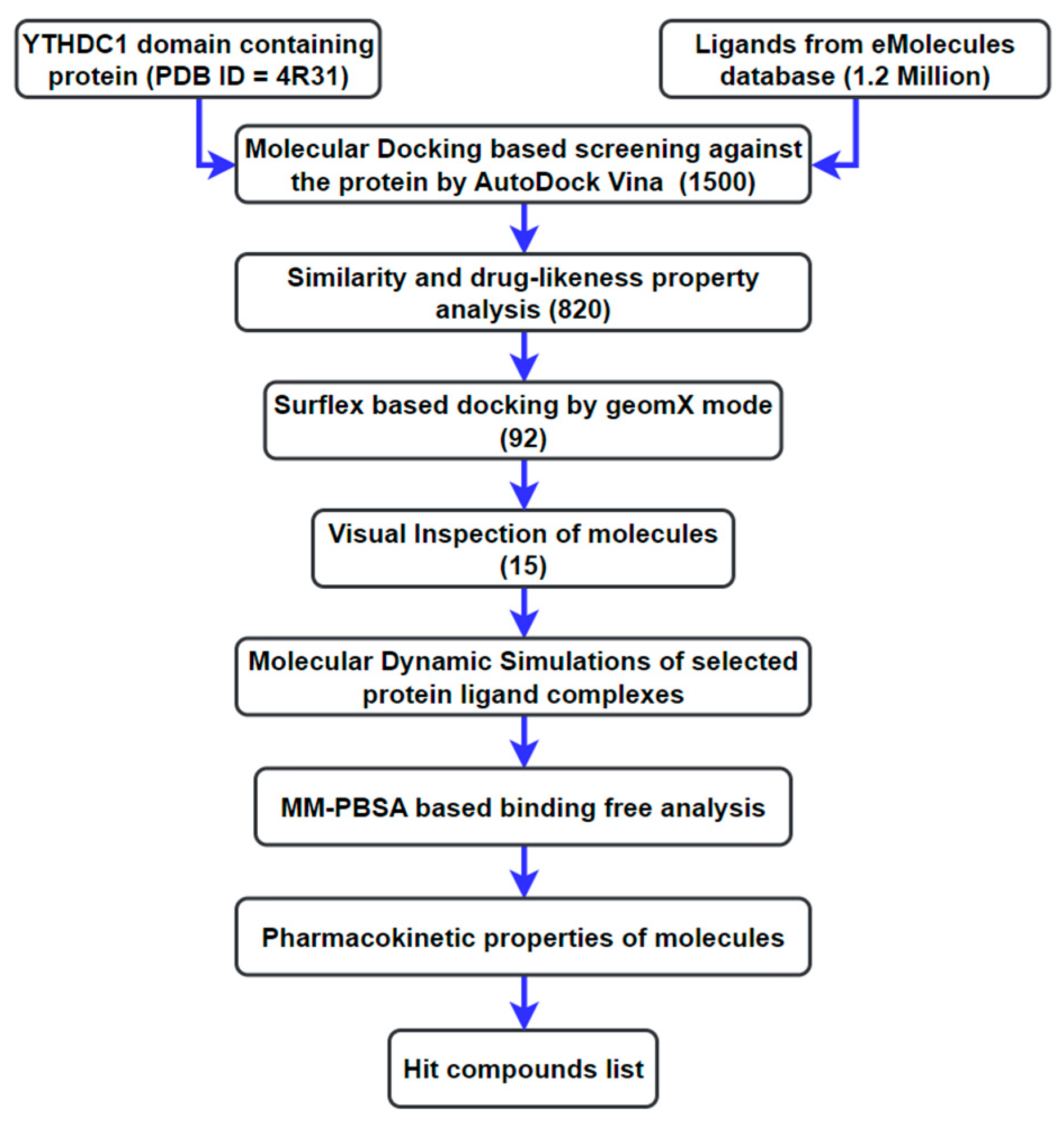
2. Materials and Methods
2.1. Preparation of Ligand Library
2.2. Preparation of the Receptor and Docking-Based Screening
2.3. MD Simulations
2.3.1. Screening
2.3.2. Calibration
2.4. Energy Calculation of the Protein–Ligand Interactions Based on MM/PBSA
2.5. ADMET Profiling
3. Results and Discussion
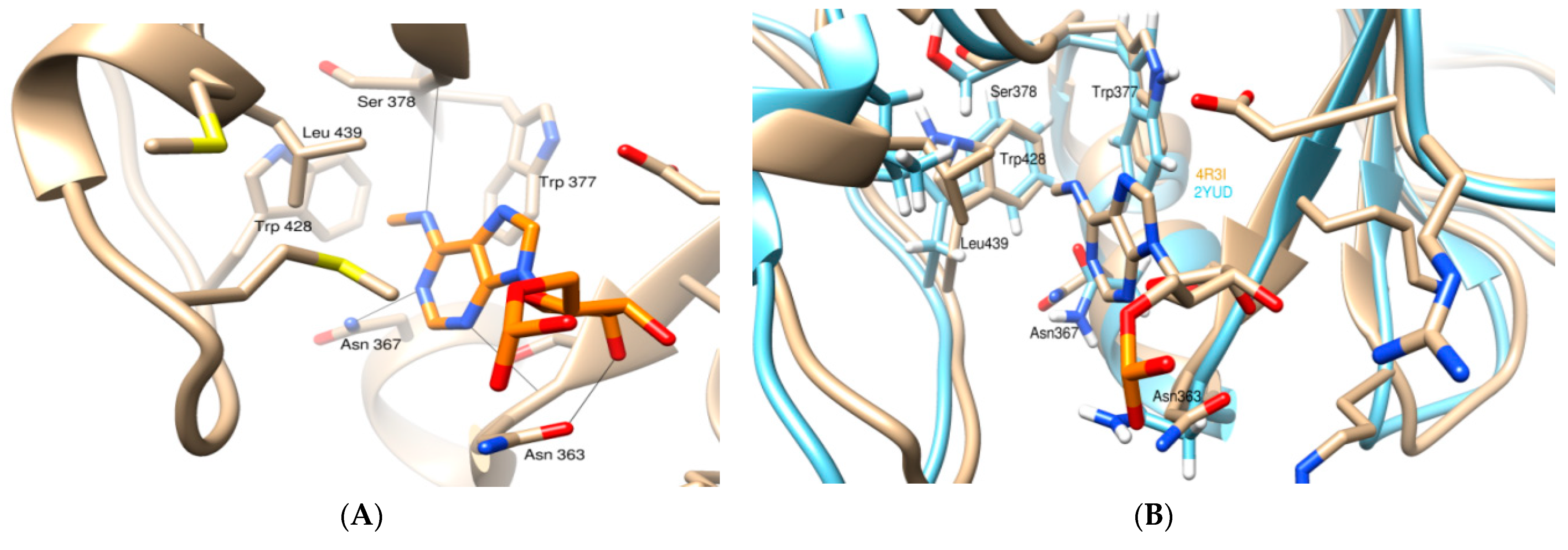
3.1. Molecular Docking Analysis
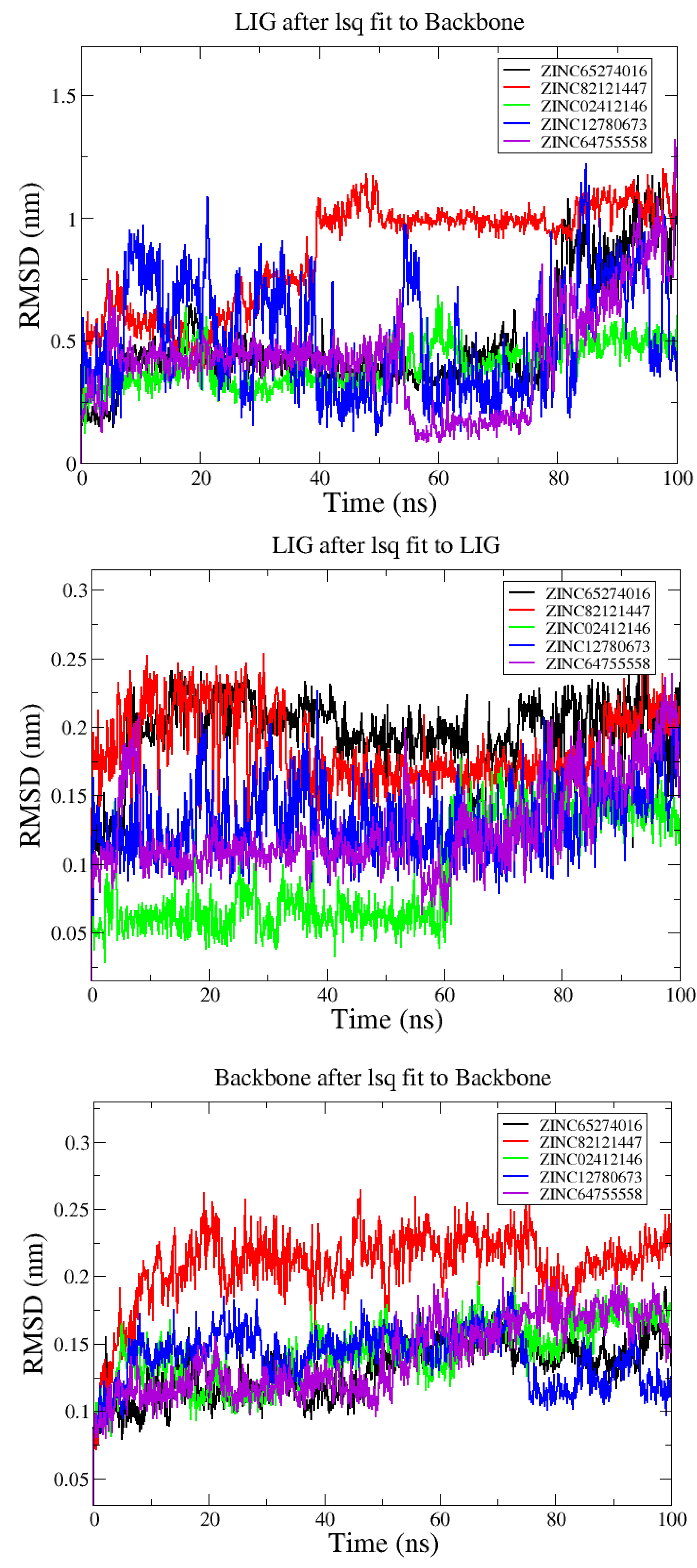
3.2. Analysis from MD Simulations
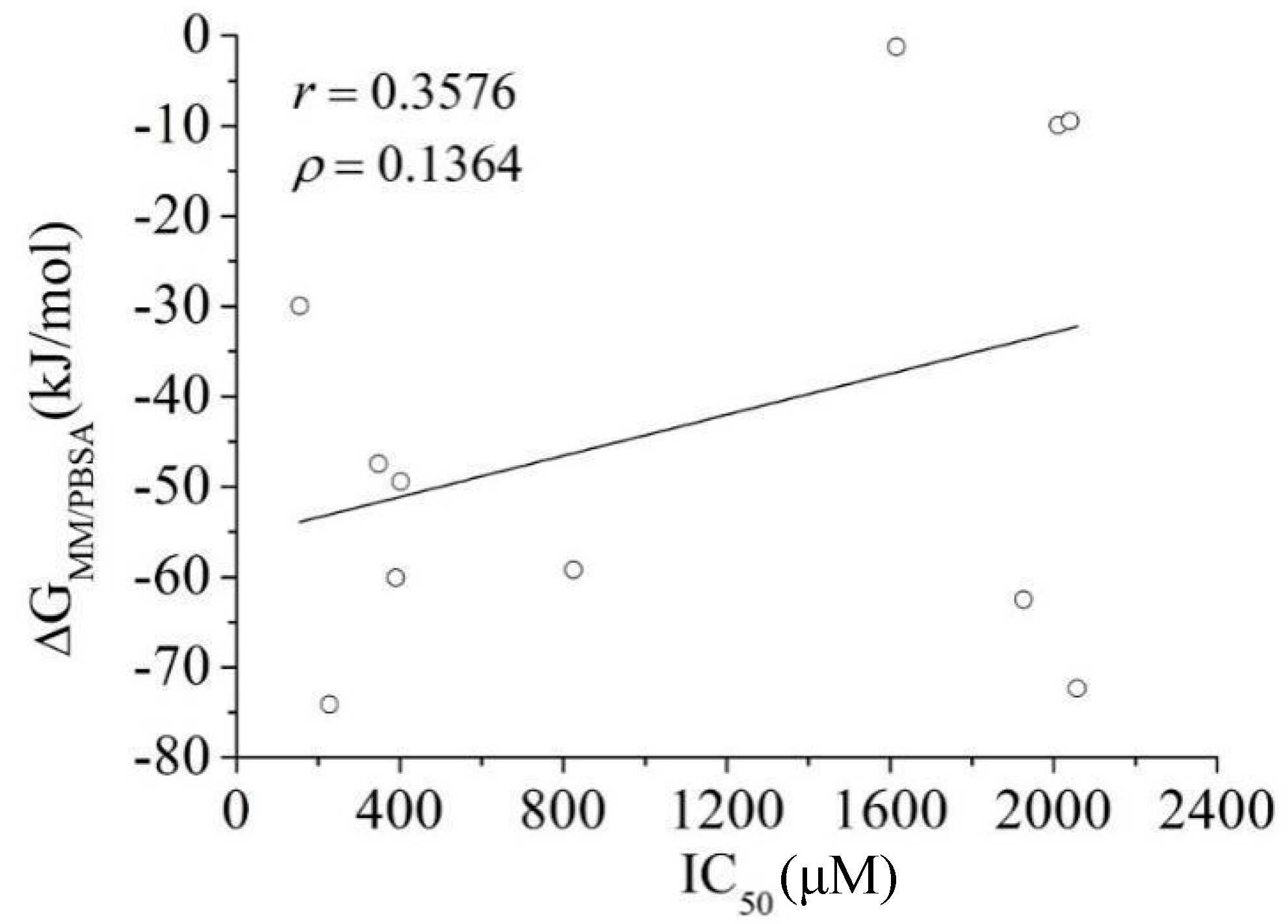
3.3. MM/PBSA Binding Free Energy Calculation: Calibration
3.4. MM/PBSA Binding Free Energy Calculation: Screening
3.5. ADMET Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, L.J.; Deng, W.Q.; Fan, S.R.; Chen, M.F.; Qi, M.; Lyu, W.Y.; Qi, Q.; Tiwari, A.K.; Chen, J.-X.; Zhang, D.-M.; et al. m6A modification: Recent advances, anticancer targeted drug discovery and beyond. Mol. Cancer 2022, 21, 52. [Google Scholar] [CrossRef] [PubMed]
- He, C. Grand challenge commentary: RNA Epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA Modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Liu, N.; Pan, T. N6-methyladenosine-encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of rna methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m(6)A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Widagdo, J.; Anggono, V. The m6A-epitranscriptomic signature in neurobiology: From neurodevelopment to brain plasticity. J. Neurochem. 2018, 147, 137–152. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.W.; Zhang, X.Q.; Jaffrey, S.R.; Qian, S.B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Do, X.Y.; Chen, C.Y.; Chen, C.; Liu, C.; Xu, M.M.; Zhao, S.Q.; Shen, S.; Gao, Y.W.; Han, D.L.; et al. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 2020, 367, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Bogerd, H.P.; Kennedy, E.W.; Emery, A.; Swanstrom, R.; Cullen, B.R. Epitranscriptomic addition of m6A regulates HIV-1 RNA stability and alternative splicing. Genes Dev. 2021, 35, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.; et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Wang, S.; Chai, P.; Jia, R.; Jia, R. Novel insights on m6A RNA methylation in tumorigenesis: A double-edged sword. Mol. Cancer 2018, 17, 101. [Google Scholar] [CrossRef]
- Liu, J.Z.; Yue, N.; Han, D.L.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.F.; Yu, M.; Lu, Z.K.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine, methylation. Nat. Chem. Biol. 2014, 10, 293–295. [Google Scholar] [CrossRef]
- Wang, T.; Kong, S.; Mei, T.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 2017, 6, e31311. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoir, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Yan, H.; Zheng, L.; Cui, X.; Zheng, X.; Li, R. Roles and mechanisms of the m6A reader YTHDC1 in biological processes and diseases. Cell Death Discov. 2022, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.C.; Shi, Y.F.; Shen, H.F.; Xie, W.X. m6A-binding proteins: The emerging crucial performers in epigenetics. J. Hematol. Oncol. 2020, 13, 35. [Google Scholar] [CrossRef]
- Xu, C.; Wang, X.; Liu, K.; Roundtree, I.A.; Tempel, W.; Li, Y.; Lu, Z.; He, C.; Min, J. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 2014, 10, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, W.; Shen, F.; Yang, X.; Liu, H.; Dai, S.; Sun, X.; Huang, J.; Guo, Q. YTH Domain Proteins: A Family of m6A Readers in Cancer progression. Front. Oncol. 2021, 11, 629560. [Google Scholar] [CrossRef]
- Rong, D.; Wu, F.; Lu, C.; Sun, G.; Shi, X.; Chen, X.; Dai, Y.; Zhong, W.; Hao, X.; Zhou, J.; et al. m6A modification of circHPS5 and hepatocellular carcinoma progression through HMGA2 expression. Mol. Ther. Nucleic Acids 2021, 26, 637–648. [Google Scholar] [CrossRef]
- Rao, X.; Lai, L.; Li, X.; Wang, L.; Li, A.; Yang, Q. N6-methyladenosine modification of circular RNA circ-ARL3 facilitates Hepatitis B virus-associated hepatocellular carcinoma via sponging miR-1305. IUBMB Life 2021, 73, 408–417. [Google Scholar] [CrossRef]
- Zhou, K.; Sun, Y.; Dong, D.; Zhao, C.; Wang, W. EMP3 negatively modulates breast cancer cell DNA replication, DNA damage repair, and stem-like properties. Cell Death Dis. 2021, 12, 844. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, T.; She, Y.; Wu, K.; Gu, S.; Li, L.; Dong, C.; Chen, C.; Zhou, Y. N6-methyladenosine-modified circIGF2BP3 inhibits CD8 T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol. Cancer 2021, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xie, W.; Pickering, B.F.; Chu, K.L.; Savino, A.M.; Yang, X.; Luo, H.; Nguyen, D.T.; Mo, S.; Barin, E.; et al. N6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 2021, 39, 958–972. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Santos, R.N.D.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Singh, N.; Ying, S.-Q.; Li, W. Identification of novel inhibitors of type-I mycobacterium tuberculosis fatty acid synthase using docking-based virtual screening and molecular dynamics simulation. Appl. Sci. 2021, 11, 6977. [Google Scholar] [CrossRef]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In Silico Models for Drug Discovery. Methods in Molecular Biology (Methods and Protocols); Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 993, pp. 95–113. [Google Scholar]
- Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. Rigorous free energy simulations in virtual screening. J. Chem. Inf. Model 2020, 60, 4153–4169. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM/PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Moroz-Omori, E.V.; Huang, D.; Bedi, R.K.; Cheriyamkunnel, S.J.; Bochenkova, E.; Dolbois, A.; Rzeczkowski, M.D.; Li, Y.; Wiedmer, L.; Caflisch, A. METTL3 inhibitors for Epitranscriptomic modulation of cellular processes. ChemMedChem 2021, 16, 3035–3043. [Google Scholar] [CrossRef]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Dolbois, A.; Bedi, R.K.; Bochenkova, E.; Müller, A.; Moroz-Omori, E.V.; Huang, D.; Caflisch, A. 1,4,9-Triazaspiro[5.5]Undecan-2-One derivatives as potent and selective METTL3 inhibitors. J. Med. Chem. 2021, 64, 12738–12760. [Google Scholar] [CrossRef] [PubMed]
- Bedi, R.K.; Huang, D.; Wiedmer, L.; Li, Y.; Dolbois, A.; Wojdyla, J.A.; Sharpe, M.E.; Caflisch, A.; Sledz, P. Selectively disrupting M6A-dependent protein–RNA interactions with fragments. ACS Chem. Biol. 2020, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bedi, R.K.; Nai, F.; von Roten, V.; Dolbois, A.; Zálešák, F.; Nachawati, R.; Huang, D.; Caflisch, A. Structure-based design of ligands of the M6A-RNA reader YTHDC1. Eur. J. Med. Chem. Rep. 2022, 5, 100057. [Google Scholar] [CrossRef]
- Li, Y.; Bedi, R.K.; Wiedmer, L.; Sun, X.; Huang, D.; Caflisch, A. Atomistic and thermodynamic analysis of N6-Methyladenosine (M6A) recognition by the reader Domain of YTHDC1. J. Chem. Theory Comput. 2021, 17, 1240–1249. [Google Scholar] [CrossRef]
- Zálešák, F.; Nai, F.; Herok, M.; Bochenkova, E.; Bedi, R.K.; Li, Y.; Errani, F.; Caflisch, A. Structure-based design of a potent and selective YTHDC1 ligand. J. Med. Chem. 2024, 67, 9516–9535. [Google Scholar] [CrossRef]
- Centore, R.C.; Charles, M.; Arora, M.; Chen, Y.; Watson, M.; Sawant, J.; Kelley, C.; Czekalska, M.; Rebmann, M.; Ghandi, M.; et al. Abstract 3231: Pharmacological inhibition of the m6A RNA reader, YTHDC1, as a novel approach to targeting biomolecular condensates in cancer. Cancer Res. 2024, 84, 3231. [Google Scholar] [CrossRef]
- Selberg, S.; Yu, L.Y.; Bondarenko, O.; Kankurim, E.; Seli, N.; Kovaleva, V.; Herodes, K.; Saarma, M.; Karelson, M. Small-molecule inhibitors of the RNA M6A demethylases FTO potently support the survival of dopamine neurons. Int. J. Mol. Sci. 2021, 22, 4537. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.E.; Hess, S.; Meyer, K.D.; Verhagen, L.A.W.; Koch, L.; Brönneke, H.S.; Dietrich, M.O.; Jordan, S.D.; Saletore, Y.; Elemento, O.; et al. The Fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci. 2013, 16, 1042–1048. [Google Scholar] [CrossRef]
- Zheng, G.; Cox, T.; Tribbey, L.; Wang, G.Z.; Iacoban, P.; Booher, M.E.; Gabriel, G.J.; Zhou, L.; Bae, N.; Rowles, J.; et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem. Neurosci. 2014, 5, 658–665. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.-F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of M6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [PubMed]
- Nai, F.; Nachawati, R.; Zálešák, F.; Wang, X.; Li, Y.; Caflisch, A. Fragment ligands of the m6A-RNA reader YTHDF2. ACS Med. Chem. Lett. 2022, 13, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- BIOVIA, DS, version 22.1; Dassault Systèmes: San Diego, CA, USA, 2022.
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM general force field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, M.; Alexov, E. DelPhiPKa web server: Predicting pKa of proteins, RNAs and DNAs. Bioinformatics 2015, 32, 614–615. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E Water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. App. Phy. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Halgren, H.T. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. 2013, 117, 8408–8421. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose-Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Yunta, M.J.R. Docking and ligand binding affinity: Uses and pitfalls. Am. J. Model. Opt. 2016, 4, 74–114. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consertium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM/PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Wright, D.W.; Hall, B.A.; Kenway, O.A.; Jha, S.; Coveney, P.V. Computing clinically relevant binding free energies of HIV-1 protease inhibitors. J. Chem. Theory Comput. 2014, 10, 1228–1241. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative assessment of scoring functions: The CASF-2016 update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Absolute binding free energy calculations for highly flexible protein MDM2 and its inhibitors. Int. J. Mol. Sci. 2020, 21, 4765. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Molecule | Hydrogen Bonds | Autodock Score (kcal/mol) | GeomX Score (kcal/mol) | Hydrogen Bonding Residues |
|---|---|---|---|---|---|
| 1 | ZINC65274016 | 4 | −10.77 | −10.94 | Asn367, Ser378, Asn363, Arg475 |
| 2 | ZINC82121447 | 2 | −9.13 | −10.20 | Ser378, Asp476 |
| 3 | ZINC02412146 | 2 | −8.95 | −10.27 | Asn363, Arg475 |
| 4 | ZINC12780673 | 3 | −8.90 | −9.87 | Asn367, Ser378, Arg475 |
| 5 | ZINC64755558 | 4 | −8.90 | −10.16 | Asn367, Ser378, Asn363, Asp476 |
| 6 | ZINC00807117 | 4 | −8.73 | −9.84 | Asn367, Ser378, Asn363, Arg475 |
| 7 | ZINC91526763 | 4 | −8.44 | −10.21 | Asn367, Asn363, Ser378, Asp476 |
| 8 | ZINC01330611 | 2 | −8.43 | −9.94 | Asn367, Asn363 |
| 9 | ZINC02170552 | 2 | −8.25 | −9.38 | Asn367, Asn363 |
| 10 | ZINC06764561 | 3 | −8.23 | −9.77 | Asn367, Asn363, Ser378 |
| 11 | ZINC10763862 | 3 | −8.10 | −10.12 | Asn367, Ser378, Lys361 |
| 12 | ZINC67726926 | 4 | −8.07 | −9.6 | Asn367, Asn363, Ser378, Asp476 |
| 13 | ZINC65384150 | 4 | −7.89 | −10.29 | Asn367, Asn363, Ser378, Asp476 |
| 14 | ZINC58341263 | 3 | −7.87 | −11.03 | Asn367, Ser378, Arg475 |
| 15 | ZINC67981980 | 3 | −7.56 | −10.24 | Asn363, Asn367, Ser378 |
| PDB ID | ∆EvdW | ∆Eelec | ∆Gpb | ∆Gnp | ∆Gbind | IC50 |
|---|---|---|---|---|---|---|
| 6T10 | −100.6 (0.6) | −56.3 (1.9) | 121.5 (2.0) | −12.1 (0.1) | −47.4 (1.5) | 348 |
| 6T11 | −103.6 (0.7) | −84.4 (1.8) | 149.7 (1.6) | −11.1 (0.0) | −49.4 (1.3) | 402 |
| 6T12 | −116.6 (0.6) | −67.6 (1.6) | 137.7 (1.9) | −12.6 (0.1) | −59.2 (1.3) | 825 |
| 6T01 | −121.9 (0.7) | −62.0 (1.7) | 123.6 (1.3) | −12.2 (0.0) | −72.4 (1.3) | 2057 |
| 6T06 | 0.0 (0.0) | −0.1 (0.0) | −9.8 (4.8) | 0.0 (0.2) | −9.9 (5.0) | 2010 |
| 6T0A | −22.0 (2.5) | −12.9 (1.9) | 28.2 (3.6) | −2.8 (0.3) | −9.5 (2.8) | 2040 |
| 6T0C | −101.3 (0.6) | −49.9 (1.9) | 132.1 (1.7) | −10.8 (0.0) | −30.0 (1.4) | 155 |
| 6T0D | −109.7 (0.8) | −72.0 (1.4) | 133.4 (1.4) | −11.7 (0.1) | −60.1 (1.4) | 391 |
| 6SZ7 | −1.3 (0.5) | −0.8 (0.5) | 1.1 (2.6) | −0.3 (0.1) | −1.2 (2.5) | 1615 |
| 6SZT | −134.4 (0.7) | −56.5 (2.6) | 141.1 (2.4) | −12.8 (0.0) | −62.5 (1.4) | 1926 |
| 6SZX | −149.4 (1.0) | −56.8 (1.8) | 146.5 (1.9) | −14.3 (0.1) | −74.1 (1.2) | 228 |
| No. | Compound ID | Binding Energy (kJ/mol) |
|---|---|---|
| 1 | ZINC82121447 | −114.03 ± 10.8 |
| 2 | ZINC02170552 | −83.26 ± 20.4 |
| 3 | ZINC65274016 | −75.24 ± 8.4 |
| 4 | ZINC10763862 | −73.36 ± 8.4 |
| 5 | ZINC02412146 | −67.71 ± 9.4 |
| 6 | ZINC12780673 | −67.54 ± 3.4 |
| 7 | ZINC65384150 | −66.7 ± 20.2 |
| 8 | ZINC00807117 | −66.6 ± 9.8 |
| 9 | ZINC01330611 | −64.4 ± 10.1 |
| 10 | ZINC06764561 | −63.8 ± 17.6 |
| 11 | ZINC64755558 | −63.7 ± 31.1 |
| 12 | ZINC67726926 | −63.07 ± 28.8 |
| 13 | ZINC67981980 | −38.6 ± 8.9 |
| 14 | ZINC58341263 | −18.1 ± 26.5 |
| 15 | ZINC91526763 | −9.99 ± 72.6 |
| Compound | ∆EvdW | ∆Eelec | ∆Gpb | ∆Gnp | ∆Gbind |
|---|---|---|---|---|---|
| ZINC82121447 | −92.04 ± 6.8 6 | −872.1 ± 1.3 | 863.04 ± 8.2 | −12.9 ± 0.2 | −114.03 ± 10.8 |
| ZINC02170552 | −132.7 ± 3.1 | 218.7 ± 11.1 | −153.5 ± 16.9 | −15.71 ± 0.1 | −83.2 ± 20.4 |
| ZINC65274016 | −165.36 ± 3.9 | −59.02 ± 5.2 | 165.36 ± 0.62 | −16.17 ± 0.08 | −75.24 ± 8.4 |
| ZINC10763862 | −168.37 ± 6.5 | −121.9 ± 5.8 | 233.28 ± 4.7 | −16.26 ± 0.3 | −73.35 ± 8.4 |
| ZINC02412146 | −158.42 ± 6.1 | −50.07 ± 3.4 | 157.87 ± 6.3 | −17.09 ± 0.2 | −67.72 ± 9.4 |
| 6MZ | −137.79 ± 14.1 | −72.43 ± 35.3 | 190.8 ± 43.8 | 15.31 ± 1.0 | −34.75 ± 16.4 |
| Ligand | LogP | Surface Area | Absorption | Distribution | Metabolism | Excretion | Toxicity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Water Solubility | Caco-2 Permeability | Intestinal Absorption | Fraction Unbound | BBB Permeability | CNS Permeability | CYP2D6 Substrate | CYP2D6 Inhibitor | Total Clearance | AMES Toxicity | Hepatoxicity | Skin Sensitization | |||
| 01 | 1.52 | 155.18 | −3.075 | 0.363 | 65.09 | 0.186 | −1.569 | −3.284 | No | No | 0.442 | No | Yes | No |
| 02 | 1.94 | 130.964 | −3.32 | 1.312 | 98.785 | 0.163 | 0.369 | −3.001 | No | No | 0.507 | Yes | Yes | No |
| 03 | 4.16 | 167.32 | −4.909 | 0.687 | 90.42 | 0.07 | −0.283 | −2.289 | No | No | 1.106 | No | Yes | No |
| 04 | 3.27 | 154.98 | −4.456 | 1.255 | 90.19 | 0 | −0.502 | −2.584 | No | No | 0.257 | Yes | Yes | No |
| 05 | 1.89 | 143.24 | −3.186 | 0.182 | 73.46 | 0.319 | −0.936 | −3.442 | No | No | −0.296 | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Singh, N.; Wang, X.; Li, W. Virtual Screening and Molecular Dynamics Simulation to Identify Inhibitors of the m6A-RNA Reader Protein YTHDC1. Appl. Sci. 2024, 14, 8391. https://doi.org/10.3390/app14188391
Aslam M, Singh N, Wang X, Li W. Virtual Screening and Molecular Dynamics Simulation to Identify Inhibitors of the m6A-RNA Reader Protein YTHDC1. Applied Sciences. 2024; 14(18):8391. https://doi.org/10.3390/app14188391
Chicago/Turabian StyleAslam, Memoona, Nidhi Singh, Xiaowen Wang, and Wenjin Li. 2024. "Virtual Screening and Molecular Dynamics Simulation to Identify Inhibitors of the m6A-RNA Reader Protein YTHDC1" Applied Sciences 14, no. 18: 8391. https://doi.org/10.3390/app14188391