

Discovery of N-Aryl-Benzimidazolone Analogs as Novel Potential HSP90 Inhibitors: A Computational Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein and Ligand Preparation
2.2. Molecular Docking
2.3. Pharmacophore Modeling and Virtual Screening
2.4. ADME-T and Drug-Likeness Properties
2.5. Molecular Dynamics Simulation
Calculating Binding Free Energy
3. Results
3.1. Target-Based Pharmacophore Modeling
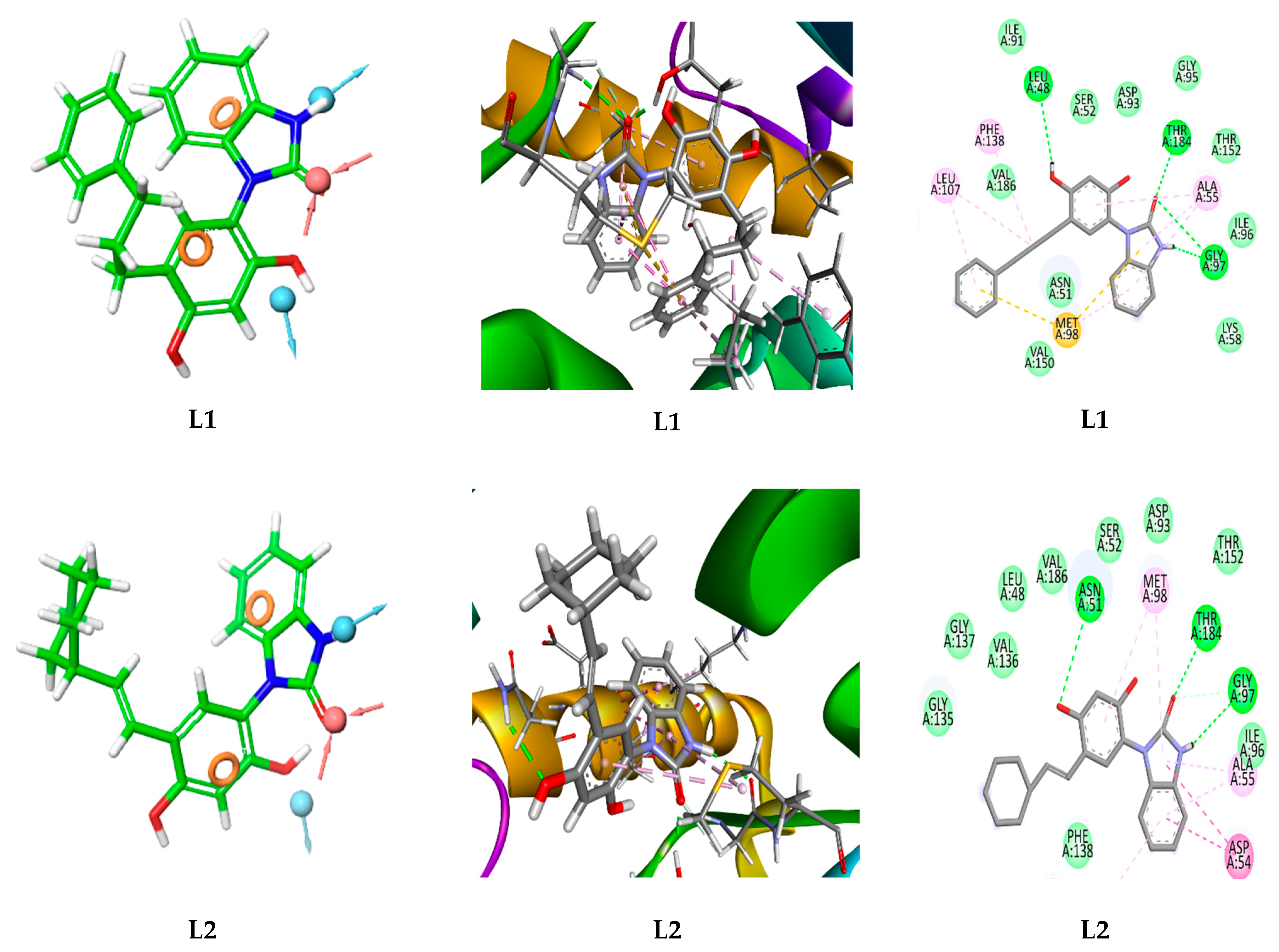
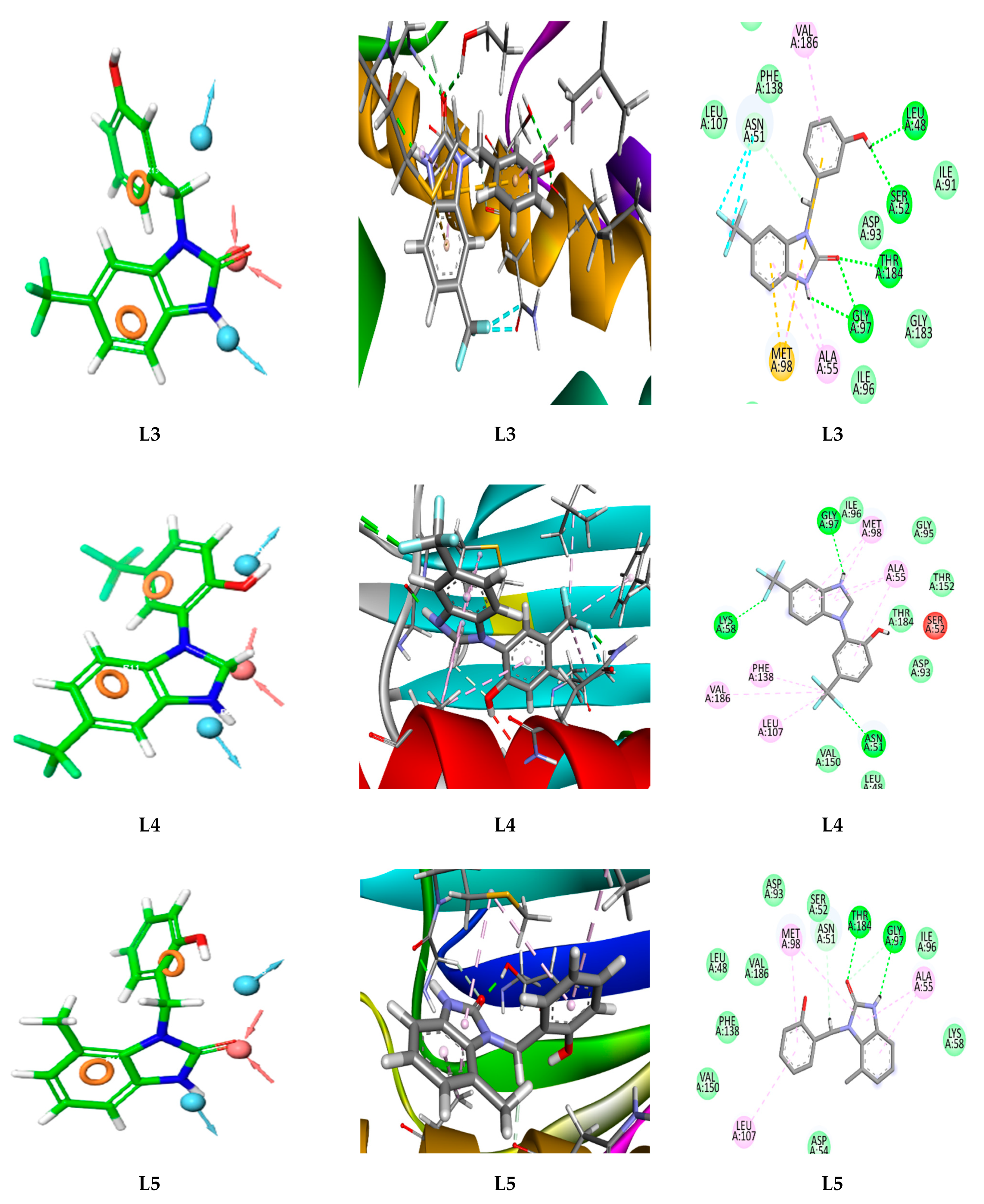
3.2. Target-Based Virtual Screening and Molecular Docking
3.3. ADMET Profiles of the Lead Compound
3.4. Physicochemical Properties
3.5. Medicinal Chemistry
3.6. Absorption and Distribution
3.7. Metabolism and Excretion
Prediction of Toxicity
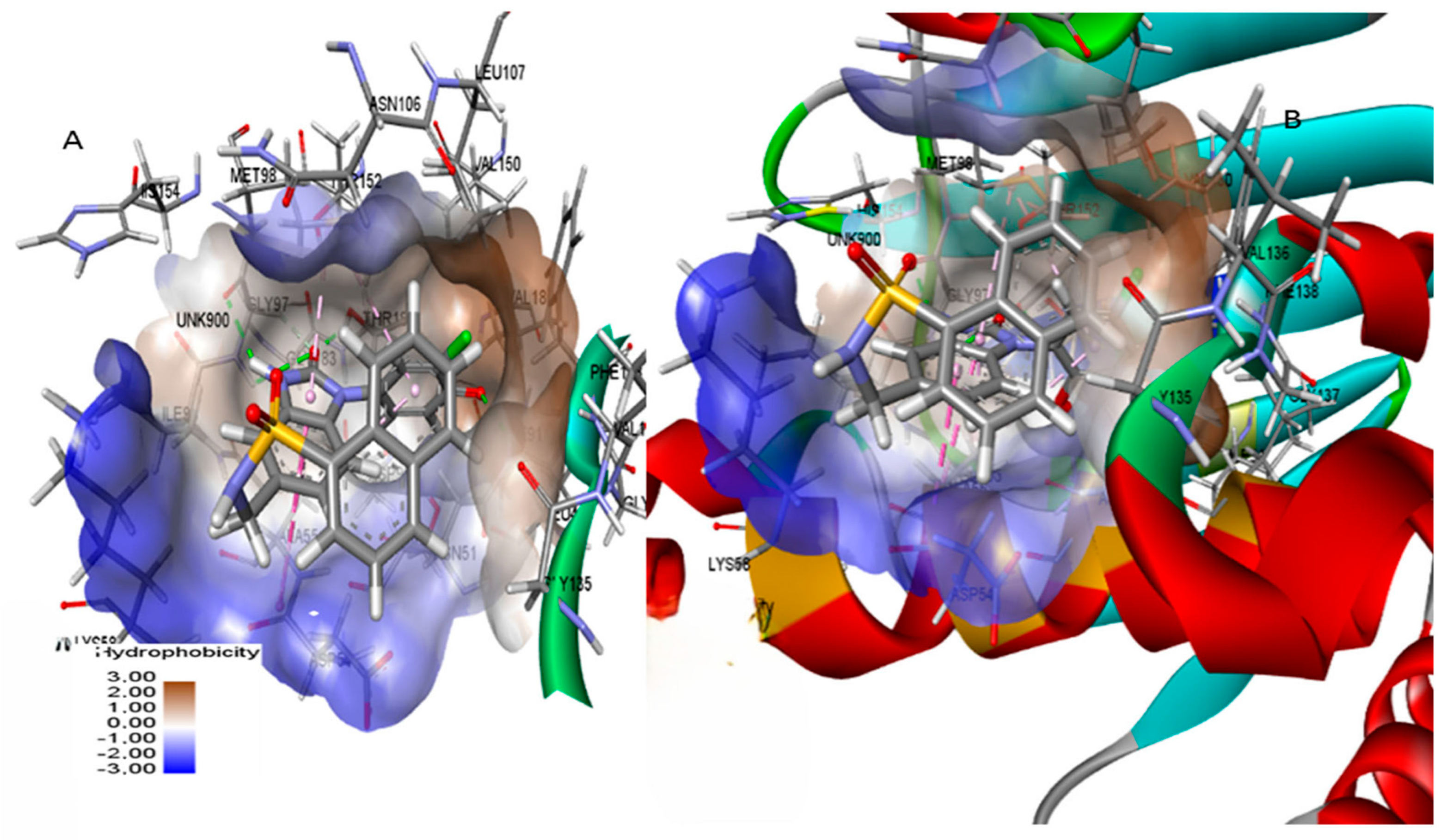
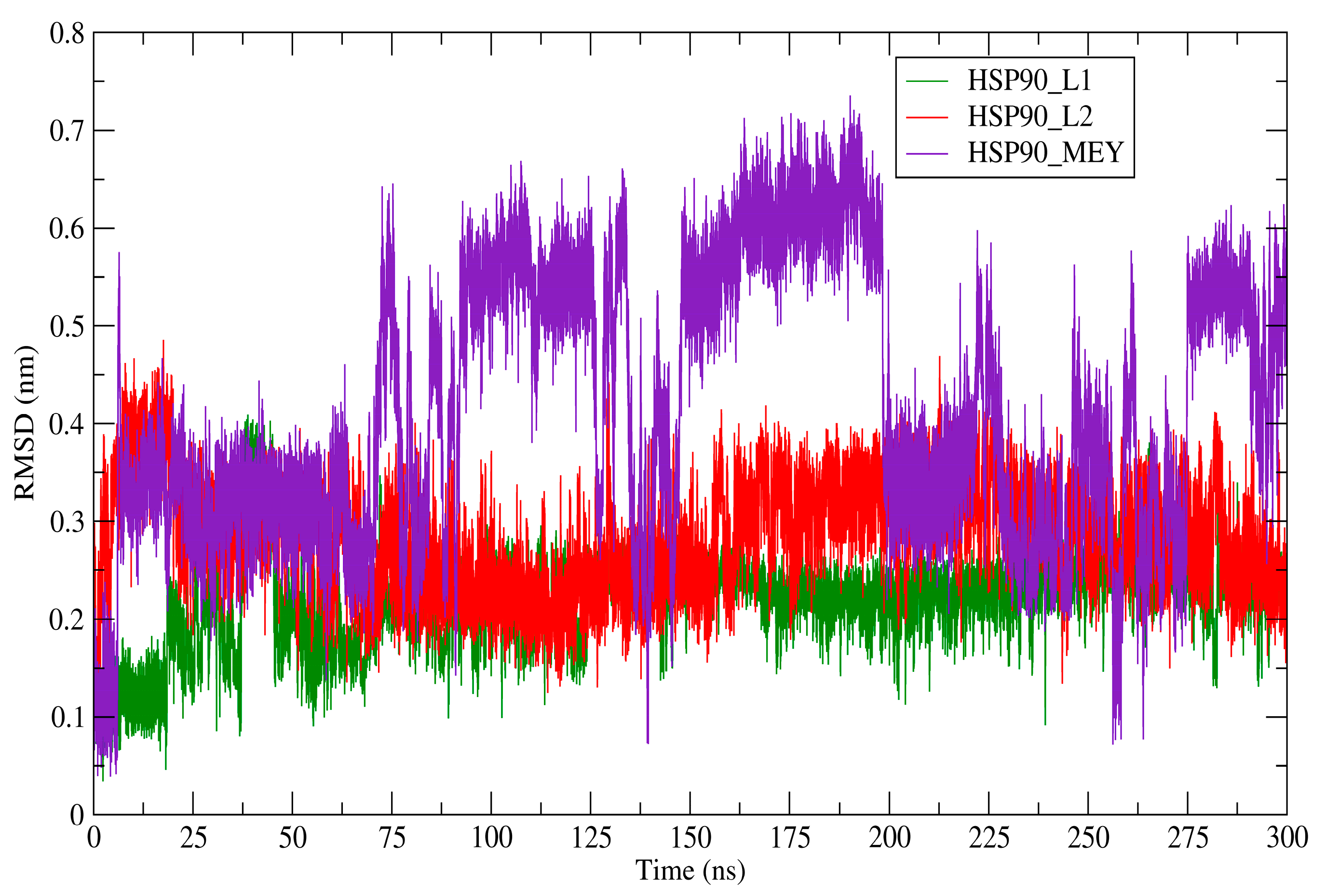
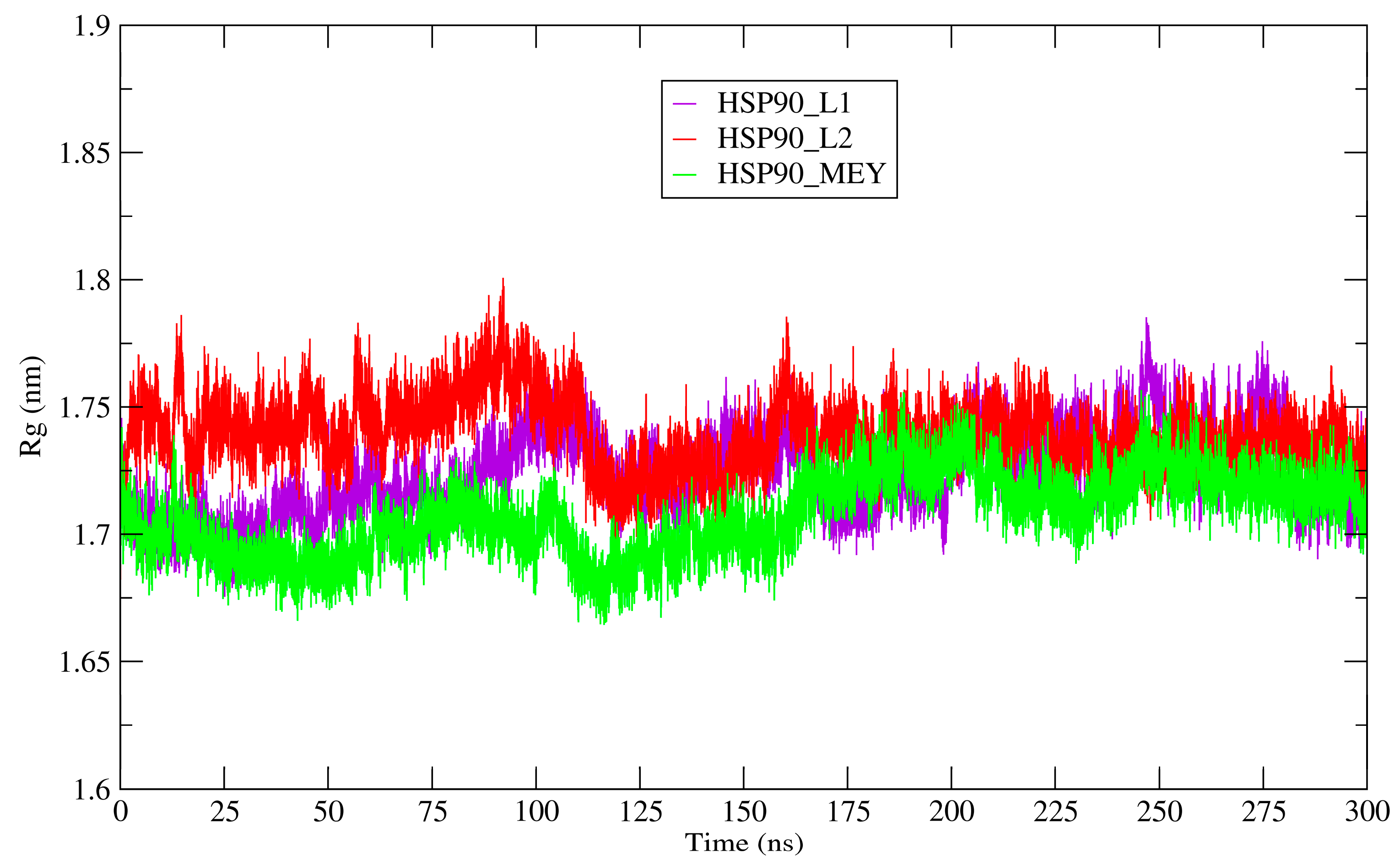
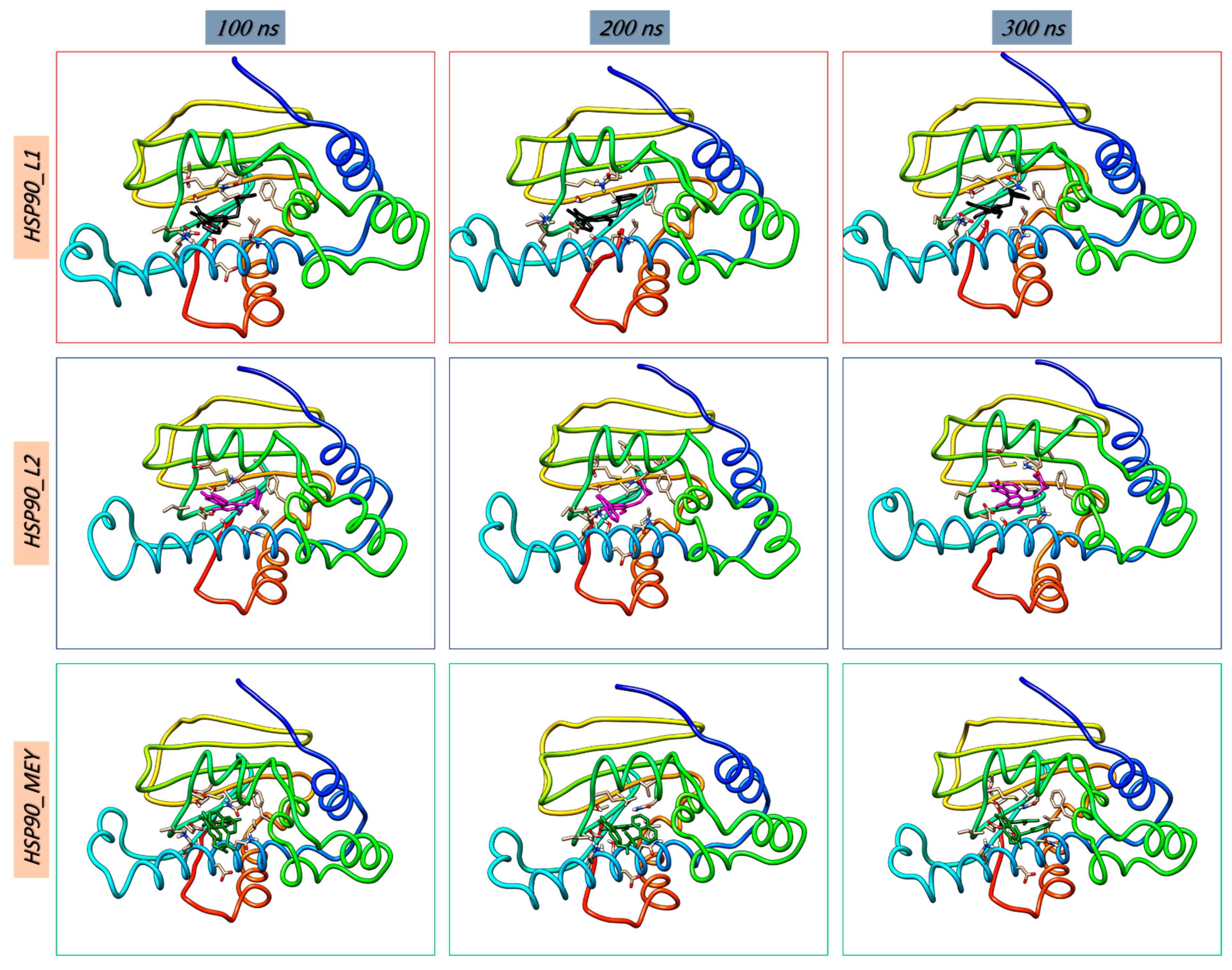
3.8. Molecular Dynamics Simulations
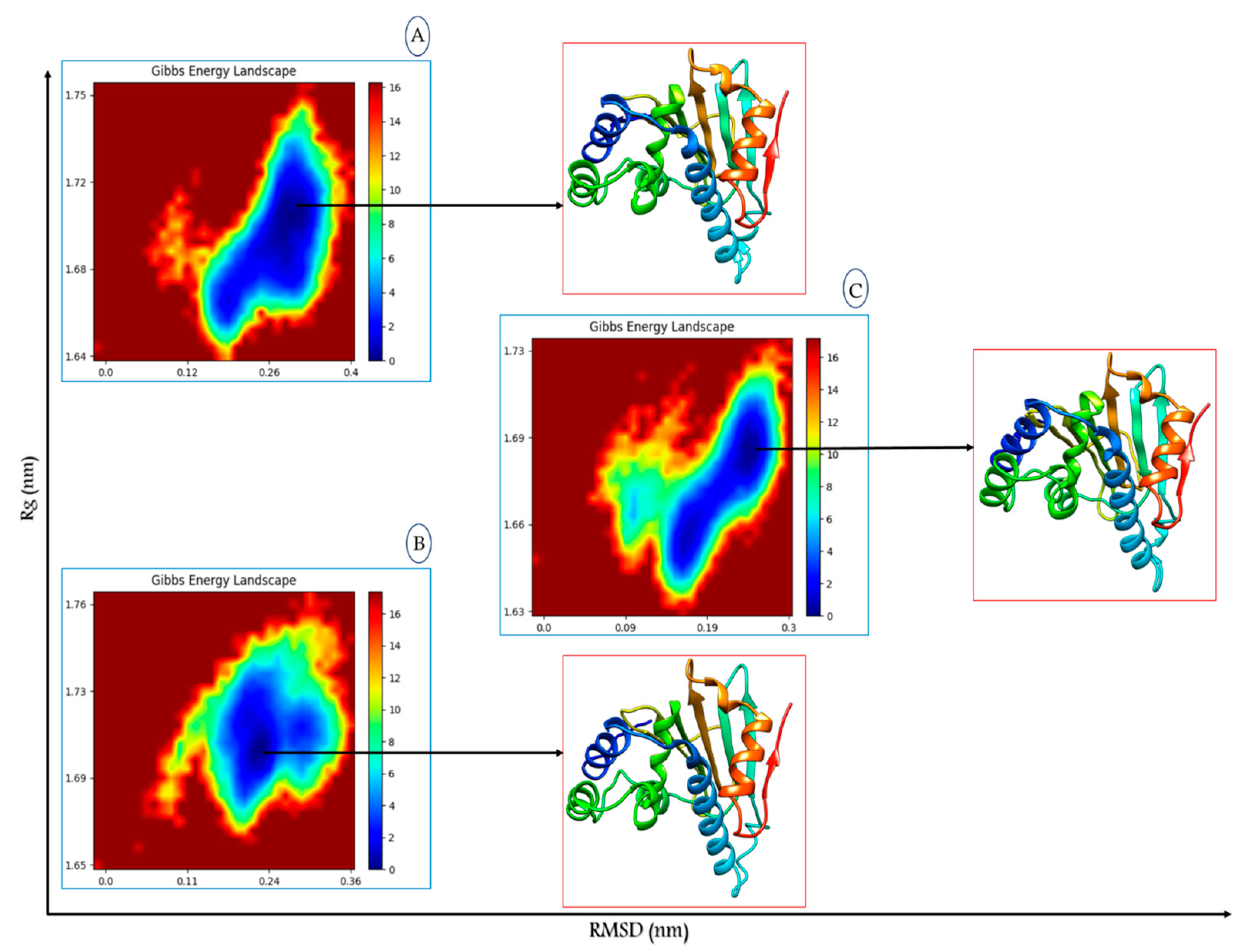
Free Energy Landscape
3.9. Binding Free Energy Calculation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. [Google Scholar] [CrossRef] [PubMed]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in Cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.; Liu, Y.; Zhao, K.; Wei, S.; Sugarman, E.T.; Liu, L.; Zhang, G. Targeting HSP90 as a Novel Therapy for Cancer: Mechanistic Insights and Translational Relevance. Cells 2022, 11, 2778. [Google Scholar] [CrossRef]
- Li, Z.-N.; Luo, Y. HSP90 Inhibitors and Cancer: Prospects for Use in Targeted Therapies (Review). Oncol. Rep. 2022, 49, 6. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, N.; Li, X.; Huang, H.; Peng, C.; Huang, W.; Foster, L.J.; He, G.; Han, B. Small-Molecule Dual Inhibitors Targeting Heat Shock Protein 90 for Cancer Targeted Therapy. Bioorganic Chem. 2023, 139, 106721. [Google Scholar] [CrossRef]
- Wright, J.B. The Chemistry of the Benzimidazoles. Chem. Rev. 1951, 48, 397–541. [Google Scholar] [CrossRef]
- Barker, H.A.; Smyth, R.D.; Weissbach, H.; Toohey, J.I.; Ladd, J.N.; Volcani, B.E. Isolation and Properties of Crystalline Cobamide Coenzymes Containing Benzimidazole or 5,6-Dimethylbenzimidazole. J. Biol. Chem. 1960, 235, 480–488. [Google Scholar] [CrossRef]
- Preethi, P.; Karthikeyan, E.; Lohita, M.; Teja, P.; Subhash, M.; Shaheena, P.; Prashanth, Y.; Sai, N. Benzimidazole: An Important Scaffold in Drug Discovery. Asian J. Pharm. Technol. 2015, 5, 138–152. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ghate, M. Substituted Benzimidazole Derivatives as Angiotensin II-AT1 Receptor Antagonist: A Review. Mini Rev. Med. Chem. 2010, 10, 1366–1384. [Google Scholar] [CrossRef]
- Ardestani, M.; Khorsandi, Z.; Keshavarzipour, F.; Iravani, S.; Sadeghi-Aliabadi, H.; Varma, R.S. Heterocyclic Compounds as Hsp90 Inhibitors: A Perspective on Anticancer Applications. Pharmaceutics 2022, 14, 2220. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Amanlou, M.; Aghaei, M.; Hassanzadeh, F.; Sadeghi-Aliabadi, H. Identification of New Hsp90 Inhibitors: Structure Based Virtual Screening, Molecular Dynamic Simulation, Synthesis and Biological Evaluation. Anti-Cancer Agents Med. Chem. 2021, 21, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; She, X.; Ou, Y.; Liu, J.; Yuan, Z.; Zhao, Q. Design, Synthesis and Biological Evaluation of a New Class of Hsp90 Inhibitors Vibsanin C Derivatives. Eur. J. Med. Chem. 2022, 244, 114844. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore Modeling and Applications in Drug Discovery: Challenges and Recent Advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Bruncko, M.; Tahir, S.K.; Song, X.; Chen, J.; Ding, H.; Huth, J.R.; Jin, S.; Judge, R.A.; Madar, D.J.; Park, C.H.; et al. N-Aryl-Benzimidazolones as Novel Small Molecule HSP90 Inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 7503–7506. [Google Scholar] [CrossRef]
- Ouassaf, M.; Belaidi, S.; Mogren Al Mogren, M.; Chtita, S.; Ullah Khan, S.; Thet Htar, T. Combined Docking Methods and Molecular Dynamics to Identify Effective Antiviral 2,5-Diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ.-Sci. 2021, 33, 101352. [Google Scholar] [CrossRef]
- Ejaz, S.A.; Aziz, M.; Fawzy Ramadan, M.; Fayyaz, A.; Bilal, M.S. Pharmacophore-Based Virtual Screening and In-Silico Explorations of Biomolecules (Curcumin Derivatives) of Curcuma longa as Potential Lead Inhibitors of ERBB and VEGFR-2 for the Treatment of Colorectal Cancer. Molecules 2023, 28, 4044. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A Platform for Systematic ADMET Evaluation Based on a Comprehensively Collected ADMET Database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, B.; Mebarka, O.; Khan, S.U.; Htar, T.T. Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Studies for the Discovery of Novel Neuraminidase Inhibitors. J. Biomol. Struct. Dyn. 2024, 42, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Akter, N.; Bourougaa, L.; Ouassaf, M.; Bhowmic, R.C.; Uddin, K.M.; Bhat, A.R.; Ahmed, S.; Kawsar, S.M.A. Molecular Docking, ADME-Tox, DFT and Molecular Dynamics Simulation of Butyroyl Glucopyranoside Derivatives against DNA Gyrase Inhibitors as Antimicrobial Agents. J. Mol. Struct. 2024, 1307, 137930. [Google Scholar] [CrossRef]
- McDonald, E.; Jones, K.; Brough, P.A.; Drysdale, M.J.; Workman, P. Discovery and Development of Pyrazole-Scaffold Hsp90 Inhibitors. Curr. Top. Med. Chem. 2006, 6, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Bourougaa, L.; Al-Mijalli, S.H.; Abdallah, E.M.; Bhat, A.R.; Kawsar, S.M.A. Marine-Derived Compounds as Potential Inhibitors of Hsp90 for Anticancer and Antimicrobial Drug Development: A Comprehensive In Silico Study. Molecules 2023, 28, 8074. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Daoui, O.; Alam, S.; Elkhattabi, S.; Belaidi, S.; Chtita, S. Pharmacophore-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Studies for the Discovery of Novel FLT3 Inhibitors. J. Biomol. Struct. Dyn. 2022, 41, 7712–7724. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of Protein-Ligand Binding Affinity by Hydrogen Bond Pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Maowa, J.; Alam, A.; Rana, K.M.; Dey, S.; Hosen, A.; Fujii, Y.; Hasan, I.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Characterization, Synergistic Antimicrobial Properties and Molecular Docking of Sugar Modified Uridine Derivatives. Ovidius Univ. Ann. Chem. 2021, 32, 6–21. [Google Scholar] [CrossRef]
- Yasmin, F.; Amin, M.R.; Hosen, M.A.; Bulbul, M.Z.H.; Dey, S.; Kawsar, S.M.A. Monosaccharide derivatives: Synthesis, antimicrobial, pass, antiviral and molecular docking studies against SARS-COV-2 Mpro inhibitors. Cellulose Chem. Technol. 2021, 55, 477–499. [Google Scholar] [CrossRef]
- Okella, H.; Okello, E.; Mtewa, A.G.; Ikiriza, H.; Kaggwa, B.; Aber, J.; Ndekezi, C.; Nkamwesiga, J.; Ajayi, C.O.; Mugeni, I.M.; et al. ADMET Profiling and Molecular Docking of Potential Antimicrobial Peptides Previously Isolated from African Catfish, Clarias Gariepinus. Front. Mol. Biosci. 2022, 9, 1039286. [Google Scholar] [CrossRef]
- George, O.A. LipinskiFilters: Computes and Visualize Lipinski’s Parameters, version 1.0.1. 2024.
- Halford, B. Wrestling with the Rule of 5. C&EN Global Enterp. 2023, 101, 16–19. [Google Scholar] [CrossRef]
- Young, R.J. Today’s Drug Discovery and the Shadow of the Rule of 5. Expert Opin. Drug Discov. 2023, 18, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to Optimize Clearance and Oral Absorption. Bioorganic Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef] [PubMed]
- Mazri, R.; Ouassaf, M.; Kerassa, A.; Alhatlani, B.Y. Exploring Potential Therapeutics: Targeting Dengue Virus NS5 through Molecular Docking, ADMET Profiling, and DFT Analysis. Chem. Phys. Impact 2024, 8, 100468. [Google Scholar] [CrossRef]
- Half Life—StatPearls—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554498/ (accessed on 29 October 2024).
- Amorim, A.M.B.; Piochi, L.F.; Gaspar, A.T.; Preto, A.J.; Rosário-Ferreira, N.; Moreira, I.S. Advancing Drug Safety in Drug Development: Bridging Computational Predictions for Enhanced Toxicity Prediction. Chem. Res. Toxicol. 2024, 37, 827. [Google Scholar] [CrossRef]
- Swanson, J.M.J.; Henchman, R.H.; McCammon, J.A. Revisiting Free Energy Calculations: A Theoretical Connection to MM/PBSA and Direct Calculation of the Association Free Energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef]
- Berkmen, Y.M.; Lande, A. Chest Roentgenography as a Window to the Diagnosis of Takayasu’s Arteritis. Am. J. Roentgenol. 1975, 125, 842–846. [Google Scholar] [CrossRef]
- Saxena, S.; Chaudhaery, S.S.; Varshney, K.; Saxena, A.K. Pharmacophore-Based Virtual Screening and Docking Studies on Hsp90 Inhibitors. SAR QSAR Environ. Res. 2010, 21, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sharma, M.; Faisal, M.; Alatar, A.A.; Kumar, R.; Ahmad, S.; Akhtar, S. Ligand-Based Pharmacophore Modeling, Molecular Docking and Simulation Studies for the Exploration of Natural Potent Antiangiogenic Inhibitors Targeting Heat Shock Protein 90. Lett. Drug Des. Discov. 2023, 20, 95–109. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The Influence of Lipophilicity in Drug Discovery and Design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CID | SP (kcal/mol) | XP (kcal/mol) |
|---|---|---|---|
| L1 | CID69438556 | −7.910 | −9.795 |
| L2 | CID69441313 | −9.373 | −9.636 |
| L3 | CID117208203 | −8.687 | −8.687 |
| L4 | CID52948538 | −8.383 | −8.383 |
| L5 | CID117209048 | −8.262 | −8.264 |
| MEY | CID 50925477 | −7.225 | −7.946 |
| Comp. | L1 | L2 | L3 | L4 | L5 | MEY |
|---|---|---|---|---|---|---|
| MW | 360.150 | 350.160 | 308.080 | 348.070 | 254.110 | 495.070 |
| Volume | 376.842 | 364.819 | 281.648 | 293.697 | 263.446 | 459.902 |
| n-Rot | 5 | 3 | 3 | 3 | 2 | 5 |
| nRing | 4 | 4 | 3 | 3 | 3 | 5 |
| nHet | 5 | 5 | 7 | 9 | 4 | 10 |
| Flexibility | 0.217 | 0.125 | 0.176 | 0.188 | 0.118 | 0.167 |
| TPSA | 78.250 | 78.250 | 58.020 | 35.500 | 58.020 | 124.420 |
| logS | −3.958 | −3.931 | −3.971 | −5.746 | −3.045 | −4.776 |
| logP | 4.422 | 5.019 | 3.464 | 4.655 | 2.836 | 4.562 |
| logD | 3.937 | 4.146 | 3.674 | 4.138 | 2.963 | 3.492 |
| Comp. | L1 | L2 | L3 | L4 | L5 | MEY |
|---|---|---|---|---|---|---|
| Absorption | ||||||
| Caco-2 Permeability | −5.108 | −5.030 | −4.849 | −4.989 | −4.815 | −5.856 |
| MDCK Permeability | 1.8 × 10−5 | 2.3 × 10−5 | 1.2 × 10−5 | 9.2 × 10−6 | 2.7 × 10−5 | 1.3 × 10−5 |
| Pgp-inhibitor | poor | excellent | excellent | medium | excellent | medium |
| HIA | excellent | excellent | excellent | excellent | excellent | medium |
| F20% | poor | poor | excellent | excellent | poor | excellent |
| Distribution | ||||||
| PPB | 97.989% | 98.221% | 95.037% | 98.178% | 95.329% | 98.622% |
| VD L/Kg | 0.501 | 0.196 | 0.781 | 4.954 | 0.424 | 0.429 |
| BBB Penetration | excellent | excellent | medium | excellent | excellent | excellent |
| System | HSP90_L1 | HSP90_L1 | HSP90_MEY |
|---|---|---|---|
| RMSD (nm) | 0.219 | 0.288 | 0.413 |
| RMSF (nm) | 0.164 | 0.175 | 0.164 |
| Rg (nm) | 1.724 | 1.739 | 1.707 |
| SASA (nm2) | 112.311 | 113.561 | 111.869 |
| HSP90_L1 | HSP90_L2 | HSP90_MEY | |
|---|---|---|---|
| ∆EVDW | −39.99 | −35.12 | −37.74 |
| ∆EEEL | −53.49 | −41.46 | −40.74 |
| ∆EPB | 63.30 | 48.18 | 51.44 |
| ΔENPOLAR | −4.14 | −3.81 | −3.93 |
| ΔGGAS | −93.49 | −76.58 | −78.48 |
| ΔGSOLV | 59.16 | 44.37 | 47.51 |
| ΔTOTAL | −34.33 | −32.21 | −30.97 |
| PubChem CID | Molecular Formula | Structure | IUPAC Name | |
|---|---|---|---|---|
| L1 | 69438556 | C22H20N2O3 | 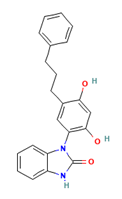 | 3-[2,4-dihydroxy-5-(3-phenylpropyl)phenyl]-1H-benzimidazol-2-one |
| L2 | 69441313 | C21H22N2O3 |  | 3-[5-[(E)-2-cyclohexylethenyl]-2,4-dihydroxyphenyl]-1H-benzimidazol-2-one |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazri, R.; Bourougaa, L.; Zekri, A.; Ouassaf, M.; Alhatlani, B.Y. Discovery of N-Aryl-Benzimidazolone Analogs as Novel Potential HSP90 Inhibitors: A Computational Approach. Appl. Sci. 2024, 14, 10817. https://doi.org/10.3390/app142310817
Mazri R, Bourougaa L, Zekri A, Ouassaf M, Alhatlani BY. Discovery of N-Aryl-Benzimidazolone Analogs as Novel Potential HSP90 Inhibitors: A Computational Approach. Applied Sciences. 2024; 14(23):10817. https://doi.org/10.3390/app142310817
Chicago/Turabian StyleMazri, Radhia, Lotfi Bourougaa, Afaf Zekri, Mebarka Ouassaf, and Bader Y. Alhatlani. 2024. "Discovery of N-Aryl-Benzimidazolone Analogs as Novel Potential HSP90 Inhibitors: A Computational Approach" Applied Sciences 14, no. 23: 10817. https://doi.org/10.3390/app142310817
APA StyleMazri, R., Bourougaa, L., Zekri, A., Ouassaf, M., & Alhatlani, B. Y. (2024). Discovery of N-Aryl-Benzimidazolone Analogs as Novel Potential HSP90 Inhibitors: A Computational Approach. Applied Sciences, 14(23), 10817. https://doi.org/10.3390/app142310817