

Lymphatic Vessel Remodeling in the Hearts of Ang II-Treated Obese db/db Mice as an Integral Component of Cardiac Remodeling
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Assessment of Obesity and Other Metabolic Anomalies
2.3. Tissue Collection
2.4. Confocal Microscopic Evaluation of Cardiac Lymphatics
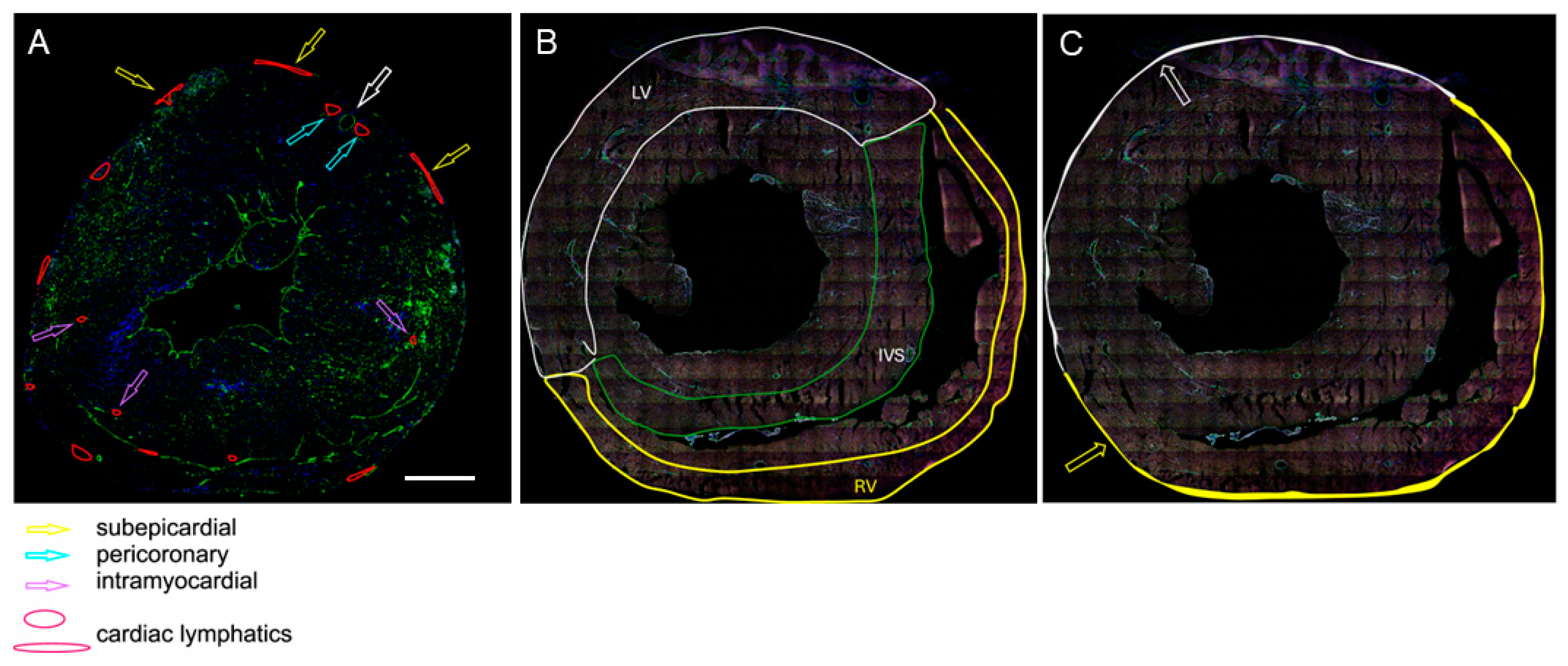
- As the ratio of the number of LyVs to the number of cardiomyocytes within the same area of tissue. The assessment was carried out at three different locations: the outer half of the left ventricular wall, the outer half of the right ventricular wall, and the half of the interventricular septum (IVS) facing the right ventricle (RV). These areas were selected because cardiac LyV in mice are only located in certain regions of the heart, as shown in the schematic drawings in Figure 1A,B.
- As the density of the subepicardial and intramyocardial LyVs, which was calculated as the number of LyVs per area of tissue (expressed in mm2) and the density of pericoronary LyVs, which was calculated as the ratio of the number of pericoronary LyVs to the number of all cross-sectioned coronary arteries in a given tissue section (a coronary artery is marked with the white-edged arrow in Figure 1A). The terms subepicardial, pericoronary, and intramyocardial were understood as follows: subepicardial LyVs (enclosed with white and yellow lines and marked with white- and yellow-edged arrows in Figure 1A,C) were located within the subepicardial area (defined as the distance between the epicardial mesothelium and the myocardial border of mesenchymal tissue); pericoronary LyVs (marked with blue-edged arrows in Figure 1A) were immediately adjacent to coronary vessels; and intramyocardial LyVs (marked with violet-edged arrows in Figure 1A) were in neither of these locations, instead being scattered in the myocardial wall.
2.5. Histology and Morphometric Analyses
2.6. Inflammatory Cell Infiltration
2.7. Ultrastructure and Morphometric Analysis
2.8. Statistics
3. Results
3.1. Metabolic Anomalies in Obese Mice
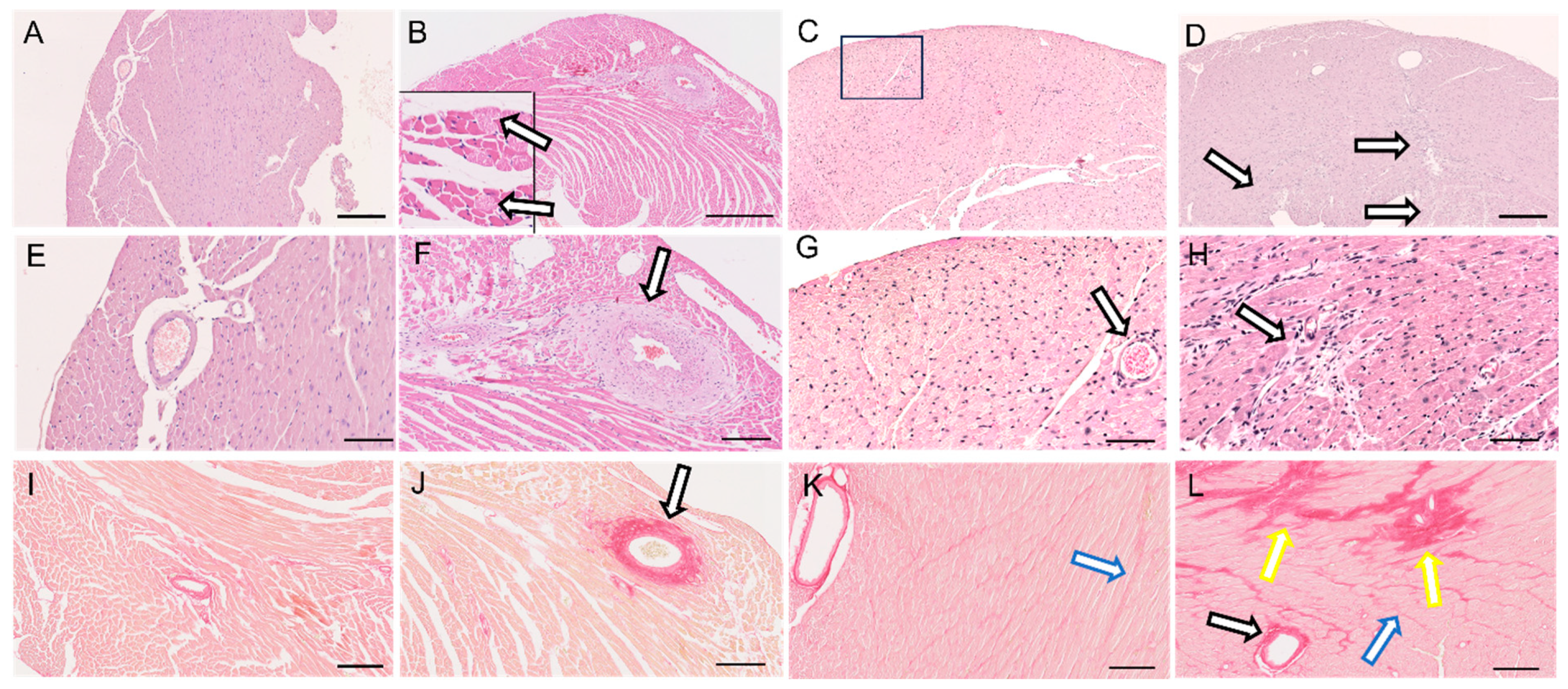
3.2. Heart Hypertrophy and Myocardial Remodeling
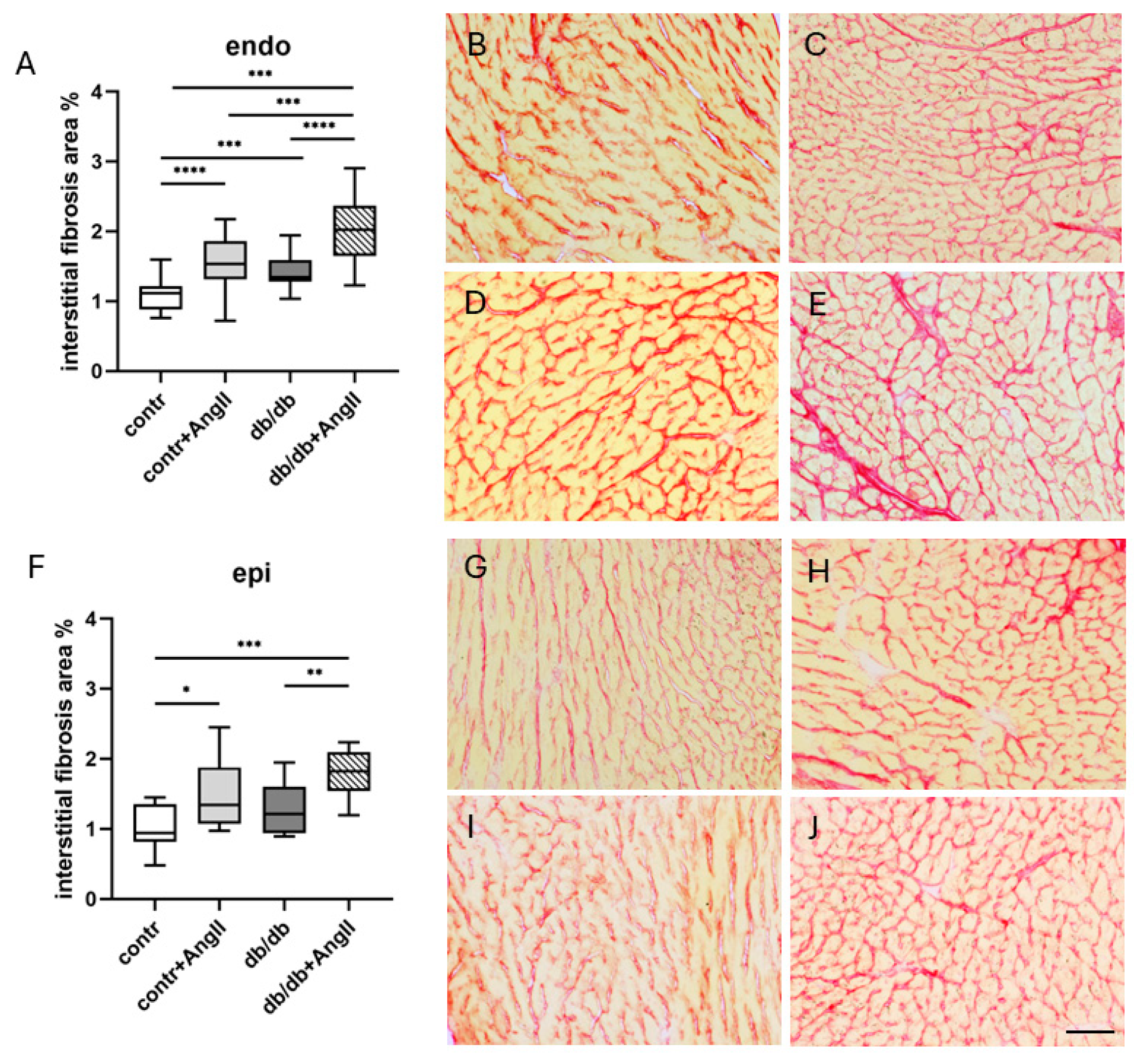
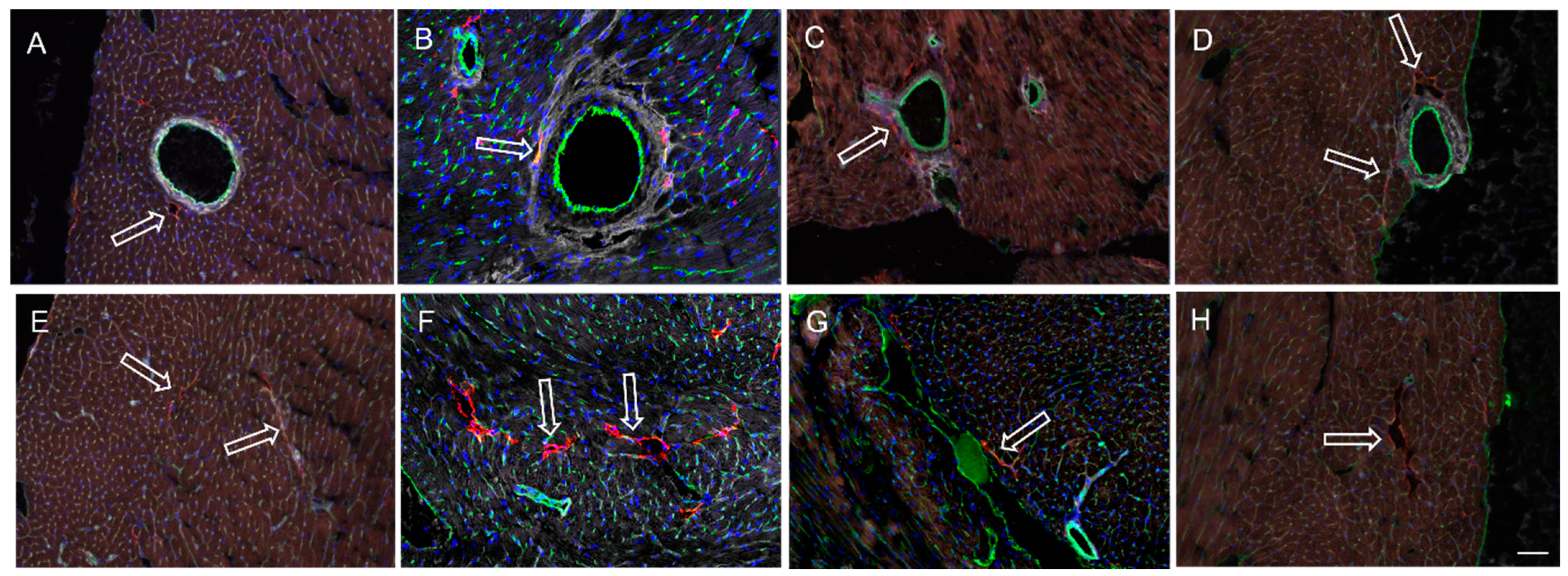
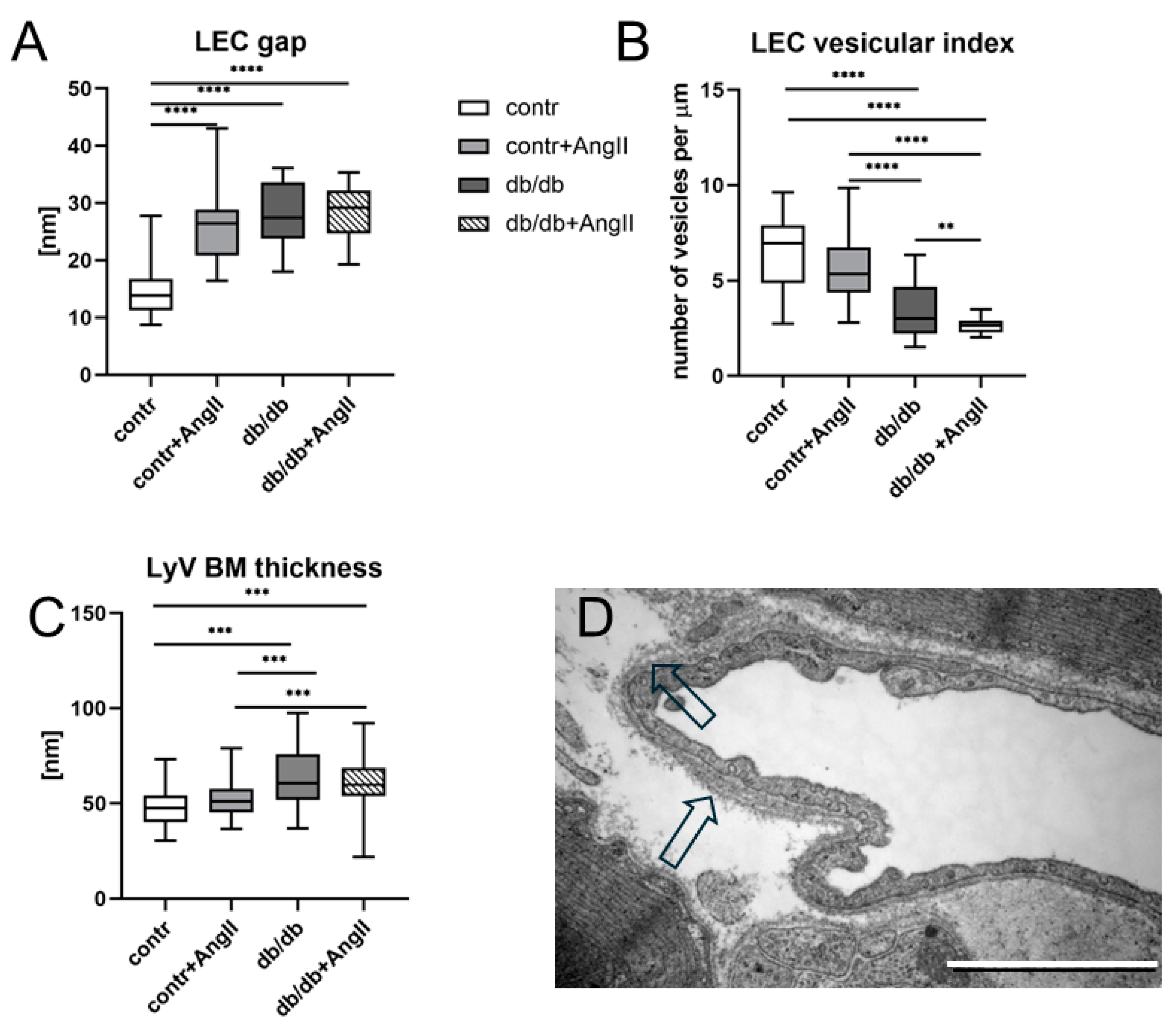
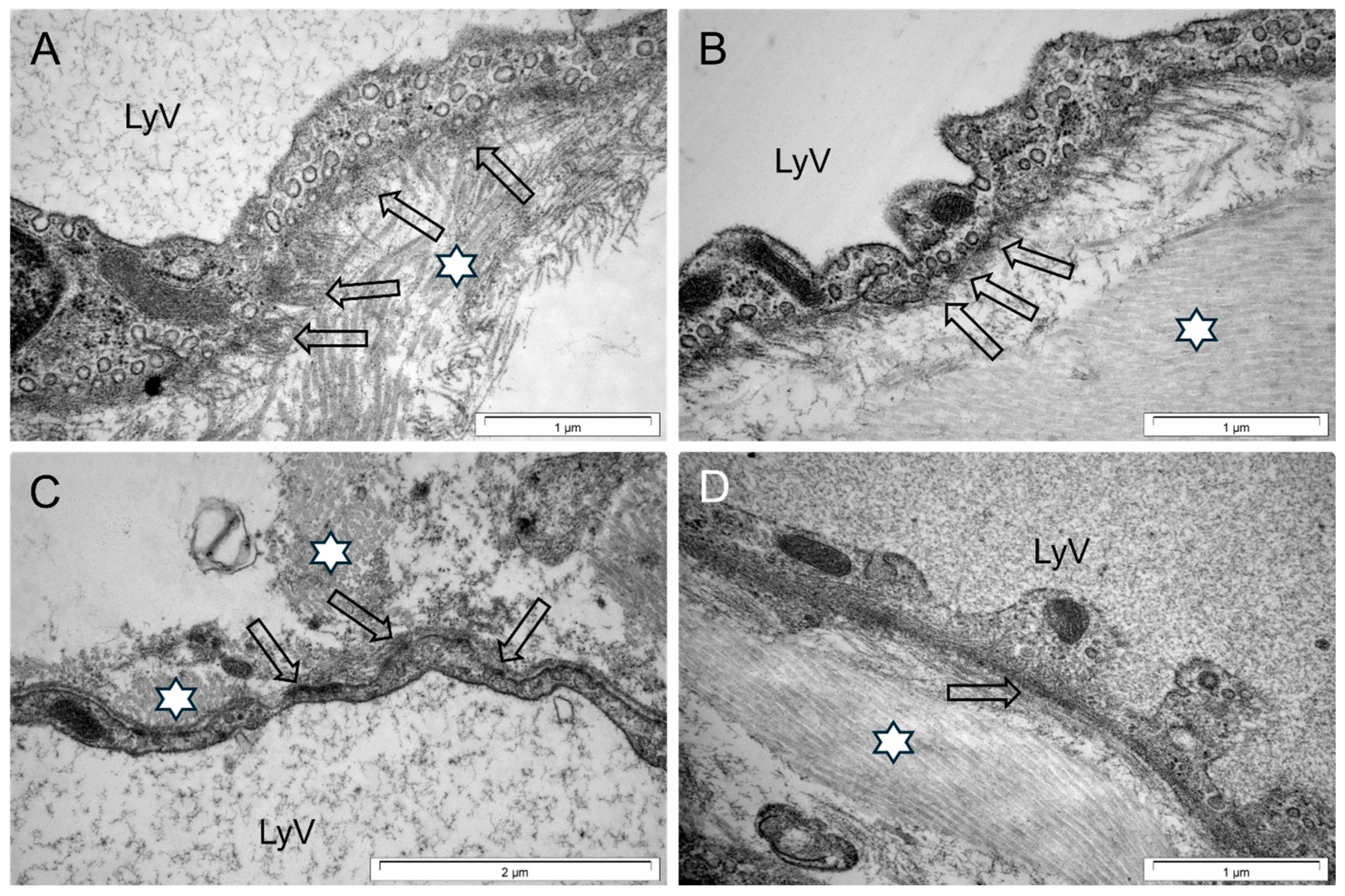
3.3. Cardiac LyV Density and Structural Remodeling
4. Discussion
4.1. Metabolic Anomalies in Obese Mice Create Specific Environment
4.2. Heart Remodeling Is Related to Ang II
4.3. Cardiac LyV Density Is Related to Obesity
4.4. LyV Remodeling Is Part of Cardiac Remodeling
5. Concluding Remarks
6. Limitations of the Study
7. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reilly, M.P.; Rader, D.J. The metabolic syndrome: More than the sum of its parts? Circulation 2003, 108, 1546–1551. [Google Scholar] [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Balasubbramanian, D.; Mitchell, B.M. Lymphatics in Cardiovascular Physiology. Cold Spring Harb. Perspect. Med. 2022, 12, a041173. [Google Scholar] [CrossRef]
- Scallan, J.P.; Hill, M.A.; Davis, M.J. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 2015, 107, 89–97. [Google Scholar] [CrossRef]
- Wu, H.; Rahman, H.N.A.; Dong, Y.; Liu, X.; Lee, Y.; Wen, A.; To, K.H.; Xiao, L.; Birsner, A.E.; Bazinet, L.; et al. Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J. Clin. Invest. 2018, 128, 4025–4043. [Google Scholar] [CrossRef]
- Cifarelli, V.; Appak-Baskoy, S.; Peche, V.S.; Kluzak, A.; Shew, T.; Narendran, R.; Pietka, K.M.; Cella, M.; Walls, C.W.; Czepielewski, R.; et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat. Commun. 2021, 12, 3350. [Google Scholar] [CrossRef]
- Zawieja, S.D.; Gasheva, O.; Zawieja, D.C.; Muthuchamy, M. Blunted flow-mediated responses and diminished nitric oxide synthase expression in lymphatic thoracic ducts of a rat model of metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H385–H393. [Google Scholar] [CrossRef]
- Nitti, M.D.; Hespe, G.E.; Kataru, R.P.; Garcia Nores, G.D.; Savetsky, I.L.; Torrisi, J.S.; Gardenier, J.C.; Dannenberg, A.J.; Mehrara, B.J. Obesity-induced lymphatic dysfunction is reversible with weight loss. J. Physiol. 2016, 594, 7073–7087. [Google Scholar] [CrossRef]
- Chakraborty, A.; Barajas, S.; Lammoglia, G.M.; Reyna, A.J.; Morley, T.S.; Johnson, J.A.; Scherer, P.E.; Rutkowski, J.M. Vascular Endothelial Growth Factor-D (VEGF-D) Overexpression and Lymphatic Expansion in Murine Adipose Tissue Improves Metabolism in Obesity. Am. J. Pathol. 2019, 189, 924–939. [Google Scholar] [CrossRef]
- Chakraborty, A.; Scogin, C.K.; Rizwan, K.; Morley, T.S.; Rutkowski, J.M. Characterizing Lymphangiogenesis and Concurrent Inflammation in Adipose Tissue in Response to VEGF-D. Front. Physiol. 2020, 11, 363. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.L.; Lin, Q.Y.; Liu, Y.; Guan, X.M.; Ma, X.L.; Cao, H.J.; Liu, Y.; Bai, J.; Xia, Y.L.; et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 2018, 39, 1818–1831. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.Y.; Bai, J.; Liu, J.Q.; Li, H.H. Angiotensin II Stimulates the Proliferation and Migration of Lymphatic Endothelial Cells Through Angiotensin Type 1 Receptors. Front. Physiol. 2020, 11, 560170. [Google Scholar] [CrossRef]
- Bai, J.; Yin, L.; Yu, W.J.; Zhang, Y.L.; Lin, Q.Y.; Li, H.H. Angiotensin II Induces Cardiac Edema and Hypertrophic Remodeling through Lymphatic-Dependent Mechanisms. Oxid. Med. Cell Longev. 2022, 2022, 5044046. [Google Scholar] [CrossRef]
- Cui, Y. The role of lymphatic vessels in the heart. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2010, 17, 307–314. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Alitalo, K. Cardiac lymphatics in health and disease. Nat. Rev. Cardiol. 2019, 16, 56–68. [Google Scholar] [CrossRef]
- Klaourakis, K.; Vieira, J.M.; Riley, P.R. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat. Rev. Cardiol. 2021, 18, 368–379. [Google Scholar] [CrossRef]
- Bai, L.; Wang, Y.; Du, S.; Si, Y.; Chen, L.; Li, L.; Li, Y. Lymphangiogenesis: A new strategy for heart disease treatment (Review). Int. J. Mol. Med. 2024, 53, 35. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Sultan, I.; Alitalo, K. Cardiac Lymphangiogenesis in CVDs. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Trzewik, J.; Mallipattu, S.K.; Artmann, G.M.; Delano, F.A.; Schmid-Schonbein, G.W. Evidence for a second valve system in lymphatics: Endothelial microvalves. FASEB J. 2001, 15, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Breslin, J.W.; Yang, Y.; Scallan, J.P.; Sweat, R.S.; Adderley, S.P.; Murfee, W.L. Lymphatic Vessel Network Structure and Physiology. Compr. Physiol. 2018, 9, 207–299. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, C.; Hu, Y.; Xu, Q.; Hu, X. Lymphatics in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e275–e283. [Google Scholar] [CrossRef]
- Null, M.; Arbor, T.C.; Agarwal, M. Anatomy, Lymphatic System; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Mendoza, E.; Schmid-Schönbein, G.W. A model for mechanics of primary lymphatic valves. J. Biomech. Eng. 2003, 125, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Wilson, J.T.; Moore, J.E., Jr. Primary and secondary lymphatic valve development: Molecular, functional and mechanical insights. Microvasc. Res. 2014, 96, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Triacca, V.; Guc, E.; Kilarski, W.W.; Pisano, M.; Swartz, M.A. Transcellular Pathways in Lymphatic Endothelial Cells Regulate Changes in Solute Transport by Fluid Stress. Circ. Res. 2017, 120, 1440–1452. [Google Scholar] [CrossRef]
- Jannaway, M.; Scallan, J.P. VE-Cadherin and Vesicles Differentially Regulate Lymphatic Vascular Permeability to Solutes of Various Sizes. Front. Physiol. 2021, 12, 687563. [Google Scholar] [CrossRef]
- Sawane, M.; Kajiya, K.; Kidoya, H.; Takagi, M.; Muramatsu, F.; Takakura, N. Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes 2013, 62, 1970–1980. [Google Scholar] [CrossRef]
- Savetsky, I.L.; Torrisi, J.S.; Cuzzone, D.A.; Ghanta, S.; Albano, N.J.; Gardenier, J.C.; Joseph, W.J.; Mehrara, B.J. Obesity increases inflammation and impairs lymphatic function in a mouse model of lymphedema. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H165–H172. [Google Scholar] [CrossRef] [PubMed]
- Westcott, G.P.; Rosen, E.D. Crosstalk between Adipose and Lymphatics in Health and Disease. Endocrinology 2022, 163, bqab224. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Rutkowski, J.M.; Helft, J.; Reddy, S.T.; Swartz, M.A.; Randolph, G.J.; Angeli, V. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am. J. Pathol. 2009, 175, 1328–1337. [Google Scholar] [CrossRef]
- Danussi, C.; Spessotto, P.; Petrucco, A.; Wassermann, B.; Sabatelli, P.; Montesi, M.; Doliana, R.; Bressan, G.M.; Colombatti, A. Emilin1 deficiency causes structural and functional defects of lymphatic vasculature. Mol. Cell Biol. 2008, 28, 4026–4039. [Google Scholar] [CrossRef]
- Danussi, C.; Del Bel Belluz, L.; Pivetta, E.; Modica, T.M.; Muro, A.; Wassermann, B.; Doliana, R.; Sabatelli, P.; Colombatti, A.; Spessotto, P. EMILIN1/alpha9beta1 integrin interaction is crucial in lymphatic valve formation and maintenance. Mol. Cell Biol. 2013, 33, 4381–4394. [Google Scholar] [CrossRef]
- Kraus, S.; Lee, E. A human initial lymphatic chip reveals distinct mechanisms of primary lymphatic valve dysfunction in acute and chronic inflammation. Lab. Chip. 2023, 23, 5180–5194. [Google Scholar] [CrossRef]
- Li, X.; Shimada, T.; Zhang, Y.; Zhou, X.; Zhao, L. Ultrastructure changes of cardiac lymphatics during cardiac fibrosis in hypertensive rats. Anat. Rec. 2009, 292, 1612–1618. [Google Scholar] [CrossRef]
- Angeli, V.; Lim, H.Y. Biomechanical control of lymphatic vessel physiology and functions. Cell. Mol. Immunol. 2023, 20, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; González, A.; Díez, J. Role of Cardiac Lymphatics in Myocardial Edema and Fibrosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 735–744. [Google Scholar] [CrossRef]
- Cuijpers, I.; Simmonds, S.J.; van Bilsen, M.; Czarnowska, E.; González Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic. Res. Cardiol. 2020, 115, 39. [Google Scholar] [CrossRef]
- Kurtz, T.W.; Griffin, K.A.; Bidani, A.K.; Davisson, R.L.; Hall, J.E. Recommendations for blood pressure measurement in humans and experimental animals. Part 2: Blood pressure measurement in experimental animals: A statement for professionals from the subcommittee of professional and public education of the American Heart Association council on high blood pressure research. Hypertension 2005, 45, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.C.; Spurgeon, H.A.; Rakusan, K.; Weisfeldt, M.L.; Lakatta, E.G. Use of tibial length to quantify cardiac hypertrophy: Application in the aging rat. Am. J. Physiol. 1982, 243, H941–H947. [Google Scholar] [CrossRef]
- Karnovsky, M.L. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J. Cell Biol. 1965, 27, 1A–149A. [Google Scholar]
- Flaht-Zabost, A.; Gula, G.; Ciszek, B.; Czarnowska, E.; Jankowska-Steifer, E.; Madej, M.; Niderla-Bielinska, J.; Radomska-Lesniewska, D.; Ratajska, A. Cardiac mouse lymphatics: Developmental and anatomical update. Anat. Rec. 2014, 297, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Dolber, P.C.; Spach, M.S. Picrosirius red staining of cardiac muscle following phosphomolybdic acid treatment. Stain. Technol. 1987, 62, 23–26. [Google Scholar] [CrossRef]
- Percival, K.R.; Radi, Z.A. A modified Verhoeff’s elastin histochemical stain to enable pulmonary arterial hypertension model characterization. Eur. J. Histochem. EJH 2016, 60, 2588. [Google Scholar] [CrossRef]
- Jankowska-Steifer, E.; Madej, M.; Niderla-Bielinska, J.; Ruminski, S.; Flaht-Zabost, A.; Czarnowska, E.; Gula, G.; Radomska-Lesniewska, D.M.; Ratajska, A. Vasculogenic and hematopoietic cellular progenitors are scattered within the prenatal mouse heart. Histochem. Cell Biol. 2015, 143, 153–169. [Google Scholar] [CrossRef]
- Johansson, B.; Mörner, S.; Waldenström, A.; Stål, P. Myocardial capillary supply is limited in hypertrophic cardiomyopathy: A morphological analysis. Int. J. Cardiol. 2008, 126, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.R.; Balint, L.; Dy, D.M.; Nielsen, N.R.; Méndez, H.G.; Aghajanian, A.; Caron, K.M. The Ebb and Flow of Cardiac Lymphatics: A Tidal Wave of New Discoveries. Physiol. Rev. 2022, 103, 391–432. [Google Scholar] [CrossRef]
- Shariq, O.A.; McKenzie, T.J. Obesity-related hypertension: A review of pathophysiology, management, and the role of metabolic surgery. Gland. Surg. 2020, 9, 80–93. [Google Scholar] [CrossRef]
- van Bilsen, M.; Daniels, A.; Brouwers, O.; Janssen, B.J.; Derks, W.J.; Brouns, A.E.; Munts, C.; Schalkwijk, C.G.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Hypertension is a conditional factor for the development of cardiac hypertrophy in type 2 diabetic mice. PLoS ONE 2014, 9, e85078. [Google Scholar] [CrossRef] [PubMed]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Zhang, Y.L.; Hernandez-Ono, A. Metabolic syndrome: Focus on dyslipidemia. Obesity 2006, 14 (Suppl. S1), 41s–49s. [Google Scholar] [CrossRef] [PubMed]
- Nishina, P.M.; Lowe, S.; Wang, J.; Paigen, B. Characterization of plasma lipids in genetically obese mice: The mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism 1994, 43, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Ran, J.; Hirano, T.; Adachi, M. Chronic ANG II infusion increases plasma triglyceride level by stimulating hepatic triglyceride production in rats. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E955–E961. [Google Scholar] [CrossRef]
- Okada, K.; Hirano, T.; Ran, J.; Adachi, M. Olmesartan medoxomil, an angiotensin II receptor blocker ameliorates insulin resistance and decreases triglyceride production in fructose-fed rats. Hypertens. Res. 2004, 27, 293–299. [Google Scholar] [CrossRef]
- Ran, J.; Hirano, T.; Adachi, M. Angiotensin II type 1 receptor blocker ameliorates overproduction and accumulation of triglyceride in the liver of Zucker fatty rats. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E227–E232. [Google Scholar] [CrossRef]
- Goncalves, A.C.; Tank, J.; Diedrich, A.; Hilzendeger, A.; Plehm, R.; Bader, M.; Luft, F.C.; Jordan, J.; Gross, V. Diabetic hypertensive leptin receptor-deficient db/db mice develop cardioregulatory autonomic dysfunction. Hypertension 2009, 53, 387–392. [Google Scholar] [CrossRef]
- Alpert, M.A.; Lavie, C.J.; Agrawal, H.; Aggarwal, K.B.; Kumar, S.A. Obesity and heart failure: Epidemiology, pathophysiology, clinical manifestations, and management. Transl. Res. J. Lab. Clin. Med. 2014, 164, 345–356. [Google Scholar] [CrossRef]
- Alpert, M.A.; Omran, J.; Mehra, A.; Ardhanari, S. Impact of obesity and weight loss on cardiac performance and morphology in adults. Prog. Cardiovasc. Dis. 2014, 56, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Carbone, S.; Lavie, C.J.; Arena, R. Obesity and Heart Failure: Focus on the Obesity Paradox. Mayo Clin. Proc. 2017, 92, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, Y.; Xiao, C.; Du, J. Angiotensin II induces inflammation leading to cardiac remodeling. Front. Biosci. 2012, 17, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef]
- González, A.; López, B.; Querejeta, R.; Díez, J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J. Mol. Cell Cardiol. 2002, 34, 1585–1593. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Oakenfull, R.; Robson, A.; Arthur, H.M. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens. Res. 2012, 35, 393–398. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef]
- Nguyen, D.V.; Shaw, L.C.; Grant, M.B. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012, 3, 170. [Google Scholar] [CrossRef]
- Romeo, G.R.; Lee, J.; Shoelson, S.E. Metabolic syndrome, insulin resistance, and roles of inflammation--mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1771–1776. [Google Scholar] [CrossRef]
- Papinska, A.M.; Mordwinkin, N.M.; Meeks, C.J.; Jadhav, S.S.; Rodgers, K.E. Angiotensin-(1-7) administration benefits cardiac, renal and progenitor cell function in db/db mice. Br. J. Pharmacol. 2015, 172, 4443–4453. [Google Scholar] [CrossRef]
- Jiang, X.; Cui, J.; Yang, C.; Song, Y.; Yuan, J.; Liu, S.; Hu, F.; Yang, W.; Qiao, S. Elevated lymphatic vessel density measured by Lyve-1 expression in areas of replacement fibrosis in the ventricular septum of patients with hypertrophic obstructive cardiomyopathy (HOCM). Heart Vessel. 2020, 35, 78–85. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Barouch, L.A. Leptin signaling and obesity: Cardiovascular consequences. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Tsuda, K.; Baba, A.; Kawabe, T.; Boh-oka, S.; Ibata, M.; Moriwaki, C.; Hano, T.; Nishio, I. Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochem. Biophys. Res. Commun. 2000, 273, 745–749. [Google Scholar] [CrossRef]
- Winters, B.; Mo, Z.; Brooks-Asplund, E.; Kim, S.; Shoukas, A.; Li, D.; Nyhan, D.; Berkowitz, D.E. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice. J. Appl. Physiol. 2000, 89, 2382–2390. [Google Scholar] [CrossRef]
- Diez, J. Mechanisms of cardiac fibrosis in hypertension. J. Clin. Hypertens 2007, 9, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Galie, P.A.; Russell, M.W.; Westfall, M.V.; Stegemann, J.P. Interstitial fluid flow and cyclic strain differentially regulate cardiac fibroblast activation via AT1R and TGF-β1. Exp. Cell Res. 2012, 318, 75–84. [Google Scholar] [CrossRef]
- Brower, G.L.; Gardner, J.D.; Forman, M.F.; Murray, D.B.; Voloshenyuk, T.; Levick, S.P.; Janicki, J.S. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2006, 30, 604–610. [Google Scholar] [CrossRef]
- Yamamoto, K.; Masuyama, T.; Sakata, Y.; Nishikawa, N.; Mano, T.; Yoshida, J.; Miwa, T.; Sugawara, M.; Yamaguchi, Y.; Ookawara, T.; et al. Myocardial stiffness is determined by ventricular fibrosis, but not by compensatory or excessive hypertrophy in hypertensive heart. Cardiovasc. Res. 2002, 55, 76–82. [Google Scholar] [CrossRef]
- Bradham, R.R.; Parker, E.F.; Barrington, B.A., Jr.; Webb, C.M.; Stallworth, J.M. The cardiac lymphatics. Ann. Surg. 1970, 171, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Bradham, R.R.; Parker, E.F. The cardiac lymphatics. Ann. Thorac. Surg. 1973, 15, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Rossitto, G.; Mary, S.; McAllister, C.; Neves, K.B.; Haddow, L.; Rocchiccioli, J.P.; Lang, N.N.; Murphy, C.L.; Touyz, R.M.; Petrie, M.C.; et al. Reduced Lymphatic Reserve in Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 76, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Garcia Nores, G.D.; Cuzzone, D.A.; Albano, N.J.; Hespe, G.E.; Kataru, R.P.; Torrisi, J.S.; Gardenier, J.C.; Savetsky, I.L.; Aschen, S.Z.; Nitti, M.D.; et al. Obesity but not high-fat diet impairs lymphatic function. Int. J. Obes. 2016, 40, 1582–1590. [Google Scholar] [CrossRef]
- Song, L.; Chen, X.; Swanson, T.A.; LaViolette, B.; Pang, J.; Cunio, T.; Nagle, M.W.; Asano, S.; Hales, K.; Shipstone, A.; et al. Lymphangiogenic therapy prevents cardiac dysfunction by ameliorating inflammation and hypertension. eLife 2020, 9, e58376. [Google Scholar] [CrossRef]
- Summer, G.; Kuhn, A.R.; Munts, C.; Miranda-Silva, D.; Leite-Moreira, A.F.; Lourenço, A.P.; Heymans, S.; Falcão-Pires, I.; van Bilsen, M. A directed network analysis of the cardiome identifies molecular pathways contributing to the development of HFpEF. J. Mol. Cell Cardiol. 2020, 144, 66–75. [Google Scholar] [CrossRef]
- Czarnowska, E.; Ratajska, A.; Jankowska-Steifer, E.; Flaht-Zabost, A.; Niderla-Bielińska, J. Extracellular matrix molecules associated with lymphatic vessels in health and disease. Histol. Histopathol. 2023, 39, 13–34. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Stritt, S.; Koltowska, K.; Mäkinen, T. Homeostatic maintenance of the lymphatic vasculature. Trends Mol. Med. 2021, 27, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Kefalides, N.A. Structure and biosynthesis of basement membranes. Int. Rev. Connect. Tissue Res. 1973, 6, 63–104. [Google Scholar] [CrossRef]
- Gatseva, A.; Sin, Y.Y.; Brezzo, G.; Van Agtmael, T. Basement membrane collagens and disease mechanisms. Essays Biochem. 2019, 63, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.E.; Johnson, P.C. Possible contribution of basement membrane to the structural rigidity of blood capillaries. Microvasc. Res. 1975, 9, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef]
- Nikolova, G.; Strilic, B.; Lammert, E. The vascular niche and its basement membrane. Trends Cell Biol. 2007, 17, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, H.P.; Dresser, K.; Hutchinson, L.; Trivedi, C.M. Pathological MAPK activation-mediated lymphatic basement membrane disruption causes lymphangiectasia that is treatable with ravoxertinib. JCI Insight 2022, 7, e153033. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Yang, X.; Liu, K.; Zhang, X.; Zuo, X.; Ye, R.; Wang, Z.; Shi, R.; Meng, Q.; et al. Signaling pathways in vascular function and hypertension: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 168. [Google Scholar] [CrossRef]
- Marshall, C.B. Rethinking glomerular basement membrane thickening in diabetic nephropathy: Adaptive or pathogenic? Am. J. Physiol. Renal Physiol. 2016, 311, F831–F843. [Google Scholar] [CrossRef]
- Lynch, P.M.; Delano, F.A.; Schmid-Schonbein, G.W. The primary valves in the initial lymphatics during inflammation. Lymphat. Res. Biol. 2007, 5, 3–10. [Google Scholar] [CrossRef]
- Gerli, R.; Solito, R.; Weber, E.; Agliano, M. Specific adhesion molecules bind anchoring filaments and endothelial cells in human skin initial lymphatics. Lymphology 2000, 33, 148–157. [Google Scholar]
- Johnson, L.A.; Banerji, S.; Lawrance, W.; Gileadi, U.; Prota, G.; Holder, K.A.; Roshorm, Y.M.; Hanke, T.; Cerundolo, V.; Gale, N.W.; et al. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat. Immunol. 2017, 18, 762–770. [Google Scholar] [CrossRef]
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- Pivetta, E.; Wassermann, B.; Del Bel Belluz, L.; Danussi, C.; Modica, T.M.; Maiorani, O.; Bosisio, G.; Boccardo, F.; Canzonieri, V.; Colombatti, A.; et al. Local inhibition of elastase reduces EMILIN1 cleavage reactivating lymphatic vessel function in a mouse lymphoedema model. Clin. Sci. 2016, 130, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Weber, E.; Sacchi, G.; Maestrini, D.; Di Cintio, F.; Gerli, R. Mechanotransduction in lymphatic endothelial cells. Lymphology 2007, 40, 102–113. [Google Scholar] [PubMed]
- Rossi, A.; Pasqui, D.; Barbucci, R.; Gerli, R.; Weber, E. The topography of microstructured surfaces differently affects fibrillin deposition by blood and lymphatic endothelial cells in culture. Tissue Eng. Part A 2009, 15, 525–533. [Google Scholar] [CrossRef]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature—Mechanisms and Consequences. Front. Immunol. 2019, 10, 471. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef]
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575. [Google Scholar] [CrossRef]
- Wiig, H.; Swartz, M.A. Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef]
- Vasques-Nóvoa, F.; Angélico-Gonçalves, A.; Alvarenga, J.M.G.; Nobrega, J.; Cerqueira, R.J.; Mancio, J.; Leite-Moreira, A.F.; Roncon-Albuquerque, R., Jr. Myocardial oedema: Pathophysiological basis and implications for the failing heart. ESC Heart Fail. 2022, 9, 958–976. [Google Scholar] [CrossRef]
- Münch, J.; Abdelilah-Seyfried, S. Sensing and Responding of Cardiomyocytes to Changes of Tissue Stiffness in the Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 642840. [Google Scholar] [CrossRef]
- Gupta, M.; Doss, B.L.; Kocgozlu, L.; Pan, M.; Mège, R.M.; Callan-Jones, A.; Voituriez, R.; Ladoux, B. Cell shape and substrate stiffness drive actin-based cell polarity. Phys. Rev. E 2019, 99, 012412. [Google Scholar] [CrossRef] [PubMed]
- Doss, B.L.; Pan, M.; Gupta, M.; Grenci, G.; Mège, R.M.; Lim, C.T.; Sheetz, M.P.; Voituriez, R.; Ladoux, B. Cell response to substrate rigidity is regulated by active and passive cytoskeletal stress. Proc. Natl. Acad. Sci. USA 2020, 117, 12817–12825. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R.; Ilan, I.S.; Lee, E. A bioengineered lymphatic vessel model for studying lymphatic endothelial cell-cell junction and barrier function. Microcirculation 2021, 28, e12730. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Teuwen, L.A.; Geldhof, V.; Carmeliet, P. How to Cross the Lymphatic Fence: Lessons From Solute Transport. Circ. Res. 2017, 120, 1376–1378. [Google Scholar] [CrossRef] [PubMed]
- Miteva, D.O.; Rutkowski, J.M.; Dixon, J.B.; Kilarski, W.; Shields, J.D.; Swartz, M.A. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ. Res. 2010, 106, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Podgrabinska, S.; Braun, P.; Velasco, P.; Kloos, B.; Pepper, M.S.; Skobe, M. Molecular characterization of lymphatic endothelial cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16069–16074. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, G.; Creed, H.A.; Cromer, W.E.; Wang, W.; Upchurch, B.D.; Smithhart, M.C.; Vadlamani, S.S.; Clark, M.C.; Busbuso, N.C.; Blais, S.N.; et al. Dichotomous effects on lymphatic transport with loss of caveolae in mice. Acta Physiol. 2021, 103, e13656. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Paulus, W.J. Unfolding Discoveries in Heart Failure. N. Engl. J. Med. 2020, 382, 679–682. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef]
- Su, W.; Guo, Z.; Randall, D.C.; Cassis, L.; Brown, D.R.; Gong, M.C. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1634–H1641. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1675–H1684. [Google Scholar] [CrossRef] [PubMed]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. J. Lab. Clin. Med. 2014, 164, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Hartog, J.W.; Voors, A.A.; Bakker, S.J.; Smit, A.J.; van Veldhuisen, D.J. Advanced glycation end-products (AGEs) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Leak, L.V.; Burke, J.F. Ultrastructural studies on the lymphatic anchoring filaments. J. Cell Biol. 1968, 36, 129–149. [Google Scholar] [CrossRef]
- Gerli, R.; Ibba, L.; Fruschelli, C. Ultrastructural cytochemistry of anchoring filaments of human lymphatic capillaries and their relation to elastic fibers. Lymphology 1991, 24, 105–112. [Google Scholar]
- Solito, R.; Alessandrini, C.; Fruschelli, M.; Pucci, A.M.; Gerli, R. An immunological correlation between the anchoring filaments of initial lymph vessels and the neighboring elastic fibers: A unified morphofunctional concept. Lymphology 1997, 30, 194–202. [Google Scholar]
- Bishop, M.A.; Malhotra, M. An investigation of lymphatic vessels in the feline dental pulp. Am. J. Anat. 1990, 187, 247–253. [Google Scholar] [CrossRef]
- Leak, L.V.; Schannahan, A.; Scully, H.; Daggett, W.M. Lymphatic vessels of the mammalian heart. Anat. Rec. 1978, 191, 183–201. [Google Scholar] [CrossRef]
- Berens von Rautenfeld, D.; Lubach, D.; Wenzel-Hora, B.; Klanke, J.; Hunneshagen, C. New techniques of demonstrating lymph vessels in skin biopsy specimens and intact skin with the scanning electron microscope. Arch. Dermatol. Res. 1987, 279, 327–334. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Taghdiri, N.; Ninh, V.K.; Mesfin, J.M.; Toomu, A.; Sehgal, R.; Lee, J.; Liang, Y.; Duran, J.M.; Adler, E.; et al. Single-cell and spatial transcriptomics of the infarcted heart define the dynamic onset of the border zone in response to mechanical destabilization. Nat. Cardiovasc. Res. 2022, 1, 1039–1055. [Google Scholar] [CrossRef] [PubMed]
- Gerner, M.Y.; Kastenmuller, W.; Ifrim, I.; Kabat, J.; Germain, R.N. Histo-cytometry: A method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 2012, 37, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Stoltzfus, C.R.; Filipek, J.; Gern, B.H.; Olin, B.E.; Leal, J.M.; Wu, Y.; Lyons-Cohen, M.R.; Huang, J.Y.; Paz-Stoltzfus, C.L.; Plumlee, C.R.; et al. CytoMAP: A Spatial Analysis Toolbox Reveals Features of Myeloid Cell Organization in Lymphoid Tissues. Cell. Rep. 2020, 31, 107523. [Google Scholar] [CrossRef] [PubMed]
- Radtke, A.J.; Kandov, E.; Lowekamp, B.; Speranza, E.; Chu, C.J.; Gola, A.; Thakur, N.; Shih, R.; Yao, L.; Yaniv, Z.R.; et al. IBEX: A versatile multiplex optical imaging approach for deep phenotyping and spatial analysis of cells in complex tissues. Proc. Natl. Acad. Sci. USA 2020, 117, 33455–33465. [Google Scholar] [CrossRef]
- Gibot, L.; Galbraith, T.; Kloos, B.; Das, S.; Lacroix, D.A.; Auger, F.A.; Skobe, M. Cell-based approach for 3D reconstruction of lymphatic capillaries in vitro reveals distinct functions of HGF and VEGF-C in lymphangiogenesis. Biomaterials 2016, 78, 129–139. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Dilution | Source, Cat. No/Clone No. |
|---|---|---|---|
| Primary antibodies | |||
| Lyve-1 | Rabbit | 1:300 | AngioBio, cat. no. 11-034; San Diego, CA, USA |
| CD31 | Rat | 1:100 | BD Biosciences, cat. no. 550274; clone no. Mec 13.3; Franklin Lakes, NJ, USA |
| CD45 | Rat | 1:50 | BD Biosciences, cat. no. 550539 |
| CD68 | Rabbit | 1:100 | Abcam cat no. 125212 |
| Secondary antibodies/lectin | |||
| Cy™3-conjugated anti-rabbit IgG | Donkey | 1:800 | Jackson ImmunoResearch, cat. no 711-165-152; Baltimore, MD, USA |
| Alexa Fluor® 647-conjugated AffiniPure Anti-Mouse IgG (H + L) | Donkey | 1:200 | Jackson ImmunoResearch, cat. no 715-605-151; Baltimore, MD, USA |
| AlexaFluor® 647-conjugated anti-rat IgG (H + L) | Donkey | 1:500 | Jackson ImmunoResearch, cat. no 712-605-153; Baltimore, MD, USA |
| Fluorescein (FITC)-conjugated anti-Goat IgG-(H + L) | Donkey | 1:250 | Jackson ImmunoResearch, cat. no 705-095-147; Baltimore, MD, USA |
| Alexa Fluor 488 Conjugated-Wheat Germ Agglutin (WGA) | lectin | 1:1800 | Thermo Fisher, cat. no. W11261; Waltham, MA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flaht-Zabost, A.; Czarnowska, E.; Jankowska-Steifer, E.; Niderla-Bielińska, J.; Żera, T.; Moskalik, A.; Bartkowiak, M.; Bartkowiak, K.; Tomczyk, M.; Majchrzak, B.; et al. Lymphatic Vessel Remodeling in the Hearts of Ang II-Treated Obese db/db Mice as an Integral Component of Cardiac Remodeling. Appl. Sci. 2024, 14, 8675. https://doi.org/10.3390/app14198675
Flaht-Zabost A, Czarnowska E, Jankowska-Steifer E, Niderla-Bielińska J, Żera T, Moskalik A, Bartkowiak M, Bartkowiak K, Tomczyk M, Majchrzak B, et al. Lymphatic Vessel Remodeling in the Hearts of Ang II-Treated Obese db/db Mice as an Integral Component of Cardiac Remodeling. Applied Sciences. 2024; 14(19):8675. https://doi.org/10.3390/app14198675
Chicago/Turabian StyleFlaht-Zabost, Aleksandra, Elżbieta Czarnowska, Ewa Jankowska-Steifer, Justyna Niderla-Bielińska, Tymoteusz Żera, Aneta Moskalik, Mateusz Bartkowiak, Krzysztof Bartkowiak, Mateusz Tomczyk, Barbara Majchrzak, and et al. 2024. "Lymphatic Vessel Remodeling in the Hearts of Ang II-Treated Obese db/db Mice as an Integral Component of Cardiac Remodeling" Applied Sciences 14, no. 19: 8675. https://doi.org/10.3390/app14198675
APA StyleFlaht-Zabost, A., Czarnowska, E., Jankowska-Steifer, E., Niderla-Bielińska, J., Żera, T., Moskalik, A., Bartkowiak, M., Bartkowiak, K., Tomczyk, M., Majchrzak, B., Kłosińska, D., Kozłowska, H., Ciszek, B., Gewartowska, M., Cudnoch-Jędrzejewska, A., & Ratajska, A. (2024). Lymphatic Vessel Remodeling in the Hearts of Ang II-Treated Obese db/db Mice as an Integral Component of Cardiac Remodeling. Applied Sciences, 14(19), 8675. https://doi.org/10.3390/app14198675