
Coaxial Fibres Incorporated with Phase Change Materials for Thermoregulation Applications

 ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Recycled Cellulose Acetate (CAt)
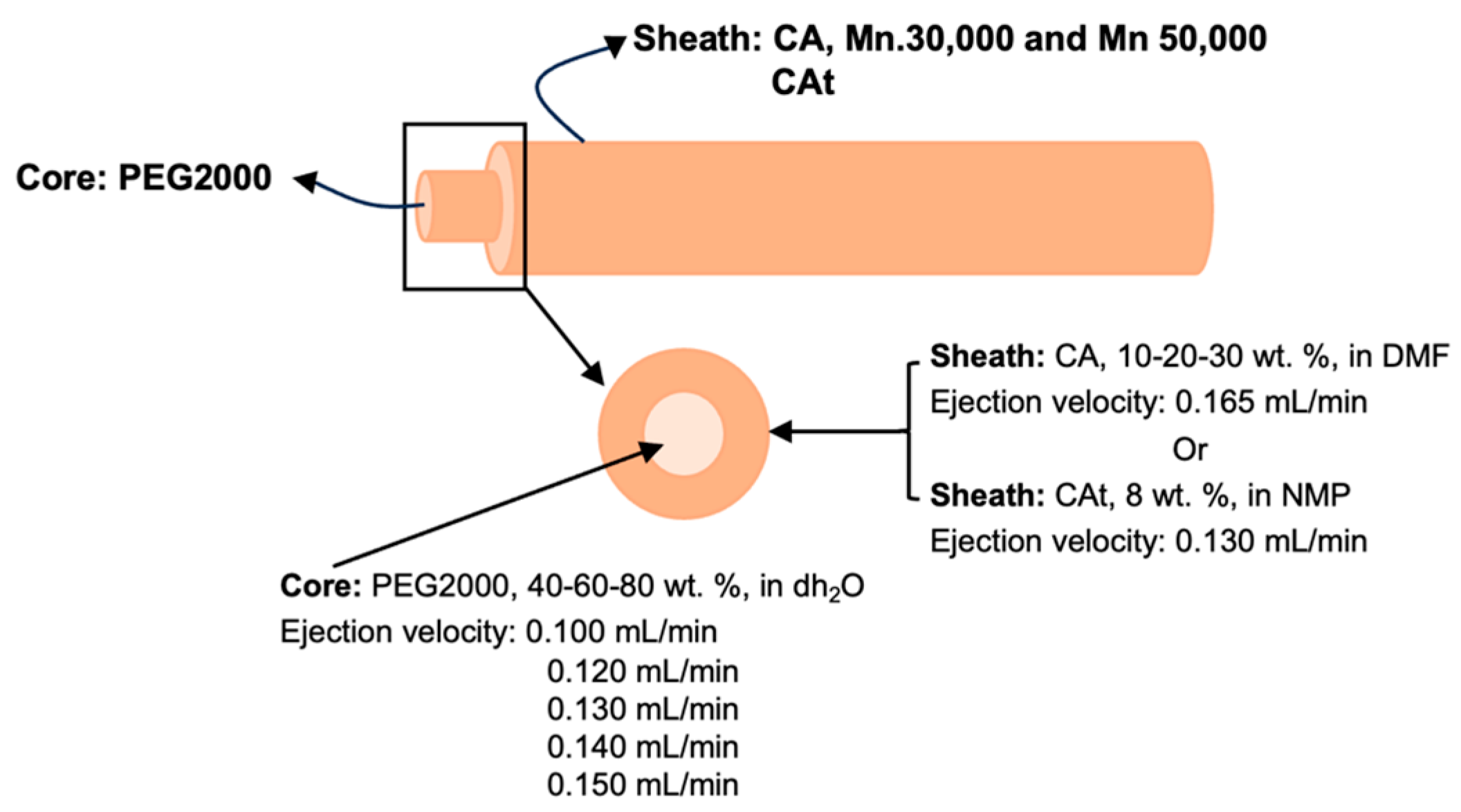
2.3. Fibres’ Production
2.4. Fibres’ Physical, Chemical, Thermal, and Mechanical Characterization
2.4.1. Bright-Field Microscopy
2.4.2. Scanning Electron Microscopy (SEM)
2.4.3. Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.4.4. Thermogravimetric Analysis (TGA)
2.4.5. Differential Scanning Calorimetry (DSC)
2.4.6. Mechanical Performance–Dynamometer
3. Results
3.1. Degree of Substitution for CAt
3.2. Fibres Production
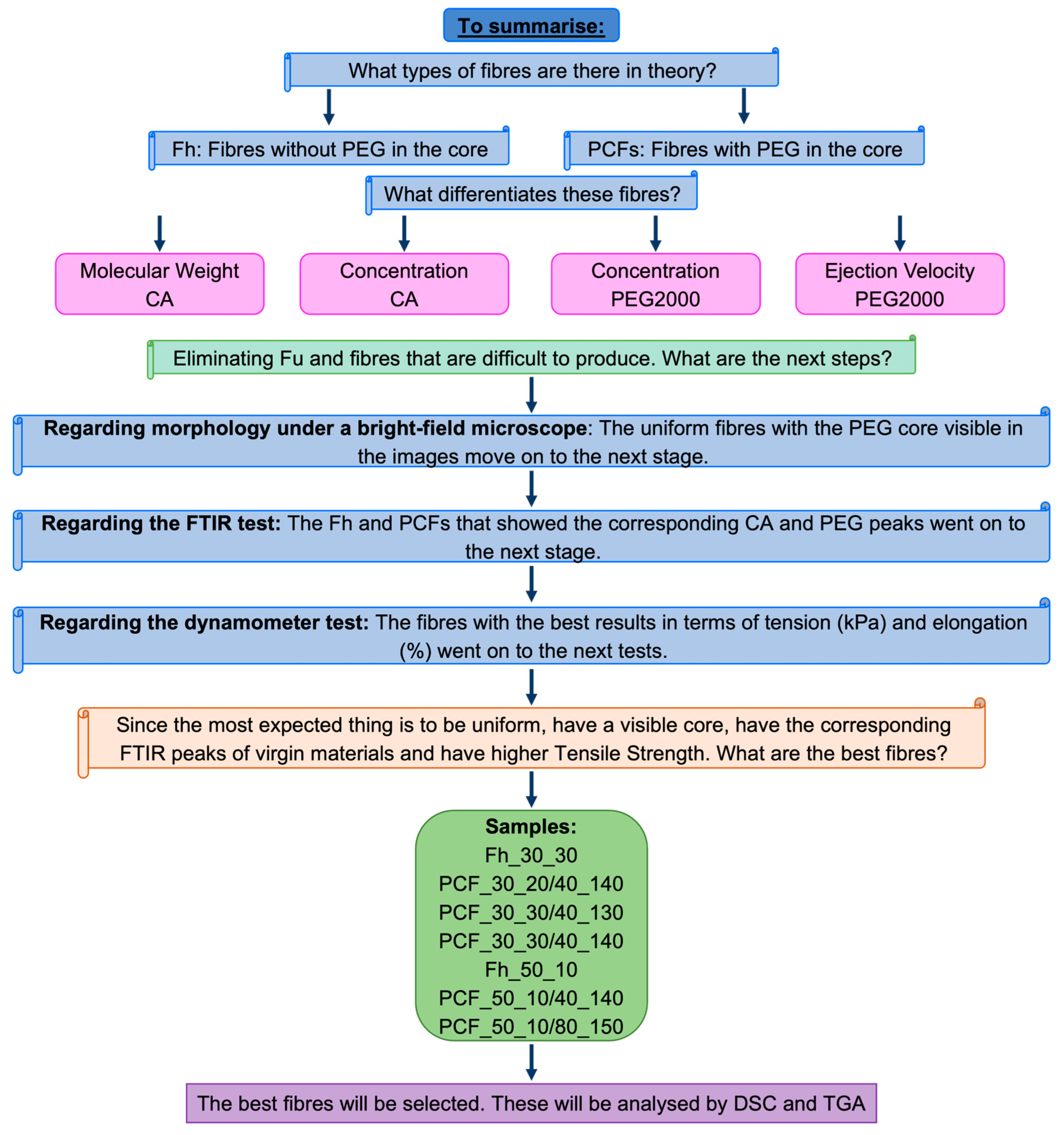
3.3. Sample Selection
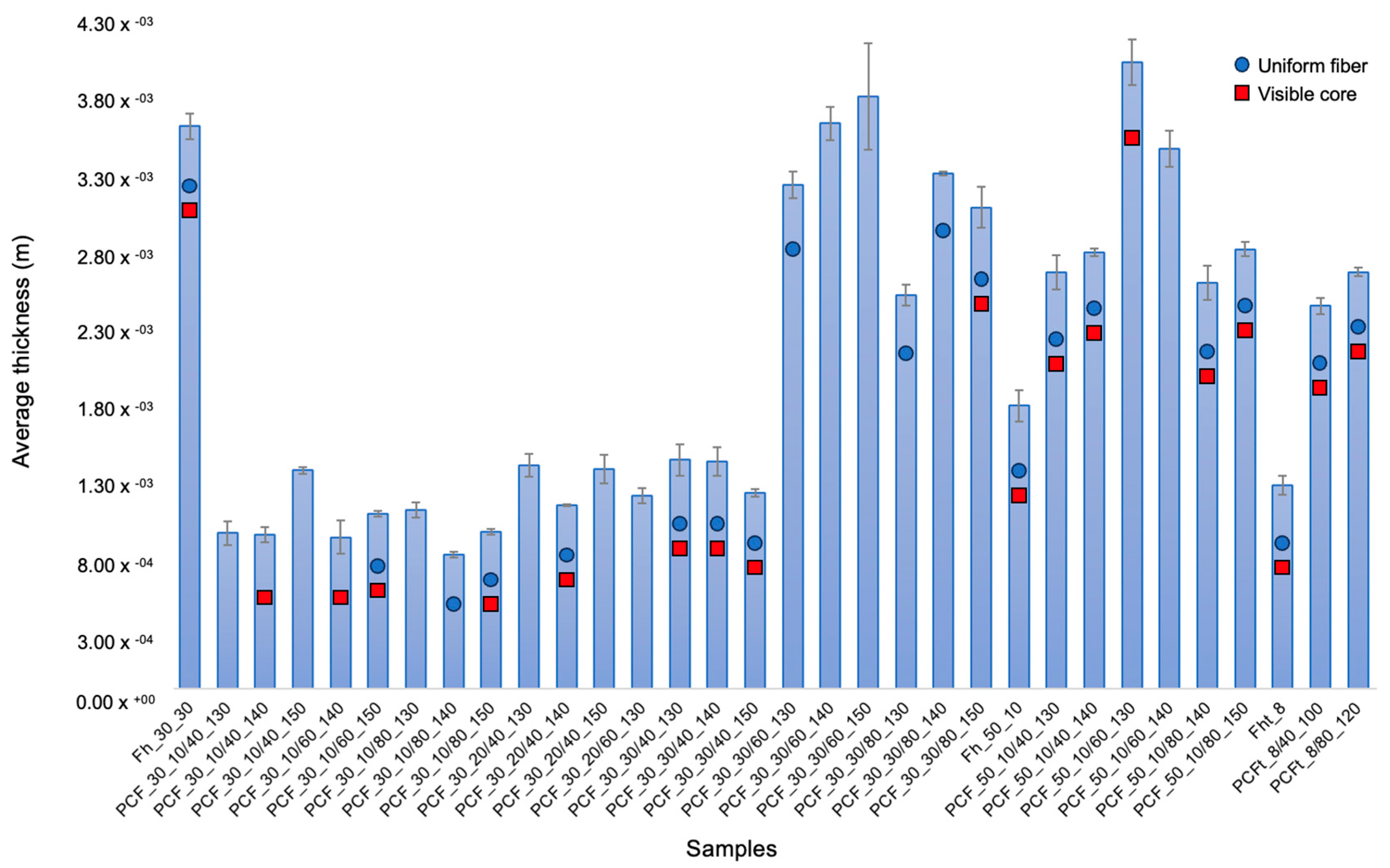
3.4. Morphology
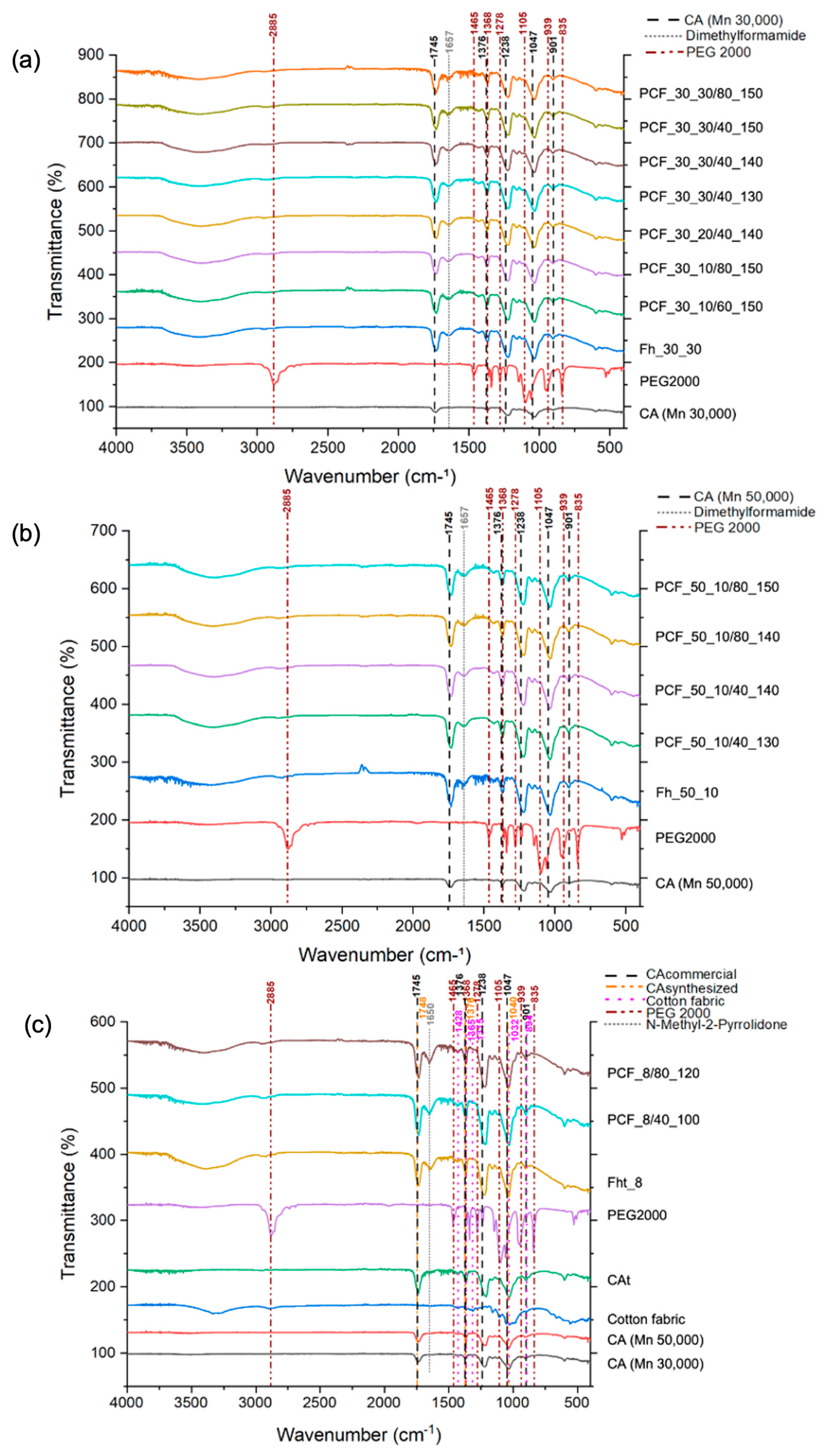
3.5. Chemical Composition (ATR-FTIR)
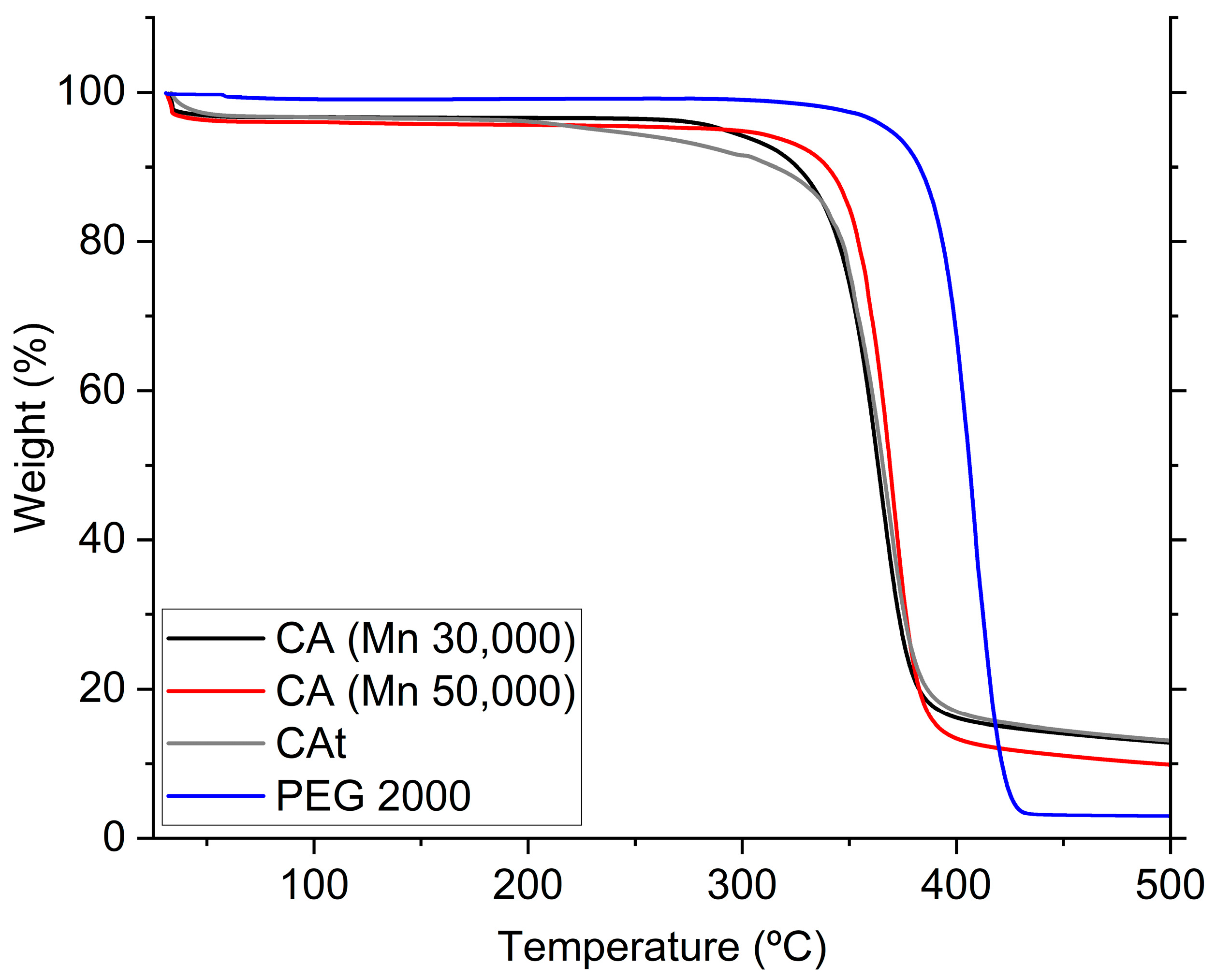
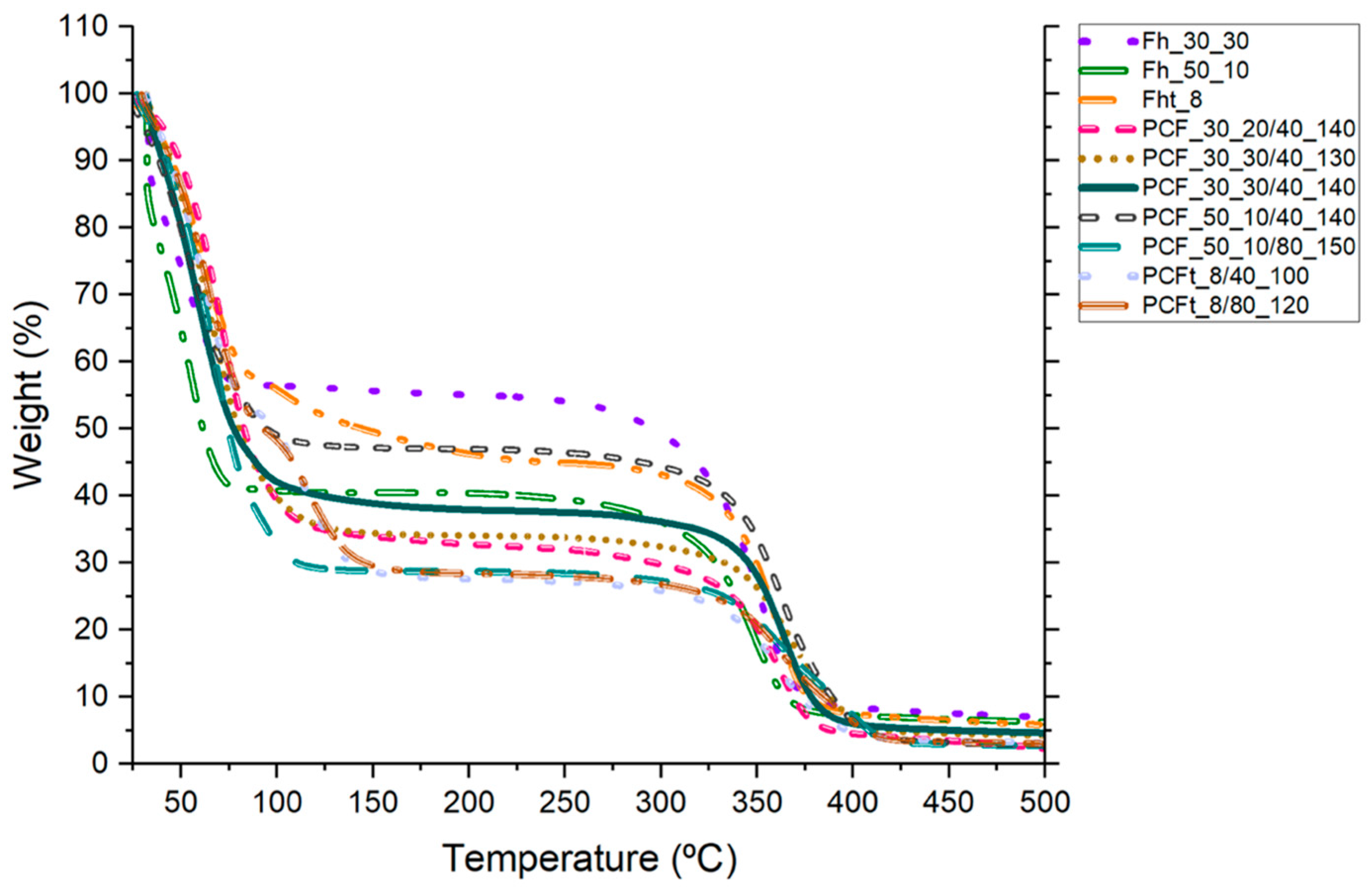
3.6. Thermogravimetric Analysis (TGA)
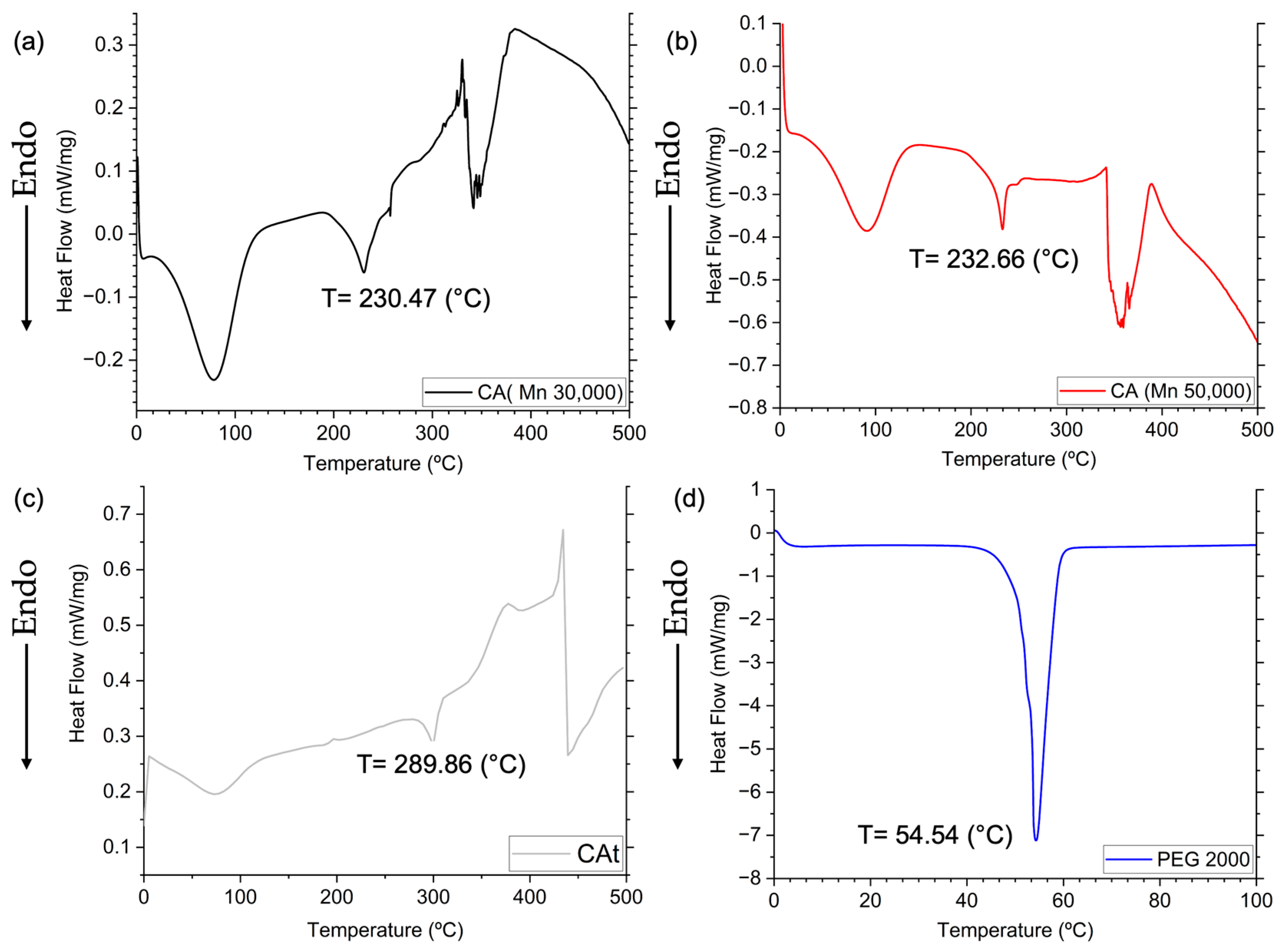
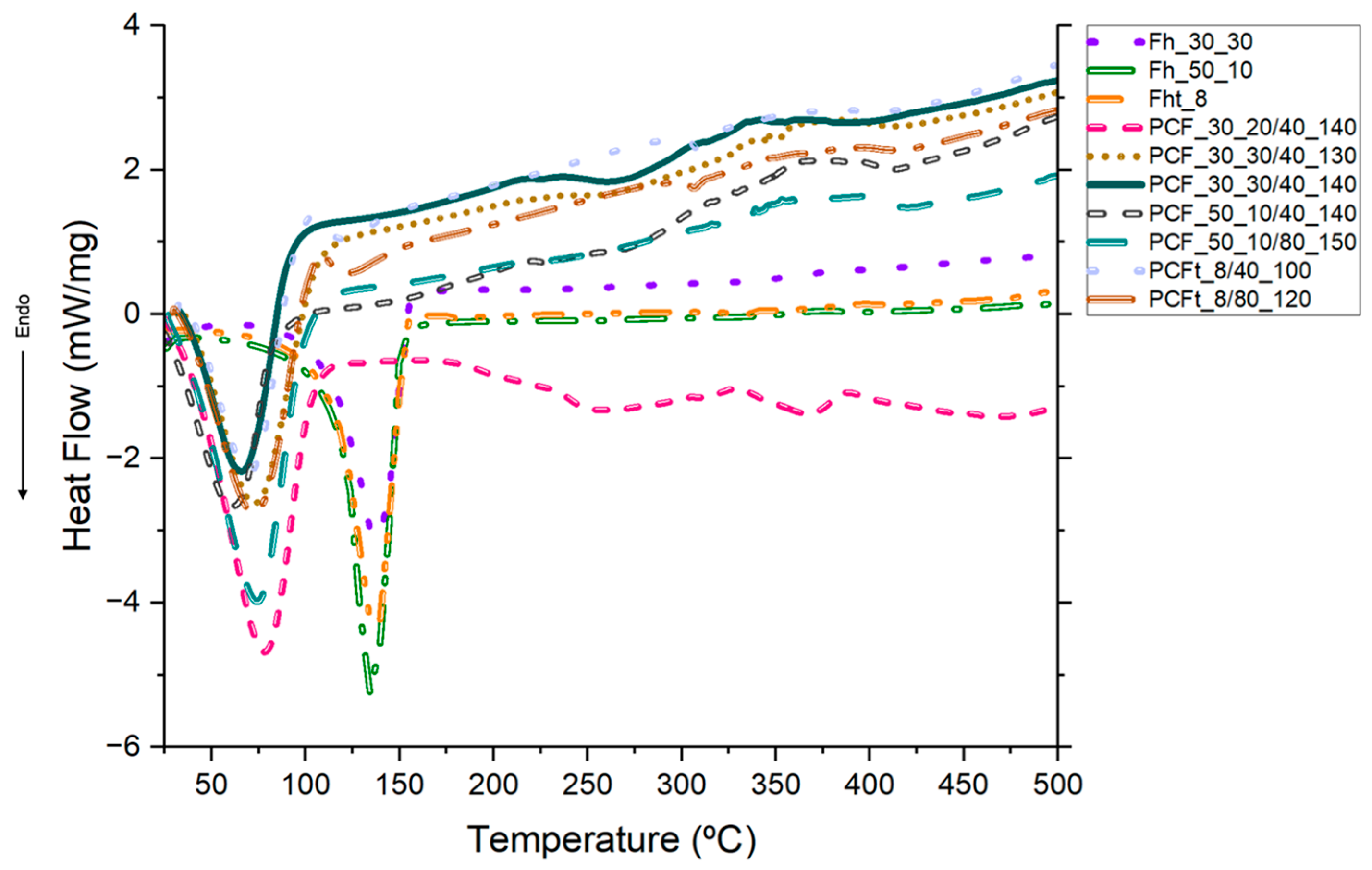
3.7. Differential Scanning Calorimetry (DSC)
3.8. Mechanical Behavior
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Segundo, I.R.; Freitas, E.; Branco, V.T.F.C.; Landi, S.; Costa, M.F.; Carneiro, J.O. Review and Analysis of Advances in Functionalized, Smart, and Multifunctional Asphalt Mixtures. Renew. Sustain. Energy Rev. 2021, 151, 111552. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Ibrahim, N.I.A.; Wahid, J.; Goh, N.A.; Koesmeri, D.R.A.; Nawi, M.N.M. The Impact of Road Pavement on Urban Heat Island (UHI) Phenomenon. Int. J. Technol. 2018, 9, 1597. [Google Scholar] [CrossRef]
- Piracha, A.; Chaudhary, M.T. Urban Air Pollution, Urban Heat Island and Human Health: A Review of the Literature. Sustainability 2022, 14, 9234. [Google Scholar] [CrossRef]
- Chen, G.; Chen, Y.; Tan, X.; Zhao, L.; Cai, Y.; Li, L. Assessing the Urban Heat Island Effect of Different Local Climate Zones in Guangzhou, China. Build. Environ. 2023, 244, 110770. [Google Scholar] [CrossRef]
- Andrade, C.; Fonseca, A.; Santos, J.A. Climate Change Trends for the Urban Heat Island Intensities in Two Major Portuguese Cities. Sustainability 2023, 15, 3970. [Google Scholar] [CrossRef]
- Vardhu, V.A.K.; Sharma, A. Classification, Mitigations and Methods to Detect UHI: A Review. Int. J. Sci. Res. Eng. Manag. 2023, 7, 2–3. [Google Scholar] [CrossRef]
- Mondal, S. Phase Change Fibers. In Handbook of Fibrous Materials; Hu, J., Kumar, B., Lu, J., Eds.; Wiley: Hoboken, NJ, USA, 2020; pp. 263–279. ISBN 978-3-527-34220-4. [Google Scholar]
- Betancourt-Jimenez, D.; Montoya, M.; Haddock, J.; Youngblood, J.P.; Martinez, C.J. Regulating Asphalt Pavement Temperature Using Microencapsulated Phase Change Materials (PCMs). Constr. Build. Mater. 2022, 350, 128924. [Google Scholar] [CrossRef]
- Chou, H.-M.; Chen, C.-R.; Nguyen, V.-L. A New Design of Metal-Sheet Cool Roof Using PCM. Energy Build. 2013, 57, 42–50. [Google Scholar] [CrossRef]
- Osterman, E.; Tyagi, V.V.; Butala, V.; Rahim, N.A.; Stritih, U. Review of PCM Based Cooling Technologies for Buildings. Energy Build. 2012, 49, 37–49. [Google Scholar] [CrossRef]
- Pinheiro, C.; Hammes, N.; Lima, O.; Landi, S.; Homem, N.; Rocha Segundo, I.; Felgueiras, H.P.; Freitas, E.; Costa, M.F.M.; Carneiro, J. Reducing the Effects of Low Albedo of Asphalt Materials Incorporating Polyethylene Glycol (PEG) 1000, 2000 and 4000 as Phase Change Materials (PCM). EPJ Web Conf. 2023, 287, 09024. [Google Scholar] [CrossRef]
- Pinheiro, C.; Landi, S.; Lima, O.; Ribas, L.; Hammes, N.; Segundo, I.R.; Homem, N.C.; Castelo Branco, V.; Freitas, E.; Costa, M.F.; et al. Advancements in Phase Change Materials in Asphalt Pavements for Mitigation of Urban Heat Island Effect: Bibliometric Analysis and Systematic Review. Sensors 2023, 23, 7741. [Google Scholar] [CrossRef]
- Baetens, R.; Jelle, B.P.; Gustavsen, A. Phase Change Materials for Building Applications: A State-of-the-Art Review. Energy Build. 2010, 42, 1361–1368. [Google Scholar] [CrossRef]
- Diaconu, B.M.; Varga, S.; Oliveira, A.C. Experimental Assessment of Heat Storage Properties and Heat Transfer Characteristics of a Phase Change Material Slurry for Air Conditioning Applications. Appl. Energy 2010, 87, 620–628. [Google Scholar] [CrossRef]
- Gil, A.; Oró, E.; Peiró, G.; Álvarez, S.; Cabeza, L.F. Material Selection and Testing for Thermal Energy Storage in Solar Cooling. Renew. Energy 2013, 57, 366–371. [Google Scholar] [CrossRef]
- Yinfei, D.; Pusheng, L.; Jiacheng, W.; Hancheng, D.; Hao, W.; Yingtao, L. Effect of Lightweight Aggregate Gradation on Latent Heat Storage Capacity of Asphalt Mixture for Cooling Asphalt Pavement. Constr. Build. Mater. 2020, 250, 118849. [Google Scholar] [CrossRef]
- Cunha, S.; Leite, P.; Aguiar, J. Characterization of Innovative Mortars with Direct Incorporation of Phase Change Materials. J. Energy Storage 2020, 30, 101439. [Google Scholar] [CrossRef]
- Kulkarni, P.; Muthadhi, A. Thermal Energy Storage Cement Mortar with Direct Incorporation of Organic and Inorganic Phase Change Materials. Innov. Infrastruct. Solut. 2021, 6, 30. [Google Scholar] [CrossRef]
- Pérez-Silva, I.; Ibarra, I.S.; Castañeda-Ovando, A.; Galán-Vidal, C.A.; Páez-Hernández, M.E. Development of Cellulose Acetate Microcapsules with Cyanex 923 for Phenol Removal from Aqueous Media. J. Chem. 2018, 2018, 9506489. [Google Scholar] [CrossRef]
- Gbekou, F.K.; Benzarti, K.; Boudenne, A.; Eddhahak, A.; Duc, M. Mechanical and Thermophysical Properties of Cement Mortars Including Bio-Based Microencapsulated Phase Change Materials. Constr. Build. Mater. 2022, 352, 129056. [Google Scholar] [CrossRef]
- Drissi, S.; Ling, T.-C.; Mo, K.H. Development of Leak-Free Phase Change Material Aggregates. Constr. Build. Mater. 2020, 230, 117029. [Google Scholar] [CrossRef]
- Rao, V.V.; Parameshwaran, R.; Ram, V.V. PCM-Mortar Based Construction Materials for Energy Efficient Buildings: A Review on Research Trends. Energy Build. 2018, 158, 95–122. [Google Scholar] [CrossRef]
- Abdullah, M.F.; Andriyana, A.; Muhamad, F.; Ang, B.C. Effect of Core-to-Shell Flowrate Ratio on Morphology, Crystallinity, Mechanical Properties and Wettability of Poly(Lactic Acid) Fibers Prepared via Modified Coaxial Electrospinning. Polymer 2021, 237, 124378. [Google Scholar] [CrossRef]
- Felgueiras, H.P.; Homem, N.C.; Teixeira, M.A.; Ribeiro, A.R.M.; Antunes, J.C.; Amorim, M.T.P. Physical, Thermal, and Antibacterial Effects of Active Essential Oils with Potential for Biomedical Applications Loaded onto Cellulose Acetate/Polycaprolactone Wet-Spun Microfibers. Biomolecules 2020, 10, 1129. [Google Scholar] [CrossRef]
- Ozipek, B.; Karakas, H. Wet Spinning of Synthetic Polymer Fibers. In Advances in Filament Yarn Spinning of Textiles and Polymers; Elsevier: Amsterdam, The Netherlands, 2014; pp. 174–186. [Google Scholar]
- Rohani Shirvan, A.; Nouri, A.; Sutti, A. A Perspective on the Wet Spinning Process and Its Advancements in Biomedical Sciences. Eur. Polym. J. 2022, 181, 111681. [Google Scholar] [CrossRef]
- Tang, Z.; Jia, S.; Wang, F.; Bian, C.; Chen, Y.; Wang, Y.; Li, B. Highly Stretchable Core–Sheath Fibers via Wet-Spinning for Wearable Strain Sensors. ACS Appl. Mater. Interfaces 2018, 10, 6624–6635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, S.; Zhang, C.; Li, C.; Xu, J.; Xia, L.; Liu, X.; Xu, W. Fabrication of Leather-like Yarns Using Waste Leather for Textile Application. Prog. Org. Coat. 2024, 186, 108053. [Google Scholar] [CrossRef]
- Quan, Q.; Zhang, Y.; Piao, H.; Zhang, H.; Zhao, J. Polybutyrolactam (PBY) Fiber: A Promising Biobased and Biodegradable Fiber Fabricated by Dry-Jet-Wet Spinning. Polymer 2022, 260, 125392. [Google Scholar] [CrossRef]
- Teo, W.E.; Ramakrishna, S. A Review on Electrospinning Design and Nanofibre Assemblies. Nanotechnology 2006, 17, R89–R106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Xu, Y.; Xu, Y.; Qiao, J.; Shi, T.; Huang, Z.; Liu, Y.; Fang, M.; Min, X.; et al. Flexible Polyethylene Glycol/Polyvinylpyrrolidone Composite Phase Change Fibres: Preparation, Characterization, and Thermal Conductivity Enhancement. Polymer 2021, 214, 123258. [Google Scholar] [CrossRef]
- Mirabedini, A. Developing Novel Spinning Methods to Fabricate Continuous Multifunctional Fibres for Bioapplications; Springer Theses; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-95377-9. [Google Scholar]
- Chen, C.; Zhao, Y.; Liu, W. Electrospun Polyethylene Glycol/Cellulose Acetate Phase Change Fibers with Core–Sheath Structure for Thermal Energy Storage. Renew. Energy 2013, 60, 222–225. [Google Scholar] [CrossRef]
- Swapnil, S.I.; Datta, N.; Mahmud, M.M.; Jahan, R.A.; Arafat, M.T. Morphology, Mechanical, and Physical Properties of Wet-spun Cellulose Acetate Fiber in Different Solvent-coagulant Systems and In-situ Crosslinked Environment. J. Appl. Polym. Sci. 2021, 138, 50358. [Google Scholar] [CrossRef]
- Santos-Sauceda, I.; Castillo-Ortega, M.M.; Del Castillo-Castro, T.; Armenta-Villegas, L.; Ramírez-Bon, R. Electrospun Cellulose Acetate Fibers for the Photodecolorization of Methylene Blue Solutions under Natural Sunlight. Polym. Bull. 2021, 78, 4419–4438. [Google Scholar] [CrossRef]
- Babapoor, A.; Karimi, G.; Golestaneh, S.I.; Mezjin, M.A. Coaxial Electro-Spun PEG/PA6 Composite Fibers: Fabrication and Characterization. Appl. Therm. Eng. 2017, 118, 398–407. [Google Scholar] [CrossRef]
- Homem, N.C.; Amorim, M.T.P. Synthesis of Cellulose Acetate Using as Raw Material Textile Wastes. Mater. Today Proc. 2020, 31, S315–S317. [Google Scholar] [CrossRef]
- Puleo, A.C.; Paul, D.R.; Kelley, S.S. The Effect of Degree of Acetylation on Gas Sorption and Transport Behavior in Cellulose Acetate. J. Membr. Sci. 1989, 47, 301–332. [Google Scholar] [CrossRef]
- Homem, N.C.; Tavares, T.D.; Miranda, C.S.; Antunes, J.C.; Amorim, M.T.P.; Felgueiras, H.P. Functionalization of Crosslinked Sodium Alginate/Gelatin Wet-Spun Porous Fibers with Nisin Z for the Inhibition of Staphylococcus Aureus-Induced Infections. Int. J. Mol. Sci. 2021, 22, 1930. [Google Scholar] [CrossRef] [PubMed]
- ISO2060:2009; Textiles—Yarns from Packages Determination of Single-End Breaking Force and Elongation at Break Using Constant Rate of Extension (CRE) Tester. Technical Committee ISO/TC 97: Geneva, Switzerland, 2009.
- Kramar, A.; González-Benito, F.J. Cellulose-Based Nanofibers Processing Techniques and Methods Based on Bottom-Up Approach—A Review. Polymers 2022, 14, 286. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gong, D.; Bratasz, L.; Zhu, Z.; Wang, C. Degradation Markers and Plasticizer Loss of Cellulose Acetate Films during Ageing. Polym. Degrad. Stab. 2019, 168, 108952. [Google Scholar] [CrossRef]
- Ahn, Y.-H.; DeWitt, S.J.A.; McGuire, S.; Lively, R.P. Incorporation of Phase Change Materials into Fibers for Sustainable Thermal Energy Storage. Ind. Eng. Chem. Res. 2021, 60, 3374–3384. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Fahmy, T.Y.; Salaheldin, E.I.; Mobarak, F.; Youssef, M.A.; Mabrook, M.R. Role of Tosyl Cellulose Acetate as Potential Carrier for Controlled Drug Release. Life Sci. J. 2015, 10, 127–133. [Google Scholar]
- Nikoomanesh, N.; Zandi, M.; Ganjloo, A. Development of Eco-Friendly Cellulose Acetate Films Incorporated with Burdock (Arctium Lappa L.) Root Extract. Prog. Org. Coat. 2024, 186, 108009. [Google Scholar] [CrossRef]
- Fei, P.; Liao, L.; Cheng, B.; Song, J. Quantitative Analysis of Cellulose Acetate with a High Degree of Substitution by FTIR and Its Application. Anal. Methods 2017, 9, 6194–6201. [Google Scholar] [CrossRef]
- Frisoni, G.; Baiardo, M.; Scandola, M.; Lednická, D.; Cnockaert, M.C.; Mergaert, J.; Swings, J. Natural Cellulose Fibers: Heterogeneous Acetylation Kinetics and Biodegradation Behavior. Biomacromolecules 2001, 2, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Huda, E.; Rahmi; Khairan. Preparation and Characterization of Cellulose Acetate from Cotton. IOP Conf. Ser. Earth Environ. Sci. 2019, 364, 012021. [Google Scholar] [CrossRef]
- Zhuang, J.; Li, M.; Pu, Y.; Ragauskas, A.; Yoo, C. Observation of Potential Contaminants in Processed Biomass Using Fourier Transform Infrared Spectroscopy. Appl. Sci. 2020, 10, 4345. [Google Scholar] [CrossRef]
- Zhu, Q.; Wu, H.; Ma, Z.; Liu, Y.; Li, J.; Zhu, L.; Zhang, X.; Wang, C.; Chen, D.; Zhu, D. Micro-Volume Blood Separation Membrane for In-Situ Biosensing. Biosensors 2022, 12, 712. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Li, S.; Wang, Z.; Yan, X.; Guo, Z.; Shao, L. A Bio-Inspired CO2-Philic Network Membrane for Enhanced Sustainable Gas Separation. J. Mater. Chem. A 2015, 3, 13758–13766. [Google Scholar] [CrossRef]
- Snyder, R.G.; Hsut, S.L.; Krimm, S. Vibrational Spectp in the C-H Stretching Region and the Structure of the Polymethylene Chain. Spectrochim. Acta Part A Mol. Spectrosc. 1978, 34, 395–406. [Google Scholar] [CrossRef]
- Hospodarova, V.; Singovszka, E.; Stevulova, N. Characterization of Cellulosic Fibers by FTIR Spectroscopy for Their Further Implementation to Building Materials. Am. J. Anal. Chem. 2018, 9, 303–310. [Google Scholar] [CrossRef]
- Das, A.M.; Ali, A.A.; Hazarika, M.P. Synthesis and Characterization of Cellulose Acetate from Rice Husk: Eco-Friendly Condition. Carbohydr. Polym. 2014, 112, 342–349. [Google Scholar] [CrossRef]
- Chen, J.; Xu, J.; Wang, K.; Cao, X.; Sun, R. Cellulose Acetate Fibers Prepared from Different Raw Materials with Rapid Synthesis Method. Carbohydr. Polym. 2016, 137, 685–692. [Google Scholar] [CrossRef]
- Lopes, S.; Bueno, L.; Aguiar Júnior, F.D.; Finkler, C. Preparation and Characterization of Alginate and Gelatin Microcapsules Containing Lactobacillus Rhamnosus. An. Acad. Bras. Ciênc. 2017, 89, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ji, T.; Niu, D.; Yang, Z. Investigation of PEG/Mixed Metal Oxides as a New Form-Stable Phase Change Material for Thermoregulation and Improved UV Ageing Resistance of Bitumen. RSC Adv. 2020, 10, 44903–44911. [Google Scholar] [CrossRef]
- Sundararajan, S.; Samui, A.B.; Kulkarni, P.S. Shape-Stabilized Poly(Ethylene Glycol) (PEG)-Cellulose Acetate Blend Preparation with Superior PEG Loading via Microwave-Assisted Blending. Sol. Energy 2017, 144, 32–39. [Google Scholar] [CrossRef]
- Faradilla, R.F.; Lee, G.; Sivakumar, P.; Stenzel, M.; Arcot, J. Effect of Polyethylene Glycol (PEG) Molecular Weight and Nanofillers on the Properties of Banana Pseudostem Nanocellulose Films. Carbohydr. Polym. 2019, 205, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, H.M.; Anis, A.; Poulose, A.M.; Al-Zahrani, S.M.; Madhar, N.A.; Alhamidi, A.; Aldeligan, S.H.; Alsubaie, F.S. Synthesis and Characterization of Cellulose Triacetate Obtained from Date Palm (Phoenix Dactylifera L.) Trunk Mesh-Derived Cellulose. Molecules 2022, 27, 1434. [Google Scholar] [CrossRef]
- Ribeiro, S.D.; Meneguin, A.B.; Barud, H.d.S.; Silva, J.M.; Oliveira, R.L.; Asunção, R.M.N.d.; Tormin, T.F.; Muñoz, R.A.A.; Filho, G.R.; Ribeiro, C.A. Synthesis and Characterization of Cellulose Acetate from Cellophane Industry Residues. Application as Acetaminophen Controlled-Release Membranes. J. Therm. Anal. Calorim. 2022, 147, 7265–7275. [Google Scholar] [CrossRef]
- Vinodhini, P.A.; Sangeetha, K.; Thandapani, G.; Sudha, P.N.; Jayachandran, V.; Sukumaran, A. FTIR, XRD and DSC Studies of Nanochitosan, Cellulose Acetate and Polyethylene Glycol Blend Ultrafiltration Membranes. Int. J. Biol. Macromol. 2017, 104, 1721–1729. [Google Scholar] [CrossRef]
- Ansu, A.K.; Pandya, M.; Sharma, R.K.; Tripathi, D. Experimental Investigation of Hybrid PCM Polyethylene Glycol with Al2O3 and CuO Nanoparticles. Res. Sq. 2022; in review. [Google Scholar] [CrossRef]
- Chen, C.; Wang, L.; Huang, Y. Electrospun Phase Change Fibers Based on Polyethylene Glycol/Cellulose Acetate Blends. Appl. Energy 2011, 88, 3133–3139. [Google Scholar] [CrossRef]
- Cai, Y.; Gao, C.; Xu, X.; Fu, Z.; Fei, X.; Zhao, Y.; Chen, Q.; Liu, X.; Wei, Q.; He, G.; et al. Electrospun Ultrafine Composite Fibers Consisting of Lauric Acid and Polyamide 6 as Form-Stable Phase Change Materials for Storage and Retrieval of Solar Thermal Energy. Sol. Energy Mater. Sol. Cells 2012, 103, 53–61. [Google Scholar] [CrossRef]
- Gonçalves, S.M.; Dos Santos, D.C.; Motta, J.F.G.; Santos, R.R.D.; Chávez, D.W.H.; Melo, N.R.D. Structure and Functional Properties of Cellulose Acetate Films Incorporated with Glycerol. Carbohydr. Polym. 2019, 209, 190–197. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | %GA | DS |
|---|---|---|
| CAt | 42.70 | 2.83 |
| CAt1 | 42.74 | 2.83 |
| CAt2 | 42.96 | 2.84 |
| Average | 42.80 ± 0.14 1 | 2.83 ± 0.01 1 |
| Samples | Fibre Type | Mn CA | wt. % CA/CAt | wt. % PEG | Ejection Velocity PEG (mL/min) |
|---|---|---|---|---|---|
| Fu_a_b | Uniaxial | 30,000 and 50,000 | 10, 20 and 30 | - | - |
| Fut_b | Uniaxial | - | 8 | - | - |
| Fh_a_b | Hollow | 30,000 and 50,000 | 10, 20 and 30 | - | - |
| Fht_b | Hollow | - | 8 | - | - |
| PCF_a_b/x_y | Phase Change Fibre | 30,000 and 50,000 | 10, 20 and 30 | 40, 60 and 80 | 0.130, 0.140 and 0.150 |
| PCFt_b/x_y | Phase Change Fibre | - | 8 | 40 and 80 | 0.100 and 0.120 |
| Samples | Peak Temperature (°C) | Melting Point (J/g) |
|---|---|---|
| Fh_30_30 | 139.84 | 111.78 |
| Fh_50_10 | 134.42 | 140.46 |
| Fht_8 | 137.52 | 134.39 |
| PCF_30_20/40_140 | 78.02 | 108.43 |
| PCF_30_30/40_130 | 72.26 | 56.38 |
| PCF_30_30/40_140 | 64.87 | 50.51 |
| PCF_50_10/40_140 | 61.61 | 70.91 |
| PCF_50_10/80_150 | 74.96 | 106.40 |
| PCFt_8/40_100 | 69.83 | 44.97 |
| PCFt_8/80_120 | 72.84 | 41.52 |
| Samples | Maximum Elongation at Break (%) | Breaking Strength (kPa) |
|---|---|---|
| Fh_30_30 | 28.86 ± 1.59 | 133.18 ± 6.57 |
| PCF_30_10/60_150 | 1.59 ± 0.14 | 105.54 ± 14.73 |
| PCF_30_10/80_150 | 2.46 ± 0.30 | 212.83 ± 52.80 |
| PCF_30_20/40_140 | 14.32 ± 0.22 | 1023.64 ± 52.56 |
| PCF_30_30/40_130 | 28.46 ± 1.08 | 1094.60 ± 150.04 |
| PCF_30_30/40_140 | 22.42 ± 0.84 | 1122.49 ± 145.56 |
| PCF_30_30/40_150 | 13.07 ± 0.93 | 920.01 ± 104.02 |
| PCF_30_30/80_150 | 25.40 ± 0.63 | 264.68 ± 22.18 |
| Fh_50_10 | 16.50 ± 0.95 | 117.28 ± 30.03 |
| PCF_50_10/40_130 | 11.71 ± 0.60 | 60.28 ± 5.86 |
| PCF_50_10/40_140 | 16.20 ± 0.99 | 100.63 ± 13.22 |
| PCF_50_10/80_140 | 12.98 ± 0.58 | 75.03 ± 17.68 |
| PCF_50_10/80_150 | 21.62 ± 0.42 | 105.69 ± 15.68 |
| Fht_8 | 4.00 ± 0.71 | 61.36 ± 18.46 |
| PCFt_8/40_100 | 7.18 ± 0.62 | 28.06 ± 14.20 |
| PCFt_8/80_120 | 9.02 ± 0.66 | 39.75 ± 9.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammes, N.; Pinheiro, C.; Segundo, I.R.; Homem, N.C.; Silva, M.M.; Felgueiras, H.P.; Soares, G.M.B.; Freitas, E.; Costa, M.F.M.; Carneiro, J.A.O. Coaxial Fibres Incorporated with Phase Change Materials for Thermoregulation Applications. Appl. Sci. 2024, 14, 2473. https://doi.org/10.3390/app14062473
Hammes N, Pinheiro C, Segundo IR, Homem NC, Silva MM, Felgueiras HP, Soares GMB, Freitas E, Costa MFM, Carneiro JAO. Coaxial Fibres Incorporated with Phase Change Materials for Thermoregulation Applications. Applied Sciences. 2024; 14(6):2473. https://doi.org/10.3390/app14062473
Chicago/Turabian StyleHammes, Nathalia, Claver Pinheiro, Iran Rocha Segundo, Natália Cândido Homem, M. M. Silva, Helena P. Felgueiras, Graça M. B. Soares, Elisabete Freitas, Manuel F. M. Costa, and Joaquim Alexandre O. Carneiro. 2024. "Coaxial Fibres Incorporated with Phase Change Materials for Thermoregulation Applications" Applied Sciences 14, no. 6: 2473. https://doi.org/10.3390/app14062473
APA StyleHammes, N., Pinheiro, C., Segundo, I. R., Homem, N. C., Silva, M. M., Felgueiras, H. P., Soares, G. M. B., Freitas, E., Costa, M. F. M., & Carneiro, J. A. O. (2024). Coaxial Fibres Incorporated with Phase Change Materials for Thermoregulation Applications. Applied Sciences, 14(6), 2473. https://doi.org/10.3390/app14062473