Abstract
Sphingolipidoses are a class of inherited lysosomal storage diseases, characterized by enzymatic deficiencies that impair sphingolipid degradation. This enzymatic malfunction results in the pathological accumulation of sphingolipids within lysosomes, leading to tissue damage across multiple organ systems. Among the various organs involved, the eye and particularly the retina, can be affected and this will be the primary focus of this study. This article will explore the various subtypes of sphingolipidoses, detailing their associated retinal abnormalities, with an emphasis on multimodal imaging findings and clinical recognition of these rare disorders.
1. Introduction
Sphingolipidoses represent a group of inherited metabolic disorders classified under lysosomal storage diseases with an incidence of approximately 1 in 10,000 individuals [1,2]. They are characterized by a pathological accumulation of a class of lipids called sphingolipids in lysosomes, resulting from deficiencies in sphingolipid hydrolases, enzymes responsible for their degradation [1]. The specific enzyme deficiency determines the subtype of sphingolipidosis, of which more than 10 subtypes have been identified, all of which, except for Fabry disease (FD), are inherited in an autosomal recessive manner [1]. These include Niemann–Pick disease types A, B, and C (NDP), Gaucher disease (GD), GM1 gangliosidoses, GM2 gangliosidoses including Sandhoff disease (SD) and Tay–Sachs disease (TSD), Farber disease, metachromatic leukodystrophy (MLD), FD, Krabbe disease (KD), and sialidosis [2].
Due to the accumulation of lipids, cellular death and organ damage occur, causing multiple systemic symptoms [2]. Certain sphingolipidoses are characterized by enzymatic deficits that lead to the pathological accumulation of specific catabolites in targeted organs or cells. For instance, FD primarily affects the liver, while GD predominantly impacts the mononuclear phagocyte system [3].
However, a common feature of nearly all sphingolipidoses is that the primarily affected area is the nervous system [2], as certain sphingolipid subtypes, such as gangliosides, are highly expressed in this tissue. This characteristic underpins the shared clinical manifestations observed across various subgroups of sphingolipid-related disorders, including progressive neurodegenerative conditions and retinal pathologies [3].
Considering the high prevalence of retinal involvement, a meticulous fundus examination is the recommended first-line diagnostic approach for sphingolipidoses, as it can facilitate early detection [4]. Additionally, it should be included as part of the annual follow-up of patients [5].
The most common retinal finding in sphingolipidoses is the appearance of a cherry-red macula, resulting from the buildup of distinct sphingolipid precursors or metabolites within retinal cells. It is observed in GM1 and GM2 gangliosidoses, including TSD and SD, and in NPD type A and B [2,3].
At fundoscopic examination, the cherry-red spot appears as a typically bright red-colored region located at the center of the macula, encircled by an area of retinal opacification [6]. Retinal ganglion cells, where ganglioside accumulation occurs, are highly concentrated in the macula but are absent in its central portion, the fovea. Therefore, the fovea, lacking retinal ganglion cells with opaque sphingolipid deposits, appears bright red in contrast to the surrounding macular opacification, creating the characteristic cherry-red spot [6]. In the ophthalmological setting, this should be differentiated from sight-threatening ischemic conditions, which can present with a similar cherry-red spot in the macula [6]. A schematic representation of a cherry-red spot in the macula is shown in Figure 1.
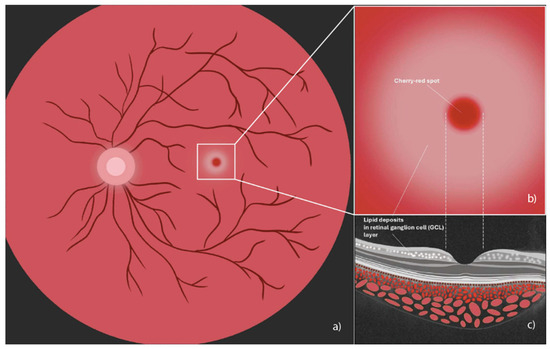
Figure 1.
Illustration of a cherry-red spot in the macula. (a) Fundus image indicating cherry red macular spot (central white box); (b) magnified image of cherry-red spot encircled by an area of retinal opacification; (c) spectral domain optical coherence tomography cross-sectional illustration where the arrow shows accumulation of hyperreflective material, indicating lipid deposits in the ganglion cell layer (graphics courtesy of Francesco Pandolfo).
Color fundus photography is recommended as an essential complementary test, serving to document distinctive ophthalmoscopic features characteristic of each sphingolipidosis, facilitating the monitoring of disease progression, and evaluating modifications in response to systemic therapy [6].
Spectral-domain optical coherence tomography (SD-OCT) is a non-invasive rapid imaging technique used in routine ophthalmologic examination that renders an in vivo simil-histological cross-sectional view of the retina layers [7]. It serves as a critical diagnostic modality for the retinal evaluation of sphingolipidoses, as it can facilitate diagnosis and the monitoring of ocular manifestations [8]. A frequently observed finding in various lipid storage disorders is hyperreflectivity of the retinal nerve fiber layer (RNFL) and the ganglion cell layer (GCL) [6]. Although in cases of suspected lipid storage disorders SD-OCT serves as a fundamental diagnostic tool for detecting retinal involvement, a definitive diagnosis requires the assessment of enzymatic deficiencies and multisystem involvement [6].
OCT angiography (OCTA) is a dye-free technique used to assess the ocular microvasculature, including the macula, choroid, and optic nerve capillaries [9]. This imaging method has been suggested as an additional technique to observe the microvasculature for the diagnostic workup of sphingolipidoses, such as FD, and facilitates the monitoring of disease progression [10]. A characteristic finding is retinal vessel tortuosity, which is observed in FD, as well as in several other sphingolipidoses [11]. The evaluation via OCTA imaging proves to be a rapid, non-invasive, and reproducible diagnostic biomarker [12].
The application of electrophysiological assessments, including the electroretinography (ERG) and visually evoked potentials (VEPs), has been shown to be essential for the diagnosis of sphingolipidoses and serves as a valuable tool in differentiating between these disorders [13]. ERG, which records the electrical response of the retina to light stimuli, enables the identification of functional retinal alterations associated with a wide range of pathologies [14]. In the context of sphingolipidoses, ERG is particularly effective in detecting subclinical functional changes, even in cases where patients remain asymptomatic from a visual standpoint [15]. For instance, in NPD and GD, regions of the retina corresponding to lipid deposition exhibit a significant depression in multifocal ERG (mfERG) responses [16,17]. VEPs are electrophysiological assessments used to evaluate the functional integrity of the optic nerves, visual pathways, and occipital cortex [17]. In demyelinating leukodystrophies, such as KD and MDL, lipid accumulation within the nervous system leads to demyelination, resulting in alterations in VEPs responses [18]. Furthermore, VEPs have been observed to be reduced or pathological in murine models of TSD and SD [19].
This review aims to increase the awareness and knowledge of retinal sphingolipidoses, particularly given their rarity. Given the paucity of the comprehensive literature systematically examining the retinal manifestations of all sphingolipidosis subtypes, we aim to address this gap by offering guidance on the most appropriate ophthalmological multimodal imaging techniques and detailing the expected findings. Furthermore, we highlight the promising role of these techniques in multidisciplinary clinical management, including diagnosis, assessment of disease progression, and evaluation of responses to systemic therapies.
2. Methods
The methodology for this targeted literature review was based on PubMed and MEDLINE. A comprehensive search was conducted for English-language publications from January 2000 to the most recent available data.
The search strategy focused on the diseases of interest (“Niemann–Pick Disease” OR “Gaucher Disease” OR “GM1 Gangliosidosis” OR “GM2 Gangliosidosis” OR “Faber Disease” OR “Tay–Sachs Disease” OR “Sandhoff Disease” OR “Metachromatic Leukodystrophy” OR “Fabry Disease” OR “Krabbe Disease” OR “sialidosis”) to explore their pathogenesis, incidence, prevalence, and systemic manifestations, including those documented in the corresponding tables. For the section addressing retinal involvement, the previously mentioned diseases were combined with ophthalmological terms of interest (“retina” OR “macula” OR “ocular involvement” OR “ophthalmological findings” OR “OCT”). Additionally, searches were conducted to identify diagnostic modalities of interest (“Fundus Photography” OR “Spectral-Domain Optical Coherence Tomography” OR “OCT Angiography” OR “Fundus Autofluorescence” OR “Adaptive Optics” OR “Electroretinography” OR “Visual Evoked Potentials”) from 2000 to the most recent available data.
3. Ophthalmological Examination: A Key Tool in the Diagnosis and Management of Sphingolipidoses
Advancements in ophthalmic imaging have significantly improved the ability to diagnose and monitor retinal diseases. A range of sophisticated instruments now enable in-depth visualization of ocular structures, enhancing both diagnostic accuracy and disease management.
3.1. Fundus Photography
Fundus imaging is a core technique for capturing detailed images of the retina. This is achieved using specialized camera systems that can be integrated into slit lamps or designed as standalone ultra-high-resolution devices. Some of these systems incorporate ultra-widefield technology, allowing for the acquisition of extensive retinal images in a single capture [20].
Fundus photography is instrumental in tracking retinal disease progression by enabling comparative analysis of structural changes over time [20,21]. The integration of artificial intelligence further enhances its capacity for detecting pathological retinal features with increased precision [21]. In sphingolipidoses, fundus photography is useful for follow-up to monitor anatomical retinal changes that may occur during the course of the disease, or to assess potential modifications during pharmacological treatment. The main limitation is that it is not significantly useful in the preclinical phase before ophthalmoscopic retinal pathologic signs appear.
3.2. Spectral-Domain Optical Coherence Tomography
SD-OCT is a noninvasive imaging method that employs low-coherence light to generate cross-sectional images of the retina, providing both qualitative and quantitative insight into its microstructure. With resolutions reaching 3–5 μm in swept-source OCT, this technology is indispensable for assessing retinal and optic nerve pathologies [22]. It has been used to identify biomarkers in neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease in evaluating the RNFL and GCL [23,24,25,26]. This instrumental examination is useful for analyzing retinal alterations related to the accumulation of sphingolipids in the retinal layers, facilitating diagnosis, monitoring disease-related changes over time, and assessing potential therapeutic responses.
SD-OCT has proven particularly useful for evaluating neurodegenerative processes at an early stage. For example, in TSD and SD, thinning of the RNFL and thickening of the macular GCL have been observed, reflecting the neurodegenerative process affecting both the retina and the central nervous system [27]. Similarly in GD, thinning of the peripapillary RNFL and thickening of the macular ganglion cell complex has been described, suggesting the potential role of SD-OCT as a non-invasive tool for the identification of early neurodegeneration biomarkers [28,29].
SD-OCT exhibits limitations in the differential diagnosis of conditions characterized by a cherry-red macula, particularly when multiple pathologies share similar findings [6]. In these instances, a thorough diagnostic approach is essential, and collaboration with a multidisciplinary medical team is recommended to enhance diagnostic accuracy and inform appropriate management strategies [30]. Additional limitations associated with this imaging modality include a decrease in image contrast in the presence of media opacities, such as those induced by cataracts or corneal opacities [31]. Furthermore, difficulty in performing the examination is often encountered in patients with compromised fixation stability, which is prevalent in conditions associated with progressive optic nerve atrophy and subsequent vision loss. Additionally, ocular motility disorders, including nystagmus, can further hinder the execution of the procedure [22,32,33,34,35,36].
3.3. OCT Angiography
An advanced adaptation of OCT, known as OCTA, enables real-time imaging of retinal and choroidal vasculature, offering critical information on blood flow dynamics. Unlike traditional fluorescein angiography (FA), OCTA eliminates the need for exogenous contrast agents, instead utilizing motion contrast from light backscattering to map vascular structures [22]. In sphingolipidoses, particularly those characterized by retinal vascular alterations, such as FD or NPD, OCTA is a valuable tool for studying the vascular changes. It is primarily useful for assessing capillary density in the macular region, vessel tortuosity, detecting potential neovascularization and identifying areas of hypoperfusion [37,38].
The limitations of OCTA are comparable to those of SD-OCT. Furthermore, imaging with FA enable the detection of vascular integrity abnormalities, identifiable as dye-leakage from the vessels, that OCTA is unable to visualize [31].
3.4. Fundus Autofluorescence (FAF)
FAF is a noninvasive technique that visualizes intrinsic fluorescence within the ocular fundus, primarily attributed to lipofuscin accumulation in the retinal pigment epithelium (RPE). By detecting abnormal autofluorescence patterns, FAF imaging provides valuable insight into RPE integrity and disease progression, making it an essential tool for diagnosing and monitoring retinal disorders [39,40]. The cherry-red spot or macular halos can be identified through FAF, as they correspond to areas of hyperautofluorescence or hypoautofluorescence in the parafoveal macular region, respectively [37].
3.5. Fluorescein Angiography
FA is employed to assess retinal and choroidal circulation, aiding in the identification of vascular abnormalities. The procedure involves intravenous injection of fluorescein dye, which reaches the ocular vasculature within seconds. The choroidal circulation fills first, followed closely by retinal perfusion, allowing for sequential imaging of blood flow dynamics. Under blue light excitation (465–490 nm), fluorescein emits a green-yellow fluorescence (520–530 nm), enabling high-contrast visualization of retinal vasculature. Serial imaging at different circulation phases provides comprehensive diagnostic data [41,42]. FA is not as commonly used as the aforementioned examinations for the diagnosis and follow-up of sphingolipidoses, but it can be useful for evaluating vascular abnormalities or RPE alterations.
FA presents several limitations when applied to sphingolipidoses, as these disorders often affect pediatric patients, where compliance may be suboptimal. Moreover, the procedure requires the intravenous administration of a contrast agent to obtain the necessary images, which is an invasive procedure and, in rare cases, may lead to undesirable adverse effects [42].
3.6. Adaptive Optics (AO)
AO is an advanced imaging technique designed to achieve high-resolution visualization of microscopic retinal structures in vivo. This technology enhances the clarity of images by correcting optical aberrations, thereby allowing precise examination of blood vessels, nerve fibers, ganglion cells, and photoreceptors [43]. AO systems optimize optical performance, achieving lateral resolutions as fine as 2 microns [43,44].
AO can be particularly beneficial in sphingolipidoses, especially FD, for evaluating vessel diameter, course, and paravascular deposits [45]. More generally, integrating AO with SD-OCT could significantly enhance the detailed analysis of retinal ultrastructure [46], whereas its combination with OCTA would facilitate a more comprehensive three-dimensional assessment of the retinal circulation [47]. This synergy could also support the development of a more refined differential diagnosis algorithm for retinal vascular pathologies.
The primary limitation of AO lies in its restricted availability in routine ophthalmological practice, attributable to its high cost and operational complexity [46].
3.7. Electroretinography
ERG records the retinal electrical response to light stimuli. A flash or patterned stimulus generates a biphasic waveform, with the initial negative a-wave originating from photoreceptors (rods and cones) and the subsequent positive b-wave arising from inner retinal layers [48].
Different ERG modalities serve distinct purposes. Full-field ERG (ffERG) is valuable for detecting widespread retinal dysfunction, aiding in the diagnosis of conditions such as diffuse retinopathies [49]. mfERG, on the other hand, provides a topographic map of retinal function, pinpointing localized defects within specific retinal layers and regions [50]. In sphingolipidoses, the ERG is useful for detecting early functional retinal impairment in the absence of structural signs, often preceding the onset of manifest anatomical alterations. In contrast, this examination may fail to reveal abnormalities even in the presence of early retinal alterations [19].
A limitation of conventional ffERG is that it provides a cumulative response from the entire retina. Consequently, unless a pathological condition affects at least 20% of the retina, the ERG results usually remain within normal thresholds [48].
3.8. Visual Evoked Potentials
VEPs measure the electrical activity of the nervous system in response to visual stimuli, enabling the detection of abnormalities affecting the optic nerve, visual pathways, or the visual cortex [51,52]. By evaluating VEPs waveforms, clinicians can assess the integrity of visual processing pathways and detect functional abnormalities [53]. Optic nerve dysfunction or abnormalities in the brain areas involved in vision can be assessed using VEPs, proving extremely useful in supporting the diagnosis of sphingolipidoses in early and late stages. Many sphingolipidoses also lead to irreversible blindness due to optic nerve atrophy. This is the case, for example, in TSD and SD [54,55], KD [18,56] and MLD [57], where optic damage is attributed to a process of retrograde degeneration secondary to abnormal myelin homeostasis [58]. In these conditions, functional alterations are already detectable in the preclinical phase through VEPs, which reveal pathological responses, as described in MLD [18].
4. Subtypes of Sphingolipidoses
4.1. Niemann–Pick Disease
4.1.1. Disease Pathogenesis and Systemic Symptoms
NPD is a sphingolipidosis with autosomal recessive inheritance divided into three types: A, B, and C. This disease is characterized by an enzymatic defect in acid sphingomyelinase, which is caused by a mutation in the sphingomyelin phosphodiesterase 1 (SMPD1) gene [59], which consequently leads to the progressive systemic accumulation of the lipid sphingomyelin [60]. Table 1 details the characteristics of NPD subtypes (Table 1).

Table 1.
Summary of the characteristics of Niemann–Pick disease.
4.1.2. Retinal Manifestations
A prevalent retinal marker across most sphingolipidoses and a distinguishing retinal feature in only NPD type A and B, is the presence of a cherry-red spot at fundus examination. Notably, this sign typically manifests only in the later stages of the disease [60].
In NPD, it is crucial to conduct an ophthalmoscopic examination, as it can reveal the cherry red spot and, also, a more specific retinal finding associated with the NPD type B, referred to as the macular halo (Figure 1). It appears as a circular yellow-white area with a possible extension up to 3.0 mm in diameter, encircling the spared fovea [3,60,67]. The macula halo syndrome was initially introduced by Cogan et al. [63] in 1983, who defined it as the presence of granular accumulations with a whitish and symmetrical appearance at the level of both maculae. They regarded the observation as a potential milder version of the cherry-red spot and a variant of NPD type B [63]. Analyzing macular halos through the use of multimodal imaging is essential for better disease characterization [67].
The use of color fundus photography is critical in NPD, as well as in other sphingolipidoses, for documenting the characteristic ophthalmoscopic features and tracking their progression over time, or potential regression in response to systemic therapy [6].
In FAF, macular halos appear as hypoautofluorescent areas [37]. SD-OCT is the primary modality for diagnosing the halo sign, proving highly effective in assessing its extent and depth [16]. The macular halo appears as a thin hyperreflective band in the inner retina corresponding to the foveal slope (foveal clivus) [63,67]. To determine the exact level within the inner retina where these granular hyperreflective deposits are localized, several hypotheses have been proposed. Some authors suggest that lipid accumulation occurs in the GCL [3], which may account for the sparing of the fovea from the macular halo, as the fovea lacks GCL [16]. These findings have been confirmed by SD-OCT, which reveals focal thickening with high reflectivity in the GCL of the fovea [3]. Further confirmation was provided by light microscopy and electron microscopy of the retina, where lipid-rich material organized into lamellar inclusions were observed at the level of the retinal ganglion cells [67].
According to other authors, lipid deposits are localized not only in the GCL but also within the inner plexiform layer and inner nuclear layer. The fact that their terminal sections are present in the foveal slope may explain why hyperreflectivity extends across this region [68]. This theory is supported by histological evidence indicating that membranous cytoplasmic bodies were observed not only in retinal ganglion cells, but also in amacrine cells, Müller cells, glial cells, and photoreceptors. However, the observation that these bodies assumed a more regular configuration specifically within the retinal ganglion cells, in contrast to a more irregular and vacuolated appearance in the other retinal layers, may provide an explanation for the stronger reflectance signal generated in the GCL that could lead to a misleading impression of exclusive GCL involvement [67].
Vertical hyperreflectivity was detected at the fovea extending from the internal limiting membrane (ILM) to the external limiting membrane (ELM) in case reports where the authors suggested that the deposits may be localized within the foveal Müller glia [39,67]. The pronounced metabolic activity of Müller glial cells could account for their predisposition to sphingomyelin accumulation in this region [67]. Additionally, lipid deposits with a distinct irregular morphology were observed on the retinal surface, projecting into the vitreous [37].
The clinical observation of perimacular gray discoloration during fundus examination has been further investigated through histological analysis in murine knockout models of the NPD Type C, revealing an elevated production of interleukin-1β in astrocytes and heightened activation of microglial cells. These findings support the hypothesis of a significant proinflammatory role of lipid deposits in retinal pathology [3]. Similar histopathological changes were identified in regions corresponding to areas of optic nerve pallor [3].
OCTA in NPD type B showed that the perimacular vessels exhibited increased tortuosity, and areas of non-flowing capillaries in the superficial retinal plexus [37]. These OCTA findings may indicate a more aggressive progression of disease [37].
Ophthalmic pathological findings generally do not lead to visual impairment [69]. However, the performance of functional tests is critical, as they may reveal alterations. mfERG provides a predictive functional assessment in NPD type B patients with ocular involvement. In areas corresponding to lipid deposition, the mfERG results appear depressed with prolonged latency [16].
Another recommended test is microperimetry, a diagnostic technique employed to assess visual sensitivity mediated by the central retina [70]. Microperimetry shows a mild reduction in retinal sensitivity in areas of retinal accumulation [63].
4.2. Gaucher Disease
4.2.1. Disease Pathogenesis and Systemic Symptoms
GD is the most prevalent lysosomal storage disorder, characterized by a deficiency of the enzyme glucocerebrosidase, which results in the pathological accumulation of glucosylsphingosine and glucosylceramide [71]. These lipid metabolites predominantly accumulate in the liver, spleen, lungs, and bone marrow [72,73,74]. Based on the degree of enzymatic activity, three clinical subtypes have been identified, with progressively reduced enzyme functionality [75]. Table 2 details the characteristics of GD (Table 2).
Table 2.
Summary of the characteristics of Gaucher disease.
4.2.2. Retinal Manifestations
Ocular manifestations in GD can affect all ocular structures. While the exact pathophysiology is still under investigation, the accumulation of Gaucher cells macrophages with a distinctive “crumpled silk” morphology due to lysosomal expansion appears to be a key factor [29,78,80]. These cells not only deposit in ocular tissues but also act as proinflammatory mediators, leading to fibrosis and microvascular compromise [29,80]. Although ocular involvement is most frequently described in Type IIIc, all GD subtypes have reported cases of ocular pathology. Notably, the retinal manifestations, when present, are highly variable, underscoring the rarity of GD and the heterogeneity of its ocular presentations [77].
Neurological ocular findings are more prevalent in neuronopathic forms and may serve as early indicators. Among the most commonly reported findings, saccadic eye movement abnormalities, including horizontal saccadic slowing and horizontal saccadic failure, typically manifest during early childhood [74,77,82]. Over time, vertical saccades may also be affected, mimicking patterns observed in progressive supranuclear palsy. Additional ocular abnormalities include the pre-retinal white dots [77,80,81,89,90]. Such white dots appear ophthalmoscopically as amorphous whitish deposits diffusely distributed in the posterior pole and retinal periphery, appearing on SD-OCT as hyperreflective pre-retinal deposits localized at the interface between the ILM and the vitreous. Their recognition has been associated with posterior vitreous detachment in correspondence of their sites of formation [91,92]. Occasionally, hyperreflective deposits are also observed within the vitreous, mimicking vitreitis. Rarely, tractional retinal detachments originating from these pre-retinal deposits have been reported [93]. However, such findings are sporadic, reflecting the limited focus on ocular evaluations in the clinical management of GD. A schematic representation of hyperreflective pre-retinal deposits is shown in Figure 2.
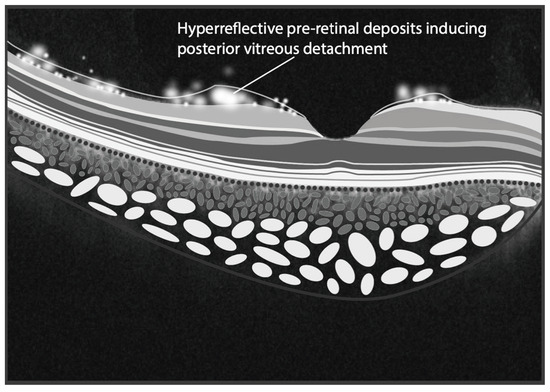
Figure 2.
The illustration depicts hyperreflective pre-retinal deposits situated at the interface between the internal limiting membrane and the vitreous, likely contributing to the induction of posterior vitreous detachment (PVD). Furthermore, some of these deposits are illustrated within the vitreous, mimicking the appearance of vitreitis. (Graphics courtesy of Dariush Rahimi).
Recent advancements suggest that SD-OCT imaging may have predictive and monitoring applications in GD beyond ocular involvement. In a study by Weill et al., where patients with GD1 were compared to healthy controls, it was reported that patients with GD1 had a higher incidence of abnormal peripapillary RNFL on SD-OCT scans. Moreover, the authors showed a significantly thinner peripapillary RNFL and macular ganglion cell complex compared to the healthy control population [28]. In a similar study involving patients with GD, carriers of the glucocerebrosidase gene (GBA) who exhibited clinical signs of possible early neurodegeneration [29] demonstrated a reduced average ganglion cell complex thickness when compared to both the control group and GD patients or carriers without such clinical indicators of potential early neurodegeneration. Thus, SD-OCT could be helpful in monitoring retinal complications and neurological progression [29].
Complementary examinations of significant value are ERG and VEPs. Studies have demonstrated that glucosylceramide accumulation in glial cells leads to impaired functionality, as evidenced by an altered ERG response characterized by a 50% reduction in the b-wave amplitude of the scotopic response and a 40% reduction in the general response [15]. These findings highlight the utility of ERG in detecting functional changes at a preclinical stage, even when the patients remain asymptomatic from a visual standpoint. Conversely, VEPs were found to be within normal parameters [15].
4.3. GM1 Gangliosidosis
4.3.1. Disease Pathogenesis and Systemic Symptoms
GM1 gangliosidosis is a lysosomal storage disorder characterized by a partial or total deficiency of the enzyme acid beta-galactosidase (β-gal) [94]. Its dysfunction leads to the progressive accumulation of GM1 ganglioside and GA1 ganglioside and other glycans, resulting in clinical manifestations in the central and peripheral nervous systems [95]. Table 3 details the characteristics of GM1 gangliosidosis (Table 3).
Table 3.
Summary of the characteristics of GM1 gangliosidosis.
4.3.2. Retinal Manifestations
Ocular involvement can affect various structures of the visual system and has been reported as early as 1959 [103]. Retinal involvement is primarily characterized by the presence of a macular cherry-red spot, which is almost always present in type 1 and type 2, along with retinal hemorrhages [84,87,88].
In a histopathological study conducted by Emery et al. analyzing both eyes from a young patient with GM1 gangliosidosis and a cherry-red macula, the authors observed ganglioside accumulations in the cytoplasm of retinal ganglion cells [99]. These accumulations are presumably responsible for this alteration, which, although not exclusive to this condition, may aid in early diagnosis [104]. Furthermore, a retinal functional study in mice demonstrated that GM1 gangliosidosis is associated with a widespread reduction in VEPs [19].
4.4. GM2 Gangliosidosis: Tay Sachs Disease and Sandhoff Disease
4.4.1. Disease Pathogenesis and Systemic Symptoms
TSD and SD are lysosomal storage disorders caused by defects in the enzymes hexosaminidase A and hexosaminidase A and B, respectively. Both diseases lead to the accumulation of GM2 gangliosides at various levels, primarily in the central nervous system, causing neurodegeneration [105,106].
These diseases present with distinctive neurological and ophthalmic manifestations, with the retina being one of the most affected areas, characterized in most cases by the distinctive cherry red spot. This is one of the most significant and earliest ophthalmic signs attributed to ganglioside accumulation, which remains a key diagnostic indicator [62]. Thus, despite the primary neurological symptoms, ophthalmic manifestations are commonly observed and provide valuable clues for early diagnosis. Table 4 and Table 5 show the systemic and ocular manifestations of SD (Table 4) and TSD (Table 5).
Table 4.
Summary of the characteristics of Sandhoff disease.
Table 5.
Summary of the characteristics of Tay–Sachs disease.
4.4.2. Retinal Manifestations
Bilateral cherry-red spots were described in a report of two pediatric patients with infantile SD [110] and in 18 patients with TSD or SD, 88% had cherry-red spots [111].
The first histopathological evaluation of human retinas in TSD and SD showed an accumulation of deposits at ganglion cell level, which had a more pleomorphic appearance in patients with SD than in those with TSD [115]. More recent studies demonstrated the presence of cytoplasmic inclusions in the retinas of mice with GM2 gangliosidosis, confirming the involvement of ganglion cells and the IPL and INL. However, no deposits were identified at the level of photoreceptor cells [116].
Retinal vessel narrowing, optic disc atrophy, retinitis pigmentosa, and macular atrophy are commonly observed in more advanced stages of the disease. These signs reflect the progressive degeneration of retinal structures as a result of the ganglioside accumulation [117].
TSD and SD result in the accumulation of GM2 gangliosides [27,112], a type of complex glycosphingolipids that are abundantly expressed within the central nervous system [118]. This accumulation at the brain level leads to early neuronal cell death and progressive neurodegeneration, which is also observed in the retina as ganglion cell loss, likely correlating with the gradual decline in visual function over time [118]. The loss of ganglion cells can be ophthalmoscopically detected by a reduction over time in the cherry-red spot, which arises from ganglion cell death at sites where gangliosides accumulate. Since the RNFL in SD-OCT is composed of unmyelinated axons of ganglion cells, its thickness is influenced by the number of ganglion cells [119]. As a result, SD-OCT findings in TSD and SD typically demonstrate RNFL thinning, indicative of ganglion cell loss, along with ballooning of the GCL [27]. These changes reflect the neurodegenerative process in the retina and central nervous system [27]. SD-OCT could be particularly useful for detecting early signs of retinal involvement before funduscopic changes become apparent. In GM2 case reports, OCT scans showed a thickening of the inner retinal layers with a shadowing effect on the outer layers in presence of the characteristic cherry red macula with a whitish ring area encircling the fovea [117,120].
While FA and FAF are not as commonly employed as fundus photography, they have offered important findings regarding the retinal alterations linked to TSD and SD. Late-phase FA images show areas of hyper-fluorescence, corresponding to areas of retinal degeneration around the fovea [121].
An additional examination in these conditions is the study of the ERG and VEPs. Denny et al. compared the retinas of mice with GM1 gangliosidosis and SD to healthy controls, finding a 44% and 40% increase in total gangliosides, respectively, along with higher levels of specific gangliosides associated with each disorder. Interestingly, the ERG were normal in all mice, with no significant differences between GM1, SD and controls, while VEPs were reduced in mice with these storage disorders [19].
4.5. Farber Disease
4.5.1. Disease Pathogenesis and Systemic Symptoms
Farber disease, also referred to as Farber’s lipogranulomatosis, is a type of lysosomal storage disorder resulting from a deficiency of the enzyme acid ceramidase (ACDase) [2]. This enzyme is crucial for the breakdown of ceramide, a lipid component involved in cell membrane structure and signaling. The deficiency leads to the pathological accumulation of ceramide in diverse tissues, including the retina, leading to inflammatory and degenerative changes [35]. Table 6 details the characteristics of Farber disease subtypes (Table 6).
Table 6.
Summary of the characteristics of Farber disease.
4.5.2. Retinal Manifestations
Fundus examination enables the identification of the cherry-red spot, which is the defining feature of this pathology [2]. SD-OCT enables the detection of a hallmark sign: hyperreflective bands localized to the GCL corresponding to ceramide deposition [35,122]. In murine models at 3 to 4 weeks of age, retinal development was reported as normal. However, alterations in the retinal layers were observed, with occasional hyperreflective spots in the vitreous, particularly near the optic nerve head [35]. At 8 to 9 weeks of age, a marked increase in hyperreflectivity, primarily along the RNFL and GCL, with hyperreflective specks, was observed in the vitreous body [35].
Multimodal imaging and electrophysiology and post-mortem histology have been used to monitor the effectiveness of emerging therapies, such as enzyme replacement or gene therapies, in mouse models [1]. A study was conducted in a mouse model of Farber disease by inducing overexpression of the mutated ASAH1 gene via a recombinant adeno-associated virus (rAAV). The efficacy of this gene therapy was then evaluated by studying the progression of retinopathy. The results demonstrated a reduction in retinal ceramide accumulation, accompanied by a decrease in central retinal thickness and fundus hyper-reflectivity. These findings suggest that the retinal evaluation of sphingolipidoses through multimodal imaging could play a promising role in monitoring therapeutic responses [1]. Notably, the latest approved gene therapies, which rely on the transplantation of genetically modified stem cells overexpressing the deficient enzyme or the direct delivery of the mutated enzyme gene via a vector into a specific organ [2], rank among the most expensive treatments in the global pharmaceutical market [1]. Furthermore, substrate reduction therapy, designed to decrease sphingolipid synthesis, may inadvertently inhibit the production of crucial sphingolipid intermediates with essential biological functions, potentially leading to significant adverse effects [123]. Consequently, retinal imaging-based monitoring of therapeutic response is critical for optimizing drug administration, ensuring an optimal dose-efficacy balance, reducing the frequency of treatment and associated costs, and minimizing adverse effects.
4.6. Metachromatic Leukodystrophy
4.6.1. Disease Pathogenesis and Systemic Symptoms
MLD is a lysosomal storage disorder which results from a deficiency of the lysosomal enzyme arylsulfatase A (ARSA) or its activator protein sphingolipid activator protein B (SapB) [124]. These enzymatic deficiencies lead to the pathological accumulation of sulfatides within the central and peripheral nervous systems, causing ongoing injury to the myelin sheath [2,125]. Clinically, this manifests as a gradual decline in both motor and cognitive function [124]. Table 7 details the characteristics of MLD (Table 7).
Table 7.
Summary of the characteristics of Metachromatic leukodystrophy.
4.6.2. Retinal Manifestations
Due to the rarity of the disease, most of the current information is derived from ophthalmoscopic observations corroborated by corresponding histological findings. Ophthalmoscopy, often normal, remains the primary diagnostic tool. Although, a distinctive feature is the presence of a pale optic disc, indicative of optic nerve atrophy [57]. This is believed to result from retrograde degeneration of the optic nerve due to abnormal myelin metabolism [58]. Histological examination revealed neuronal storage at the level of the optic nerve, a key factor in the disease’s pathogenesis [57,67]. Additional ophthalmoscopic findings may include areas of RPE degeneration [127] and a grayish macula caused by the accumulation of lamellar lysosomes in retinal ganglion cells [57]. These ganglion cell deposits, a hallmark of the disease, have been histologically confirmed as containing metachromatic material [40]. The exclusive involvement of ganglion cells in adult MLD has not yet been fully explained. It could be because ganglion cells are more actively involved in sulfatide metabolism, thus making them more susceptible to sulfatide accumulation [40].
In MLD, cells in the bipolar and photoreceptor layers, as well as RPE cells, are typically spared from lysosomal material accumulation [57]. However, in some cases, elevated lipofuscin levels have been observed both in the RPE and ganglion cells, suggesting that this accumulation may be driven by a mechanism akin to that observed in infantile Type II glycogenosis [40,128]. The process of lipofuscin formation, occurring within the lysosomal compartment, may be compounded by the concurrent accumulation of sulfatides, characteristic of MLD. This leads to the formation of residual bodies containing both lipofuscin and disease-specific metachromatic material [40]. This is further supported by the fact that the RPE normally contains high levels of arylsulfatase A, which likely makes it particularly vulnerable to morphofunctional alterations when these levels are deficient, as is the case in MLD [41].
Bettinger et al. in 2024 observed that, in a cohort of 50 patients with MLD, 1 patient exhibited visual function impairment, 11 showed a pathological light reflex, and 2 presented with nystagmus. The authors used SD-OCT to evaluate the involvement of the RNFL, hypothesizing that the thickness of the RNFL reflects the integrity of the unmyelinated axons in the anterior visual pathway, making it a potential surrogate marker for tracking disease progression; however, the RNFL was found to be normal [18]. Bettinger et al. observed that, among a cohort of 19 MLD patients, 58% demonstrated altered VEPs, a finding corroborated by a separate previous cohort of 12 MLD patients reported by the same authors, 78% of whom exhibited similar results. This supports that VEPs should be performed as an additional diagnostic test, even in the absence of overt visual impairment or negative SD-OCT findings, as it may provide early evidence of disease progression [18].
4.7. Fabry Disease
4.7.1. Disease Pathogenesis and Systemic Symptoms
FD is an X-linked lysosomal storage disease caused by a diminished or absent function of the enzyme alpha-galactosidase A. This deficiency leads to the progressive accumulation of sphingolipids, particularly globotriaosylceramide (GL-3), in multiple organs [129]. Table 8 details the characteristics of FD (Table 8).
Table 8.
Summary of the characteristics of Fabry disease.
4.7.2. Retinal Manifestations
Symptoms of FD are often nonspecific or subtle, resulting in frequent diagnostic delay [137]. Among ocular signs, cornea verticillata is the most commonly noted manifestation [11]. Other findings include anterior capsular or posterior subcapsular cataracts. A hallmark feature of FD is vessel tortuosity and venous vascular aneurysmal dilation in conjunctival and retinal vessels [131].
Posterior segment involvement includes optic neuropathies and retinal vasculopathies, such as retinal vein and retinal artery occlusion [10], which can result in retinal ischemia and are considered medical emergencies [138,139]. Vascular occlusions occur due to the deposition of accumulated material within smooth muscle and vascular endothelial cells, leading to vascular luminal narrowing. Moreover, an increase in pro-aggregatory factors and a reduction in fibrinolytic activity contribute to an elevated risk of vascular occlusion [139]. In FD, retinal vascular occlusion may represent an early clinical manifestation, even in the absence of other systemic or ophthalmologic findings. Consequently, some authors recommend screening for FD in patients presenting with such occlusive events [139]. A comprehensive understanding of these pathophysiological mechanisms is crucial for early diagnosis and timely intervention, aiming to prevent severe retinal complications that may lead to irreversible visual impairment. Additionally, macular choroidal neovascularization has been reported [11,135].
OCT-A is useful in analyzing retinal vascular changes, allowing quantification of the retinal vascular plexuses density and peripapillary capillaries. Retinal vessel density (VD) values were found to be significantly reduced in both the macular superficial capillary plexus and deep capillary plexus when compared to those of a healthy control group. Furthermore, the foveal avascular zone (FAZ) was notably larger in affected individuals. The authors speculated that a reduced macular blood supply due to structural retinal vessel alterations could lead to increased thrombotic risk in FD [10]. Another study found larger FAZ areas and lower superficial capillary plexus and deep capillary plexus VD values in FD patients compared to controls [137]. However, no significant discrepancies in FAZ size between FD patients and controls were found in one study, where the authors observed a higher foveal VD in patients with FD. The authors proposed a correlation between the presence of cornea verticillata, plasma lyso-Gb3 levels (a biomarker of FD), and foveal VD, suggesting that this association could potentially serve as a biomarker for FD [140]. A schematic representation of the FAZ enlargement in a patient with FD is shown in Figure 3.
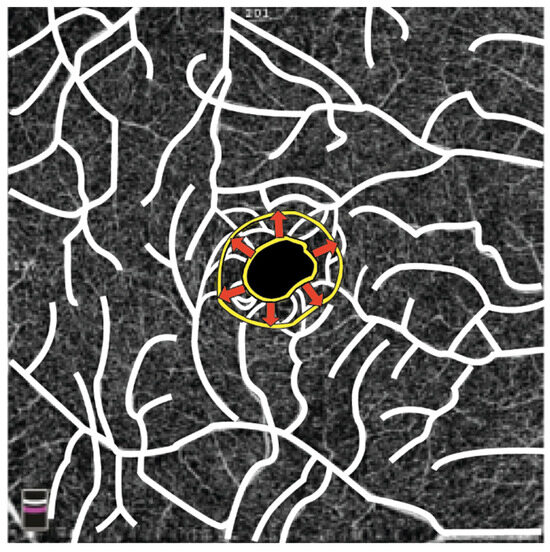
Figure 3.
The illustration depicts a yellow ring outlining the contour of the FAZ. In patients with FD, the FAZ extends from the inner yellow ring to the outer yellow ring in the area indicated by the arrows due to structural changes in retinal vessels. (Graphics courtesy of Dariush Rahimi).
Through SD-OCT it was shown that hyperreflective foci (HRF) in the inner retinal layers are prevalent among FD patients [141]. HRFs are defined as isolated point-like artifacts (10–30 μm) with high-intermediate reflectivity and no shadowing cone [142]. The authors found a direct relationship between the number of HRF and serum lyso-Gb3 levels, with significantly more HRF observed in the foveal and parafoveal regions of FD patients compared to controls [141]. These findings suggest that the presence of HRF could be a novel biomarker of intraretinal inflammation. Similar observations were made in other systemic diseases, such as multiple sclerosis, reinforcing the potential of HRFs as indicators of inflammation [142]. A schematic representation of HRFs within the inner retinal layers is shown in Figure 4.
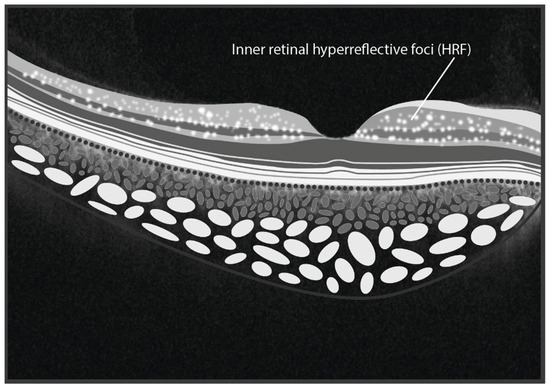
Figure 4.
The illustration depicts hyperreflective foci situated within the inner retinal layers in Fabry Disease. (graphics courtesy of Dariush Rahimi).
Retinal vessel dimensions measured in FD patients were smaller and thinner than those of healthy controls [143]. Using AO [144], sphingolipid deposits in the paravascular zone were identified and graded in increasing severity from 0 to 4. Despite normal fundus examination findings, AO revealed punctiform opacities (5–10 μm) or diffuse opacifications along both veins and arteries in 14 enrolled patients. These deposits increased in density and continuity with disease severity, highlighting their potential utility as diagnostic tools for staging and monitoring FD [45].
The use of fERG, in conjunction with OCTA, can serve as a tool to support the diagnosis, evaluate retinal function, and detect potential microangiopathies that might otherwise go unrecognized. Indeed, fERG performed in patients with FD exhibit a significantly lower mean amplitude compared to healthy controls [137].
Multimodal ophthalmological imaging offers valuable insights into potential biomarkers for FD, aiding in the analysis and study of this pathology. A combined use of these biomarkers can deepen our understanding of FD-related retinal imaging and improve diagnostic and monitoring strategies.
4.8. Krabbe Disease
4.8.1. Disease Pathogenesis and Systemic Symptoms
KD is a lysosomal storage disorder, also known as globoid cell leukodystrophy. It is characterized by mutations in the beta-galactocerebrosidase gene [145], leading to the inability to degrade certain lipids and their subsequent accumulation, which disrupts the turnover of myelin in neuronal cells [146].
The symptoms specifically affect the central and peripheral nervous systems [56], with severity closely linked to the residual function of the enzyme. However, the infantile form is the most common presentation [147]. Table 9 details the characteristics of KD (Table 9).
Table 9.
Summary of the characteristics of Krabbe disease.
4.8.2. Retinal Manifestations
Ocular involvement is indirectly related to the neurological damage that underpins this disease. Both in infantile forms and in late-onset variants, vision loss—often complete—has been reported [147,154]. In a postmortem histopathological specimens of a 3-year-old patient, ultrastructural analysis of the optic nerve revealed widespread ganglion cell loss, thinning of the RNFL, optic disc atrophy, and the presence of globoid inclusions within the optic nerve [155]. Bettinger et al., in KD reported fixational deficits, visual function alterations, pathological light reflex, and VEPs alterations have been reported and RNFL thinning in two of three patients [18]. In KD, thinning of the RNFL occurs because the enzymatic defect leads to the accumulation of psychosine, an intermediate in the catabolism of monoglycosylceramides, which is toxic to oligodendrocytes [6]. Damage to oligodendrocytes compromises their ability to produce myelin, resulting in demyelination and progressive neurodegeneration that advances in a retrograde fashion. Ultimately, this process culminates in the thinning of both the RNFL and the GCL of the retina, as confirmed by postmortem ocular analyses [6]. These anatomical changes mirror the clinical manifestations of the disease. Early neurodegeneration typically presents with irritability and progressive motor and cognitive decline. As the disease continues in a retrograde manner, later stages involve the afferent visual pathways, affecting both the optic radiation and the optic nerve, leading to blindness, which emerges as a late clinical symptom [6].
4.9. Sialidosis
4.9.1. Disease Pathogenesis and Systemic Symptoms
Sialidosis is a lysosomal storage disease caused by a deficiency of the enzyme lysosomal sialidase, NEU1, which is responsible for the cleavage of terminal sialic acid residues of sialoglycoconjugates. These enzymatic deficiencies lead to the pathological accumulation of sialoglycoproteins or sialyloligosaccharides in tissues, particularly in visceral organs, muscles, and the nervous system [156]. Table 10 details the characteristics of Sialodosis (Table 10).
Table 10.
Summary of the characteristics of sialidosis.
4.9.2. Retinal Manifestations
Sialidosis types 1 and 2 can present with ocular involvement, characterized by manifestations such as nystagmus, corneal clouding resulting from the accumulation of metabolic byproducts, cataracts and optic nerve atrophy [32,33,36,159]. In the congenital form of type 2 sialidosis, bilateral congenital cataracts are frequently observed, along with foveal hypoplasia [152]. From a retinal perspective, the most distinctive finding in both type 1 and type 2 sialidosis is the presence of bilateral macular cherry-red spots, attributed to the pathological accumulation of lipidic material within the retinal ganglion cells. This feature is considered a key hallmark of the disease [33].
SD-OCT imaging corroborates these findings, revealing thickening of GCL with areas of increased reflectivity [158]. Over time, a paradoxical reduction in this ophthalmoscopic sign may occur, likely due to progressive ganglion cell degeneration [151].
The accumulation of sialyloligosaccharides has been identified in type 1 sialidosis, within the peripapillary RNFL, where an increase in thickness may be observed [33]. Some authors suggested that, in mild forms of type 1 sialidosis, the cherry-red macula represents the earliest clinical manifestation, highlighting its significance as a key diagnostic marker for early detection [163]. However, this claim is contradicted by other studies indicating that this finding appears only after several years of symptomatic disease progression [164]. SD-OCT based assessment of accumulated pathological material may serve as a valuable tool for monitoring disease progression [163].
A single case of Bergmeister’s papilla was reported in type 1 sialidosis, characterized by an epipapillary glial membrane that appeared as a whitish area of vitreous thickening on fundoscopic examination. Although typically asymptomatic, its presence required clinical monitoring, as contraction of the membrane may, in rare cases, lead to tractional retinal detachment [158].
A potential association between type 1 sialidosis and choroidal hyperpermeability has been proposed. In a patient presenting with bilateral cherry-red macula and retinal pigment epithelial detachment (PED) with subretinal fluid detected via SD-OCT, indocyanine green angiography revealed a hypocyanescent parafovea, potentially linked to inner retinal layer hyperreflectivity, along with a hypercyanescent fovea, possibly associated with choroidal hyperpermeability. The hypothesized underlying mechanism suggested that lysosomal dysfunction in sialidosis leads to RPE damage, compromising the integrity of the blood-retinal barrier. This disruption may result in increased choroidal permeability, ultimately contributing to the development of PED and subretinal fluid accumulation [165].
5. Current Challenges and Future Directions
Sphingolipidoses are rare lysosomal storage disorders characterized by intrinsic variability, primarily due to the residual enzymatic activity of the affected enzymes. This variability poses a significant challenge in the initial diagnostic phase, largely due to phenotypic overlap with other neurometabolic disorders and the occasional inconclusiveness of laboratory findings. Moreover, it may occur that, despite the presence of enzymatic deficiencies or their activators, diagnostic tests fail to detect an accumulation of the substrates that these enzymes are supposed to degrade. This may lead to a missed diagnosis, as the apparent absence of substrate accumulation could mask the underlying pathology [166].
However, an even greater challenge lies in the longitudinal assessment of disease progression and therapeutic response. Consequently, the identification of reliable biomarkers for patient stratification is crucial for optimizing clinical management. The need for early and non-invasively obtainable biomarkers stems from the presence of unmet needs, primarily due to the reliance on time-consuming and costly tests, which are not always conclusive [167].
Ocular biomarkers could constitute a valuable asset in evaluating the sphingolipidoses. The eye, owing to its accessibility and the availability of high-resolution, non-invasive imaging modalities, such as SD-OCT and OCT-A, serves as an ideal model for investigating microstructural and vascular alterations associated with sphingolipid accumulation. Previous studies have demonstrated that pathological sphingolipid deposition is associated with quantifiable changes in retinal architecture, particularly in the RNFL and the GCL [18,27,28,35], as well as alterations in retinal microvascular integrity [37,140]. Notably, lipid accumulation at intraretinal level also appears to play a pro-inflammatory role, as hypothesized in both NPD and GD. In the latter, lipid deposition can, over time, lead to anatomical and functional alterations of the retina, progressing towards fibrosis and microvascular impairment [29,80]. In FD, the inflammatory process is so pronounced that HRFs have been observed, similar to those described in other systemic diseases such as multiple sclerosis, and have been proposed as new biomarkers of intraretinal inflammation. These alterations can cause functional damage even in a preclinical stage, as seen in NPD, GD, and FD, where mfERG results appear depressed [16,17,50,162]. Analyzing sphingolipidoses at retinal level represents an essential tool for the early identification of anatomical and functional alterations, with a significant impact on clinical management. Case-control studies, particularly in more prevalent disorders such as FD and GD, have highlighted the diagnostic utility of these ocular biomarkers in differentiating affected individuals from healthy controls [12,28,140].
A critical future direction in the application of ocular diagnostics lies in the ability to subclassify disease phenotypes, enabling the assessment of disease activity and progression in correlation with systemic biomarkers. This necessitates the development of advanced image analysis methodologies capable of high-output, quantitative assessments of microscopic structures. A compelling example is the study conducted by Atiskova et al. [12], in which an image-processing algorithm was employed to calculate the retinal vessel tortuosity index (RVTI) in patients with FD [12]. This parameter exhibited a strong correlation with systemic disease burden, including cardiovascular involvement and plasma lyso-Gb3 levels, suggesting its potential utility as a predictive marker for disease severity and therapeutic efficacy [12]. Similar findings were reported by San Romàn et al., further supporting the role of retinal vascular biomarkers in disease monitoring [168].
The primary limitation in integrating these advanced imaging methodologies into routine clinical practice stems from the rarity of sphingolipidoses, which limits the feasibility of conducting large-scale, statistically powered studies. However, emerging fields such as “oculomics”—the systematic identification of ocular biomarkers for systemic diseases—hold promise for overcoming this challenge [169,170]. Standardization of imaging protocols, coupled with the establishment of large, multi-center datasets, could facilitate the identification of robust, reproducible biomarkers [171]. Furthermore, the application of artificial intelligence (AI)-driven analytical approaches to these datasets may enhance the sensitivity and specificity of diagnostic algorithms, enabling the automated extraction of statistically significant biomarkers for each individual sphingolipidoses subtype [172,173].
Among advanced imaging analysis techniques, AO can be recognized as a superior modality compared to OCTA in distinguishing retinal vascular structures [174], which are often involved in sphingolipidosis. Furthermore, with the progressive advancement of technology, the application of novel investigative methodologies, may become feasible. For example, elastography, allows for the indirect evaluation of the biomechanical properties of retinal layers [175], which, as demonstrated, often serve as sites for metabolite accumulation in these pathologies. Such advancements have the potential to refine both diagnostic accuracy and therapeutic monitoring, ultimately improving patient outcome in these complex metabolic disorders.
6. Conclusions
Due to the limited studies in the scientific literature and the rarity of the sphingolipidoses, a comprehensive review was conducted to provide an overview of the ophthalmological features and relevant novel retinal diagnostic examinations with multimodal imaging.
At fundus examination, the most characteristic and frequently observed finding is the cherry-red macula, which is typically seen in GM1 and GM2 gangliosidoses, NPD (Type A and B), Farber disease and sialidosis. Optic disc pallor or atrophy has been identified in NPD, GM2, MLD, and KD. Other more specific findings include the macular halo in NPD type B, perimacular gray discoloration in NPD type C, and retinal vein occlusion and retinal artery occlusion in FD.
FAF is a crucial imaging modality to confirm ophthalmoscopic findings. Particularly, the presence of a cherry-red spot in the macula appears as areas of hyperautofluorescence around the fovea, and macular halo are seen as hypoautofluorescent areas.
SD-OCT is an essential diagnostic tool, particularly useful for detecting early signs of retinal involvement prior to the appearance of funduscopic changes. In NPD, GM1, Farber disease, and MLD, SD-OCT reveals a hallmark feature of hyperreflectivity of the GCL, which is associated with lipid deposits.
In KD, TSD, SD, and GD, SD-OCT typically shows thinning of the RNFL. This finding reflects the neurodegenerative processes affecting the retina and the central nervous system, making it a potential surrogate marker for tracking disease progression. Atrophy of the RPE and chorioretinal degeneration have been observed in the later stages of these diseases.
OCTA in FD demonstrates typical vascular changes, including reduced retinal VD and increased vessel tortuosity, which are also observed in NPD type B. Vascular alterations in FD have been further investigated using AO, which identified intracellular lipid deposits in retinal vessels.
Electrophysiological tests are recommended to detect early functional changes during the preclinical stage and provide evidence of disease progression. The ERG showed depression in NPD Type B, GD and FD, while VEPs were reduced in GM1 and SD, and altered in MLD.
Despite the promising potential of retinal imaging, the existing literature on this topic is limited, often outdated, and based on histological studies or animal models. For this reason, in addition to SD-OCT, AO may emerge as a promising tool, offering the advantage of visualizing retinal structures in vivo at a cellular level.
Longitudinal studies are necessary to improve our understanding of the temporal relationship between the progression of sphingolipidoses and retinal changes. By optimizing its use, multimodal retinal imaging could become an indispensable tool for ophthalmologists, enabling them to use retinal findings for early diagnosis and to assess disease progression. In this regard, multimodal retinal imaging could serve as a promising additional tool for assessing response to systemic therapy within a multidisciplinary clinical framework.
Author Contributions
Conceptualization, C.C., D.F. (Daniele Fumi), D.F. (Davide Fasciolo) and S.A.; methodology, C.C., D.F. (Daniele Fumi) and D.F. (Davide Fasciolo); formal analysis, C.C., D.F. (Daniele Fumi), D.F. (Davide Fasciolo) and S.A.; resources C.C., D.F. (Daniele Fumi), D.F. (Davide Fasciolo), M.D.P., F.D.T. and S.F.; data curation, C.C., D.F. (Daniele Fumi), D.F., (Davide Fasciolo) M.D.P., F.D.T., S.F. and M.D.P.; writing—original draft preparation, C.C., D.F. (Daniele Fumi), D.F. (Davide Fasciolo), M.D.P., S.F., F.D.T. and S.A.; writing—review and editing, C.C., D.F. (Daniele Fumi), D.F. (Davide Fasciolo), S.F., M.D.P. and S.A.; supervision S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to Dariush Rahimi and Francesco Pandolfo for the illustrations of the figures.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Shaimardanova, A.A.; Solovyeva, V.V.; Issa, S.S.; Rizvanov, A.A. Gene Therapy of Sphingolipid Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3627. [Google Scholar] [CrossRef] [PubMed]
- Abed Rabbo, M.; Khodour, Y.; Kaguni, L.S.; Stiban, J. Sphingolipid Lysosomal Storage Diseases: From Bench to Bedside. Lipids Health Dis. 2021, 20, 44. [Google Scholar] [CrossRef]
- Chen, H.; Chan, A.Y.; Stone, D.U.; Mandal, N.A. Beyond the Cherry-Red Spot: Ocular Manifestations of Sphingolipid-Mediated Neurodegenerative and Inflammatory Disorders. Surv. Ophthalmol. 2013, 59, 64–76. [Google Scholar] [CrossRef]
- Park, J.H.; Ko, J.M.; Kim, M.S.; Kim, M.J.; Seong, M.-W.; Yoo, T.; Lim, B.C.; Chae, J.-H. Novel HEXA Variants in Korean Children with Tay-Sachs Disease with Regression of Neurodevelopment from Infancy. Mol. Genet. Genom. Med. 2021, 9, e1677. [Google Scholar] [CrossRef]
- Sawicka-Gutaj, N.; Machaczka, M.; Kulińska-Niedziela, I.; Bernardczyk-Meller, J.; Gutaj, P.; Sowiński, J.; Ruchała, M. The Appearance of Newly Identified Intraocular Lesions in Gaucher Disease Type 3 despite Long-Term Glucocerebrosidase Replacement Therapy. Ups. J. Med. Sci. 2016, 121, 192–195. [Google Scholar] [CrossRef]
- Tripathy, K.; Patel, B.C. Cherry Red Spot. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Cujbă, L.; Banc, A.; Stan, C.; Drugan, T.; Nicula, C. Macular OCT’s Proficiency in Identifying Retrochiasmal Visual Pathway Lesions in Multiple Sclerosis—A Pilot Study. Diagnostics 2024, 14, 1221. [Google Scholar] [CrossRef]
- Rani, P.K.; Prabha, D.; Jakati, S.; Nalawade, R. Seeing beyond Gaucher Disease: Early Detection and Treatment of Ocular Complications. Indian J. Ophthalmol. 2023, 71, 3424–3425. [Google Scholar] [CrossRef]
- Shiga, Y.; Nishida, T.; Jeoung, J.W.; Di Polo, A.; Fortune, B. Optical Coherence Tomography and Optical Coherence Tomography Angiography: Essential Tools for Detecting Glaucoma and Disease Progression. Front. Ophthalmol. 2023, 3, 1217125. [Google Scholar] [CrossRef]
- Cakmak, A.I.; Atalay, E.; Cankurtaran, V.; Yaşar, E.; Turgut, F.H. Optical Coherence Tomography Angiography Analysis of Fabry Disease. Int. Ophthalmol. 2020, 40, 3023–3032. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, W.; Yao, X.; Song, W.; Zhao, Y.; Yuan, Y.; Zhang, W. Investigation of Ocular Involvement in Patients with Fabry Disease. Ann. Med. 2023, 55, 2226909. [Google Scholar] [CrossRef]
- Atiskova, Y.; Wildner, J.; Spitzer, M.S.; Aries, C.; Muschol, N.; Dulz, S. Retinal Vessel Tortuosity as a Prognostic Marker for Disease Severity in Fabry Disease. Orphanet J. Rare Dis. 2021, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Pampiglione, G.; Harden, A. Neurophysiological Investigations in GM1 and GM2 Gangliosidoses. Neuropediatrics 1984, 15 (Suppl. S1), 74–84. [Google Scholar] [CrossRef] [PubMed]
- Kulyabin, M.; Zhdanov, A.; Dolganov, A.; Ronkin, M.; Borisov, V.; Maier, A. Enhancing Electroretinogram Classification with Multi-Wavelet Analysis and Visual Transformer. Sensors 2023, 23, 8727. [Google Scholar] [CrossRef] [PubMed]
- Seidova, S.-F.; Kotliar, K.; Foerger, F.; Klopfer, M.; Lanzl, I. Functional Retinal Changes in Gaucher Disease. Doc. Ophthalmol. 2009, 118, 151–154. [Google Scholar] [CrossRef]
- Kim, C.; Jeong, J.; Yu, H.G. Diagnostic and Predictive Methods for a Niemann-Pick Disease Type B Patient with Ocular Involvement. J. Inherit. Metab. Dis. 2010, 33, 633–634. [Google Scholar] [CrossRef]
- Creel, D.J. Visually Evoked Potentials. Handb. Clin. Neurol. 2019, 160, 501–522. [Google Scholar] [CrossRef]
- Bettinger, C.M.; Dulz, S.; Atiskova, Y.; Guerreiro, H.; Schön, G.; Guder, P.; Maier, S.L.; Denecke, J.; Bley, A.E. Overview of Neuro-Ophthalmic Findings in Leukodystrophies. J. Clin. Med. 2024, 13, 5114. [Google Scholar] [CrossRef]
- Denny, C.A.; Alroy, J.; Pawlyk, B.S.; Sandberg, M.A.; d’Azzo, A.; Seyfried, T.N. Neurochemical, Morphological, and Neurophysiological Abnormalities in Retinas of Sandhoff and GM1 Gangliosidosis Mice. J. Neurochem. 2007, 101, 1294–1302. [Google Scholar] [CrossRef]
- Ghasemi Falavarjani, K.; Tsui, I.; Sadda, S.R. Ultra-Wide-Field Imaging in Diabetic Retinopathy. Vis. Res. 2017, 139, 187–190. [Google Scholar] [CrossRef]
- Li, Y.; Foo, L.-L.; Wong, C.W.; Li, J.; Hoang, Q.V.; Schmetterer, L.; Ting, D.S.W.; Ang, M. Pathologic Myopia: Advances in Imaging and the Potential Role of Artificial Intelligence. Br. J. Ophthalmol. 2023, 107, 600–606. [Google Scholar] [CrossRef]
- Kashani, A.H.; Chen, C.-L.; Gahm, J.K.; Zheng, F.; Richter, G.M.; Rosenfeld, P.J.; Shi, Y.; Wang, R.K. Optical Coherence Tomography Angiography: A Comprehensive Review of Current Methods and Clinical Applications. Prog. Retin. Eye Res. 2017, 60, 66–100. [Google Scholar] [CrossRef] [PubMed]
- Di Pippo, M.; d’Agostino, S.; Ruggeri, F.; Carrozzi, C.; Fasciolo, D.; Abdolrahimzadeh, S. Parkinson’s Disease: What Can Retinal Imaging Tell Us? J. Integr. Neurosci. 2024, 23, 23. [Google Scholar] [CrossRef]
- Snyder, P.J.; Alber, J.; Alt, C.; Bain, L.J.; Bouma, B.E.; Bouwman, F.H.; DeBuc, D.C.; Campbell, M.C.W.; Carrillo, M.C.; Chew, E.Y.; et al. Retinal Imaging in Alzheimer’s and Neurodegenerative Diseases. Alzheimers Dement. 2021, 17, 103–111. [Google Scholar] [CrossRef]
- Cipollini, V.; Abdolrahimzadeh, S.; Troili, F.; De Carolis, A.; Calafiore, S.; Scuderi, L.; Giubilei, F.; Scuderi, G. Neurocognitive Assessment and Retinal Thickness Alterations in Alzheimer Disease: Is There a Correlation? J. Neuroophthalmol. 2020, 40, 370–377. [Google Scholar] [CrossRef]
- Kashani, A.H.; Asanad, S.; Chen, J.; Singer, M.B.; Zhang, J.; Sharifi, M.; Khansari, M.M.; Abdolahi, F.; Shi, Y.; Biffi, A.; et al. Past, Present and Future Role of Retinal Imaging in Neurodegenerative Disease. Prog. Retin. Eye Res. 2021, 83, 100938. [Google Scholar] [CrossRef]
- Brownstein, S.; Carpenter, S.; Polomeno, R.C.; Little, J.M. Sandhoff’s Disease (GM2 Gangliosidosis Type 2). Histopathology and Ultrastructure of the Eye. Arch. Ophthalmol. 1980, 98, 1089–1097. [Google Scholar] [CrossRef]
- Weill, Y.; Zimran, A.; Zadok, D.; Wasser, L.M.; Revel-Vilk, S.; Hanhart, J.; Dinur, T.; Arkadir, D.; Becker-Cohen, M. Macular Ganglion Cell Complex and Peripapillary Retinal Nerve Fiber Layer Thinning in Patients with Type-1 Gaucher Disease. Int. J. Mol. Sci. 2020, 21, 7027. [Google Scholar] [CrossRef]
- McNeill, A.; Roberti, G.; Lascaratos, G.; Hughes, D.; Mehta, A.; Garway-Heath, D.F.; Schapira, A.H.V. Retinal Thinning in Gaucher Disease Patients and Carriers: Results of a Pilot Study. Mol. Genet. Metab. 2013, 109, 221–223. [Google Scholar] [CrossRef]
- Sivley, M.D. Fabry Disease: A Review of Ophthalmic and Systemic Manifestations. Optom. Vis. Sci. 2013, 90, e63–e78. [Google Scholar] [CrossRef]
- Spaide, R.F.; Fujimoto, J.G.; Waheed, N.K.; Sadda, S.R.; Staurenghi, G. Optical Coherence Tomography Angiography. Prog. Retin. Eye Res. 2018, 64, 1–55. [Google Scholar] [CrossRef]
- Sobral, I.; Cachulo, M.d.L.; Figueira, J.; Silva, R. Sialidosis Type I: Ophthalmological Findings. BMJ Case Rep. 2014, 2014, bcr2014205871. [Google Scholar] [CrossRef] [PubMed]
- Hm, K.; Rh, R.; Hv, D.-M.; Do, H. Optical Coherence Tomography Findings in a Patient with Type 1 Sialidosis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2016, 31, 199–201. [Google Scholar] [CrossRef]
- Kern, J.; Böhringer, J.; Timmann, D.; Trollmann, R.; Stendel, C.; Kamm, C.; Röbl, M.; Santhanakumaran, V.; Groeschel, S.; Beck-Wödl, S.; et al. Clinical, Imaging, Genetic, and Disease Course Characteristics in Patients with GM2 Gangliosidosis: Beyond Age of Onset. Neurology 2024, 102, e207898. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid Ceramidase Deficiency: Farber Disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Khan, A.; Sergi, C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics 2018, 8, 29. [Google Scholar] [CrossRef]
- Bolukbasi, S.; Dogan, C.; Kiykim, E.; Cakir, A.; Erden, B.; Bayat, A.H.; Elcioglu, M.N.; Aktuglu Zeybek, A.C. Multimodal Imaging Including Optical Coherence Tomography Angiography in Patients with Type B Niemann-Pick Disease. Int. Ophthalmol. 2019, 39, 2545–2552. [Google Scholar] [CrossRef]
- Hufendiek, K.; Lindziute, M.; Kaufeld, J.; Volkmann, I.; Brockmann, D.; Hosari, S.; Hohberger, B.; Mardin, C.; Framme, C.; Tode, J.; et al. Investigation of OCTA Biomarkers in Fabry Disease: A Long Term Follow-Up of Macular Vessel Area Density and Foveal Avascular Zone Metrics. Ophthalmol. Ther. 2023, 12, 2713–2727. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Fundus Autofluorescence. In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; Volume 1085, pp. 15–16. ISBN 978-3-319-95045-7. [Google Scholar]
- Xu, A.; Chen, C. Clinical Application of Ultra-Widefield Fundus Autofluorescence. Int. Ophthalmol. 2021, 41, 727–741. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Fluorescein Angiography. In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; Volume 1085, pp. 7–10. ISBN 978-3-319-95045-7. [Google Scholar]
- Cavallerano, A.A. Ophthalmic Fluorescein Angiography. Optom. Clin. 1996, 5, 1–23. [Google Scholar]
- Szewczuk, A.; Zaleska-Żmijewska, A.; Dziedziak, J.; Szaflik, J.P. Clinical Application of Adaptive Optics Imaging in Diagnosis, Management, and Monitoring of Ophthalmological Diseases: A Narrative Review. Med. Sci. Monit. 2023, 29, e941926. [Google Scholar] [CrossRef]
- Akyol, E.; Hagag, A.M.; Sivaprasad, S.; Lotery, A.J. Adaptive Optics: Principles and Applications in Ophthalmology. Eye 2021, 35, 244–264. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Germain, D.P.; Bacherini, D.; Finocchio, L.; Pacini, B.; Marziali, E.; Lenzetti, C.; Tanini, I.; Koraichi, F.; Coriat, C.; et al. In Vivo Observation of Retinal Vascular Deposits Using Adaptive Optics Imaging in Fabry Disease. Retina 2020, 40, 1623–1629. [Google Scholar] [CrossRef]
- Lombardo, M.; Serrao, S.; Devaney, N.; Parravano, M.; Lombardo, G. Adaptive Optics Technology for High-Resolution Retinal Imaging. Sensors 2012, 13, 334–366. [Google Scholar] [CrossRef]
- Li, Y.; Tang, J. Blood Vessel Tail Artifacts Suppression in Optical Coherence Tomography Angiography. Neurophotonics 2022, 9, 021906. [Google Scholar] [CrossRef]
- Creel, D.J. Electroretinograms. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 160, pp. 481–493. ISBN 978-0-444-64032-1. [Google Scholar]
- Bhatt, Y.; Hunt, D.M.; Carvalho, L.S. The Origins of the Full-Field Flash Electroretinogram b-Wave. Front. Mol. Neurosci. 2023, 16, 1153934. [Google Scholar] [CrossRef]
- Hood, D.C.; Odel, J.G.; Chen, C.S.; Winn, B.J. The Multifocal Electroretinogram. J. Neuro-Ophthalmol. 2003, 23, 225–235. [Google Scholar] [CrossRef]
- Baiano, C.; Zeppieri, M. Visual Evoked Potential. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- International Society for Clinical Electrophysiology of Vision; Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P. ISCEV Standard for Clinical Visual Evoked Potentials: (2016 Update). Doc. Ophthalmol. 2016, 133, 1–9. [Google Scholar] [CrossRef]
- Marmoy, O.R.; Tekavčič Pompe, M.; Kremers, J. Chromatic Visual Evoked Potentials: A Review of Physiology, Methods and Clinical Applications. Prog. Retin. Eye Res. 2024, 101, 101272. [Google Scholar] [CrossRef]
- A Case Refort of Sandhoff Disease—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/15929490/ (accessed on 25 January 2025).
- Larsen, H.W.; Ehler, N. Ocular Manifestations in Tay-Sachs’ and Niemann-Pick’s Diseases. (A Clinical, Pathological, Histochemical and Biochemical Investigation). Acta Ophthalmol. 1965, 43, 285–293. [Google Scholar] [CrossRef]
- Graziano, A.C.E.; Cardile, V. History, Genetic, and Recent Advances on Krabbe Disease. Gene 2015, 555, 2–13. [Google Scholar] [CrossRef]
- Goebel, H.H.; Busch-Hettwer, H.; Bohl, J. Ultrastructural Study of the Retina in Late Infantile Metachromatic Leukodystrophy. Ophthalmic Res. 1992, 24, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Green, W.R. Clinical and Ultrastructural Ocular Histopathologic Studies of Adult-Onset Metachromatic Leukodystrophy. Am. J. Ophthalmol. 1976, 82, 472–479. [Google Scholar] [CrossRef]
- Pfrieger, F.W. The Niemann-Pick Type Diseases—A Synopsis of Inborn Errors in Sphingolipid and Cholesterol Metabolism. Prog. Lipid Res. 2023, 90, 101225. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick Diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Escobar, B.; Solano, M.H.; Zarabanda, L.; Casas, C.P.; Castro, C. Niemann-Pick Disease: An Approach for Diagnosis in Adulthood. Cureus 2019, 11, e4767. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Schuchman, E.H. Acid Sphingomyelinase Deficiency. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Onur, İ.U.; Aşula, M.F.; Ekinci, C.; Mert, M. Macula Halo Syndrome. Int. Ophthalmol. 2019, 39, 1391–1395. [Google Scholar] [CrossRef]
- Servín Muñoz, I.V.; Ortuño-Sahagún, D.; Griñán-Ferré, C.; Pallàs, M.; González-Castillo, C. Alterations in Proteostasis Mechanisms in Niemann-Pick Type C Disease. Int. J. Mol. Sci. 2024, 25, 3806. [Google Scholar] [CrossRef]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High Incidence of Unrecognized Visceral/Neurological Late-Onset Niemann-Pick Disease, Type C1, Predicted by Analysis of Massively Parallel Sequencing Data Sets. Genet. Med. 2016, 18, 41–48. [Google Scholar] [CrossRef]
- Platt, F.M. The Expanding Boundaries of Sphingolipid Lysosomal Storage Diseases; Insights from Niemann-Pick Disease Type C. Biochem. Soc. Trans. 2023, 51, 1777–1787. [Google Scholar] [CrossRef]
- Li, Y.; Cherepanoff, S.; Fung, A.T. Bilateral Macular Halo and Full-Thickness Macular Hole Repair in Niemann-Pick Disease Type B. J. Vitr. Dis. 2024, 8, 462–465. [Google Scholar] [CrossRef]
- Rudich, D.S.; Curcio, C.A.; Wasserstein, M.; Brodie, S.E. Inner Macular Hyperreflectivity Demonstrated by Optical Coherence Tomography in Niemann-Pick Disease. JAMA Ophthalmol. 2013, 131, 1244–1246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Angeli, O.; Nagy, Z.; Schneider, M. Ocular manifestation of an adult Niemann-Pick disease type B. Orv. Hetil. 2023, 164, 1838–1844. [Google Scholar] [CrossRef]
- Bagdonaite-Bejarano, L.; Hansen, R.M.; Fulton, A.B. Microperimetry in Three Inherited Retinal Disorders. Semin. Ophthalmol. 2019, 34, 334–339. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Mikosch, P.; Hughes, D. An Overview on Bone Manifestations in Gaucher Disease. Wien. Med. Wochenschr. 2010, 160, 609–624. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s Disease Registry: Clinical Characteristics, Complications and Treatment of 562 Patients. Orphanet J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef]
- Alaei, M.R.; Tabrizi, A.; Jafari, N.; Mozafari, H. Gaucher Disease: New Expanded Classification Emphasizing Neurological Features. Iran. J. Child. Neurol. 2019, 13, 7–24. [Google Scholar]
- Sidransky, E. Gaucher Disease: Complexity in a “Simple” Disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef]
- Donald, A.; Tan, C.Y.; Chakrapani, A.; Hughes, D.A.; Sharma, R.; Cole, D.; Bardins, S.; Gorges, M.; Jones, S.A.; Schneider, E. Eye Movement Biomarkers Allow for the Definition of Phenotypes in Gaucher Disease. Orphanet J. Rare Dis. 2020, 15, 349. [Google Scholar] [CrossRef]
- Eghbali, A.; Hassan, S.; Seehra, G.; FitzGibbon, E.; Sidransky, E. Ophthalmological Findings in Gaucher Disease. Mol. Genet. Metab. 2019, 127, 23–27. [Google Scholar] [CrossRef]
- Abu-Asab, M.S.; Yeung, I.Y.L.; Ardeljan, C.; Gonzalez, A.N.; Sidransky, E.; Chan, C.-C. Ocular Implications of Gaucher Disease. In Advances in Vision Research, Volume I: Genetic Eye Research in Asia and the Pacific; Prakash, G., Iwata, T., Eds.; Springer: Tokyo, Japan, 2017; pp. 413–423. ISBN 978-4-431-56511-6. [Google Scholar]
- Gupta, N.; Oppenheim, I.; Kauvar, E.; Tayebi, N.; Sidransky, E. Type 2 Gaucher Disease: Phenotypic Variation and Genotypic Heterogeneity. Blood Cells Mol. Dis. 2011, 46, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Shrier, E.M.; Barr, C.C.; Grabowski, G.A. Vitreous Opacities and Retinal Vascular Abnormalities in Gaucher Disease. Arch. Ophthalmol. 2004, 122, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Fujiwaki, T.; Yamaguchi, S.; Tasaka, M.; Takayanagi, M.; Isobe, M.; Taketomi, T. Evaluation of Sphingolipids in Vitreous Bodies from a Patient with Gaucher Disease, Using Delayed Extraction Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 806, 47–51. [Google Scholar] [CrossRef]
- Hopf, S.; Pfeiffer, N.; Liesenfeld, M.; Mengel, K.-E.; Hennermann, J.B.; Schmidtmann, I.; Pitz, S. A Comprehensive Monocentric Ophthalmic Study with Gaucher Disease Type 3 Patients: Vitreoretinal Lesions, Retinal Atrophy and Characterization of Abnormal Saccades. Orphanet J. Rare Dis. 2019, 14, 257. [Google Scholar] [CrossRef] [PubMed]
- Seehra, G.K.; Eghbali, A.; Sidransky, E.; FitzGibbon, E. White Vitreous Opacities in Five Patients with Gaucher Disease Type. Am. J. Med. Genet. A 2020, 182, 808–812. [Google Scholar] [CrossRef]
- Anand, S.; Kidd, D.; Hughes, D. Photo Essay: Retinal Changes in Type 3 Gaucher Disease. Neuroophthalmology 2018, 42, 402–403. [Google Scholar] [CrossRef]
- Grabowski, G.A. Phenotype, Diagnosis, and Treatment of Gaucher’s Disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Patterson, M.C.; Horowitz, M.; Abel, R.B.; Currie, J.N.; Yu, K.T.; Kaneski, C.; Higgins, J.J.; O’Neill, R.R.; Fedio, P.; Pikus, A. Isolated Horizontal Supranuclear Gaze Palsy as a Marker of Severe Systemic Involvement in Gaucher’s Disease. Neurology 1993, 43, 1993–1997. [Google Scholar] [CrossRef]
- Wang, J.S.; Koch, R.L.; Kenney-Jung, D.; Huggins, E.; Sodhi, S.S.; Landstrom, A.P.; Grewal, D.S.; Kishnani, P.S. Gaucher Disease Type 3c: Expanding the Clinical Spectrum of an Ultra-Rare Disease. JIMD Rep. 2024, 65, 313–322. [Google Scholar] [CrossRef]
- Kurolap, A.; Del Toro, M.; Spiegel, R.; Gutstein, A.; Shafir, G.; Cohen, I.J.; Barrabés, J.A.; Feldman, H.B. Gaucher Disease Type 3c: New Patients with Unique Presentations and Review of the Literature. Mol. Genet. Metab. 2019, 127, 138–146. [Google Scholar] [CrossRef]
- Wollstein, G.; Elstein, D.; Strassman, I.; Seelenfreund, M.; Zylbermann, R.; Zimran, A. Preretinal White Dots in Adult-Type Gaucher Disease. Retina 1999, 19, 570. [Google Scholar] [CrossRef]
- Hsing, Y.E.; Foster, A. Preretinal and Posterior Vitreous Deposits in Gaucher Disease. JAMA Ophthalmol. 2014, 132, 992. [Google Scholar] [CrossRef]
- Hua, H.-U.; Haghighi, A.; Shillingford, N.; Lee, T.C.; Nagiel, A. Vitreous Hemorrhage in Type 3 Gaucher Disease: An Angiographic and Pathologic Analysis. Retin. Cases Brief. Rep. 2022, 16, 414–418. [Google Scholar] [CrossRef]
- Sheck, L.H.N.; Wilson, C.J.; Vincent, A.L. Analysis of the Pre-Retinal Opacities in Gaucher Disease Using Spectral Domain Optical Coherent Tomography. Ophthalmic Genet. 2012, 33, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Gekka, T.; Arai, K.; Tsuneoka, H. A Case of Traction Retinal Detachment in a Patient with Gaucher Disease. Ophthalmic Genet. 2017, 38, 273–276. [Google Scholar] [CrossRef]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 Gangliosidosis and Morquio B Disease: An Update on Genetic Alterations and Clinical Findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Scaglia, F. GM1 Gangliosidosis: Review of Clinical, Molecular, and Therapeutic Aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef]
- Tanaka, R.; Momoi, T.; Yoshida, A.; Okumura, M.; Yamakura, S.; Takasaki, Y.; Kiyomasu, T.; Yamanaka, C. Type 3 GM1 Gangliosidosis: Clinical and Neuroradiological Findings in an 11-Year-Old Girl. J. Neurol. 1995, 242, 299–303. [Google Scholar] [CrossRef]
- Roze, E.; Paschke, E.; Lopez, N.; Eck, T.; Yoshida, K.; Maurel-Ollivier, A.; Doummar, D.; Caillaud, C.; Galanaud, D.; Billette de Villemeur, T.; et al. Dystonia and Parkinsonism in GM1 Type 3 Gangliosidosis. Mov. Disord. 2005, 20, 1366–1369. [Google Scholar] [CrossRef]
- Emery, J.M.; Green, W.R.; Wyllie, R.G.; Howell, R.R. GM1-Gangliosidosis. Ocular and Pathological Manifestations. Arch. Ophthalmol. 1971, 85, 177–187. [Google Scholar] [CrossRef]
- Sorcinelli, R.; Sitzia, A.; Loi, M. Cherry-Red Spot, Optic Atrophy and Corneal Cloudings in a Patient Suffering from GM1 Gangliosidosis Type I. Metab. Pediatr. Syst. Ophthalmol. 1987, 10, 62–63. [Google Scholar]
- Goebel, H.H.; Shimokawa, K.; Argyrakis, A.; Pilz, H. The Ultrastructure of the Retina in Adult Metachromatic Leukodystrophy. Am. J. Ophthalmol. 1978, 85, 841–849. [Google Scholar] [CrossRef]
- D’Souza, P.; Farmer, C.; Johnston, J.M.; Han, S.T.; Adams, D.; Hartman, A.L.; Zein, W.; Huryn, L.A.; Solomon, B.; King, K.; et al. GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study. Genet. Med. 2024, 26, 101144. [Google Scholar] [CrossRef]
- Norman, R.M.; Urich, H.; Tingey, A.H.; Goodbody, R.A. Tay-Sachs’ Disease with Visceral Involvement and Its Relationship to Niemann-Pick’s Disease. J. Pathol. Bacteriol. 1959, 78, 409–421. [Google Scholar] [CrossRef]
- Padhi, T.R.; Pattnaik, S.; Kesarwani, S.; Das, T. Macular Cherry-Red Spot Helps Diagnose Rare Storage Disorder in an Infant with Repeated Respiratory Tract Infections: Case Report. Semin. Ophthalmol. 2015, 30, 224–226. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef]
- Gort, L.; de Olano, N.; Macías-Vidal, J.; Coll, M.A.J. Spanish GM2 Working Group GM2 Gangliosidoses in Spain: Analysis of the HEXA and HEXB Genes in 34 Tay-Sachs and 14 Sandhoff Patients. Gene 2012, 506, 25–30. [Google Scholar] [CrossRef]
- Tavasoli, A.R.; Parvaneh, N.; Ashrafi, M.R.; Rezaei, Z.; Zschocke, J.; Rostami, P. Clinical Presentation and Outcome in Infantile Sandhoff Disease: A Case Series of 25 Patients from Iranian Neurometabolic Bioregistry with Five Novel Mutations. Orphanet J. Rare Dis. 2018, 13, 130. [Google Scholar] [CrossRef]
- Xiao, C.; Tifft, C.; Toro, C. Sandhoff Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Hendriksz, C.J.; Corry, P.C.; Wraith, J.E.; Besley, G.T.N.; Cooper, A.; Ferrie, C.D. Juvenile Sandhoff Disease—Nine New Cases and a Review of the Literature. J. Inherit. Metab. Dis. 2004, 27, 241–249. [Google Scholar] [CrossRef]
- Liu, M.; Huang, D.; Wang, H.; Zhao, L.; Wang, Q.; Chen, X. Clinical and Molecular Characteristics of Two Chinese Children with Infantile Sandhoff Disease and Review of the Literature. J. Mol. Neurosci. 2020, 70, 481–487. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Jafari, N.; Nejad Biglari, H.; Jabbeh Dari, S.; Ahmad Abadi, F.; Alaee, M.-R.; Nemati, H.; Saket, S.; Tonekaboni, S.H.; Taghdiri, M.-M.; et al. GM2-Gangliosidosis (Sandhoff and Tay Sachs Disease): Diagnosis and Neuroimaging Findings (An Iranian Pediatric Case Series). Iran. J. Child. Neurol. 2014, 8, 55–60. [Google Scholar]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Ebrahimi, M.; Etemad, K.; Ahmad Abadi, F.; Hosseini Nezhad, Z. GM1 and GM2-Gangliosidosis: Clinical Features, Neuroimaging Findings and Electroencephalography. Iran. J. Child. Neurol. 2024, 18, 127–140. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neuro-Ophthalmology of Late-Onset Tay-Sachs Disease (LOTS)—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/15557512/ (accessed on 25 January 2025).
- Garner, A. Ocular Pathology of GM2 Gangliosidosis—Type 2 (Sandhoff’s Disease). Br. J. Ophthalmol. 1973, 57, 514–520. [Google Scholar] [CrossRef][Green Version]
- Sango, K.; Yamanaka, S.; Ajiki, K.; Arai, N.; Takano, M. Involvement of Retinal Neurons and Pigment Epithelial Cells in a Murine Model of Sandhoff Disease. Ophthalmic Res. 2008, 40, 241–248. [Google Scholar] [CrossRef]
- Udwadia-Hegde, A.; Hajirnis, O. Temporary Efficacy of Pyrimethamine in Juvenile-Onset Tay-Sachs Disease Caused by 2 Unreported HEXA Mutations in the Indian Population. Child. Neurol. Open 2017, 4, 2329048X16687887. [Google Scholar] [CrossRef]
- Toro, C.; Zainab, M.; Tifft, C.J. The GM2 Gangliosidoses: Unlocking the Mysteries of Pathogenesis and Treatment. Neurosci. Lett. 2021, 764, 136195. [Google Scholar] [CrossRef]
- Alasil, T.; Wang, K.; Keane, P.A.; Lee, H.; Baniasadi, N.; de Boer, J.F.; Chen, T.C. Analysis of Normal Retinal Nerve Fiber Layer Thickness by Age, Sex, and Race Using Spectral Domain Optical Coherence Tomography. J. Glaucoma 2013, 22, 532–541. [Google Scholar] [CrossRef]
- Gahramanova, L.; Galbinur, T.; Mammadkhanova, A. LATE-ONSET LYSOSOMAL STORAGE DISORDER WITH MACULAR CHERRY-RED SPOT. Retin. Cases Brief. Rep. 2021, 15, 602–604. [Google Scholar] [CrossRef]
- Abalem, M.F.; Francischini, S.; Carricondo, P.C.; Graziano, R.M. Fundus Autofluorescence in Tay-Sachs Disease. JAMA Ophthalmol. 2014, 132, 876. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A.; Green, W.R.; Moser, A.B.; Tiffany, C. Increased Levels of Ceramide in the Retina of a Patient with Farber’s Disease. Arch. Ophthalmol. 1988, 106, 1163. [Google Scholar] [CrossRef]
- Beraza-Millor, M.; Rodríguez-Castejón, J.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A.; Solinís, M.Á. Systematic Review of Genetic Substrate Reduction Therapy in Lysosomal Storage Diseases: Opportunities, Challenges and Delivery Systems. BioDrugs 2024, 38, 657–680. [Google Scholar] [CrossRef]
- Martina, J.A.; Raben, N.; Puertollano, R. SnapShot: Lysosomal Storage Diseases. Cell 2020, 180, 602–602.e1. [Google Scholar] [CrossRef]
- Van Rappard, D.F.; Boelens, J.J.; Wolf, N.I. Metachromatic Leukodystrophy: Disease Spectrum and Approaches for Treatment. Best. Pr. Res. Clin. Endocrinol. Metab. 2015, 29, 261–273. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Karnati, R.; Talla, V.; Peterson, K.; Laurie, G.W. Lacritin and Other Autophagy Associated Proteins in Ocular Surface Health. Exp. Eye Res. 2016, 144, 4–13. [Google Scholar] [CrossRef]
- Weiter, J.J.; Feingold, M.; Kolodny, E.H.; Raghaven, S.S. Retinal Pigment Epithelial Degeneration Associated with Leukocytic Arylsulfatase A Deficiency. Am. J. Ophthalmol. 1980, 90, 768–772. [Google Scholar] [CrossRef]
- El-Abassi, R.; Singhal, D.; England, J.D. Fabry’s Disease. J. Neurol. Sci. 2014, 344, 5–19. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Nilsson, M.; Kolagari, H.T.; Epstein, D.; Samolov, B.; Olsson, M.; Naess, K.; Oscarson, M.; Teaer Fahnehjelm, K. Visual Outcome, Ocular Findings, and Visual Quality of Life in Patients with Fabry Disease. Ophthalmic Genet. 2022, 43, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Kalkum, G.; Pitz, S.; Karabul, N.; Beck, M.; Pintos-Morell, G.; Parini, R.; Rohrbach, M.; Bizjajeva, S.; Ramaswami, U. Paediatric Fabry Disease: Prognostic Significance of Ocular Changes for Disease Severity. BMC Ophthalmol. 2016, 16, 202. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Hughes, D.A. Fabry Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Torregrosa, J.-V. Current Aspects of Fabry’s Disease. Med. Clin. 2018, 151, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Bini, A.; Mignani, R.; Minuti, B.; Menchini, U. Subfoveal Choroidal Neovascularization in a Patient with Fabry’s Disease. Int. Ophthalmol. 2009, 29, 435–437. [Google Scholar] [CrossRef]
- Samiy, N. Ocular Features of Fabry Disease: Diagnosis of a Treatable Life-Threatening Disorder. Surv. Ophthalmol. 2008, 53, 416–423. [Google Scholar] [CrossRef]
- Minnella, A.M.; Barbano, L.; Verrecchia, E.; Martelli, F.; Pagliei, V.; Gambini, G.; Placidi, G.; Falsini, B.; Caporossi, A.; Manna, R. Macular Impairment in Fabry Disease: A Morpho-Functional Assessment by Swept-Source OCT Angiography and Focal Electroretinography. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2667–2675. [Google Scholar] [CrossRef]
- Kuriyama, A.; Nakamura, S.; Inokuchi, Y.; Abe, H.; Yasuda, H.; Hidaka, Y.; Nagaoka, K.; Soeda, T.; Shimazawa, M.; Hara, H. The Protective Effect of Anti-VEGF-A/Ang-2 Bispecific Antibody on Retinal Vein Occlusion Model Mice. Eur. J. Pharmacol. 2024, 976, 176691. [Google Scholar] [CrossRef]
- Ersoz, M.G.; Ture, G. Cilioretinal Artery Occlusion and Anterior Ischemic Optic Neuropathy as the Initial Presentation in a Child Female Carrier of Fabry Disease. Int. Ophthalmol. 2018, 38, 771–773. [Google Scholar] [CrossRef]
- Lindziute, M.; Kaufeld, J.; Hufendiek, K.; Volkmann, I.; Brockmann, D.; Hosari, S.; Hohberger, B.; Christian, M.; Framme, C.; Jan, T.; et al. Correlation of Retinal Vascular Characteristics with Laboratory and Ocular Findings in Fabry Disease: Exploring Ocular Diagnostic Biomarkers. Orphanet J. Rare Dis. 2023, 18, 314. [Google Scholar] [CrossRef]
- Atiskova, Y.; Rassuli, R.; Koehn, A.F.; Golsari, A.; Wagenfeld, L.; du Moulin, M.; Muschol, N.; Dulz, S. Retinal Hyperreflective Foci in Fabry Disease. Orphanet J. Rare Dis. 2019, 14, 296. [Google Scholar] [CrossRef]
- Puthenparampil, M.; Torresin, T.; Franciotta, S.; Marin, A.; De Napoli, F.; Mauceri, V.A.; Miante, S.; Pilotto, E.; Midena, E.; Gallo, P. Hyper-Reflecting Foci in Multiple Sclerosis Retina Associate with Macrophage/Microglia-Derived Cytokines in Cerebrospinal Fluid. Front. Immunol. 2022, 13, 852183. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Nicolosi, C.; Vicini, G.; Lenzetti, C.; Virgili, G.; Rizzo, S. Computer-Assisted Retinal Vessel Diameter Evaluation in Fabry Disease. Eur. J. Ophthalmol. 2021, 31, 173–178. [Google Scholar] [CrossRef]
- Roorda, A. Adaptive Optics Ophthalmoscopy. J. Refract. Surg. 2000, 16, S602–S607. [Google Scholar] [CrossRef]
- Orsini, J.J.; Escolar, M.L.; Wasserstein, M.P.; Caggana, M. Krabbe Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Wenger, D.A.; Rafi, M.A.; Luzi, P. Molecular Genetics of Krabbe Disease (Globoid Cell Leukodystrophy): Diagnostic and Clinical Implications. Hum. Mutat. 1997, 10, 268–279. [Google Scholar] [CrossRef]
- Hagberg, B.; Kollberg, H.; Sourander, P.; Akesson, H.O. Infantile Globoid Cell Leucodystrophy (Krabbe’s Disease). A Clinical and Genetic Study of 32 Swedish Cases 1953. Neuropädiatrie 1969, 1, 74–88. [Google Scholar] [CrossRef]
- Barczykowski, A.L.; Foss, A.H.; Duffner, P.K.; Yan, L.; Carter, R.L. Death Rates in the U.S. Due to Krabbe Disease and Related Leukodystrophy and Lysosomal Storage Diseases. Am. J. Med. Genet. A 2012, 158A, 2835–2842. [Google Scholar] [CrossRef]
- Greco, M.R.; Lopez, M.A.; Beltran-Quintero, M.L.; Tuc Bengur, E.; Poe, M.D.; Escolar, M.L. Infantile Krabbe Disease (0–12 Months), Progression, and Recommended Endpoints for Clinical Trials. Ann. Clin. Transl. Neurol. 2024, 11, 3064–3080. [Google Scholar] [CrossRef]
- Pavuluri, P.; Vadakedath, S.; Gundu, R.; Uppulety, S.; Kandi, V. Krabbe Disease: Report of a Rare Lipid Storage and Neurodegenerative Disorder. Cureus 2017, 9, e949. [Google Scholar] [CrossRef]
- Bascou, N.; DeRenzo, A.; Poe, M.D.; Escolar, M.L. A Prospective Natural History Study of Krabbe Disease in a Patient Cohort with Onset between 6 Months and 3 Years of Life. Orphanet J. Rare Dis. 2018, 13, 126. [Google Scholar] [CrossRef]
- Jain, M.; De Jesus, O. Krabbe Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Durães, J.; Salsano, E.; Macário, M.D.C. Adult-Onset Krabbe Disease. Neurol. Clin. Pr. 2021, 11, e15–e17. [Google Scholar] [CrossRef]
- Lyon, G.; Hagberg, B.; Evrard, P.; Allaire, C.; Pavone, L.; Vanier, M. Symptomatology of Late Onset Krabbe’s Leukodystrophy: The European Experience. Dev. Neurosci. 1991, 13, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.; Ashton, N. Ultrastructure of the Optic Nerve in Krabbe’s Leucodystrophy. Br. J. Ophthalmol. 1973, 57, 885–891. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van de Vlekkert, D.; Hu, H.; Weesner, J.A.; Fremuth, L.E.; Brown, S.A.; Lu, M.; Gomero, E.; Campos, Y.; Sheppard, H.; d’Azzo, A. AAV-Mediated Gene Therapy for Sialidosis. Mol. Ther. 2024, 32, 2094–2112. [Google Scholar] [CrossRef]
- Diagnosis and Management of Type 1 Sialidosis: Clinical Insights from Long-Term Care of Four Unrelated Patients—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/32752208/ (accessed on 17 February 2025).
- Rossi, S.; Gesualdo, C.; Tartaglione, A.; Bilo, L.; Coppola, A.; Simonelli, F. Bergmeister’s Papilla in a Young Patient with Type 1 Sialidosis: Case Report. BMC Ophthalmol. 2020, 20, 356. [Google Scholar] [CrossRef] [PubMed]
- Daich Varela, M.; Zein, W.M.; Toro, C.; Groden, C.; Johnston, J.; Huryn, L.A.; d’Azzo, A.; Tifft, C.J.; FitzGibbon, E.J. A Sialidosis Type I Cohort and a Quantitative Approach to Multimodal Ophthalmic Imaging of the Macular Cherry-Red Spot. Br. J. Ophthalmol. 2021, 105, 838–843. [Google Scholar] [CrossRef]
- Sergi, C.; Beedgen, B.; Kopitz, J.; Zilow, E.; Zoubaa, S.; Otto, H.F.; Cantz, M.; Linderkamp, O. Refractory Congenital Ascites as a Manifestation of Neonatal Sialidosis: Clinical, Biochemical and Morphological Studies in a Newborn Syrian Male Infant. Am. J. Perinatol. 1999, 16, 133–141. [Google Scholar] [CrossRef]
- NEU1 Mutation in a Korean Infant with Type 2 Sialidosis Presenting as Isolated Fetal Ascites—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25223955/ (accessed on 20 February 2025).
- Ries, M.; Deeg, K.H.; Wölfel, D.; Ibel, H.; Maier, B.; Buheitel, G. Colour Doppler Imaging of Intracranial Vasculopathy in Severe Infantile Sialidosis. Pediatr. Radiol. 1992, 22, 179–181. [Google Scholar] [CrossRef]
- Rosenberg, R.; Halimi, E.; Mention-Mulliez, K.; Cuisset, J.-M.; Holder, M.; Defoort-Dhellemmes, S. Five Year Follow-up of Two Sisters with Type II Sialidosis: Systemic and Ophthalmic Findings Including OCT Analysis. J. Pediatr. Ophthalmol. Strabismus 2013, 50, e33–e36. [Google Scholar] [CrossRef]
- Palmeri, S.; Villanova, M.; Malandrini, A.; van Diggelen, O.P.; Huijmans, J.G.; Ceuterick, C.; Rufa, A.; DeFalco, D.; Ciacci, G.; Martin, J.J.; et al. Type I Sialidosis: A Clinical, Biochemical and Neuroradiological Study. Eur. Neurol. 2000, 43, 88–94. [Google Scholar] [CrossRef]
- Chen, Y.-K.; Lai, C.-H.; Chen, N.-N.; Wu, W.-C.; Ling, X.-C.; Lai, S.-C.; Kao, L.-Y.; Chen, C.-Y. Possible Choroidal Hyperpermeability in Cherry-Red Spot: A Connection to Transretinal Hyperreflective Foveola in Type 1 Sialidosis. Retina 2024, 44, e14–e15. [Google Scholar] [CrossRef]
- Yuan, L. Unraveling the Diagnostic Enigma: Laboratory Diagnosis of Sphingolipid Activator Protein Deficiencies. J. Lab. Precis. Med. 2024, 9, 22. [Google Scholar] [CrossRef]
- Mahlovanyi, B.; Król, N.; Lopushansky, A.; Shpotyuk, Y.; Boussard-Pledel, C.; Bureau, B.; Szmuc, K.; Gruzeł, G.; Łach, K.; Kowal, A.; et al. Diagnostic and Prognostic Perspectives of Fabry Disease via Fiber Evanescent Wave Spectroscopy Advanced by Machine Learning. Biosens. Bioelectron. 2025, 273, 117139. [Google Scholar] [CrossRef]
- San Román, I.; Rodríguez, M.-E.; Caporossi, O.; Zoppetti, C.; Sodi, A.; Mecocci, A.; López, D.; Rodríguez, B.; Gimeno, J.-R. Computer Assisted Retinal Vessel Tortuosity Evaluation in Novel Mutation Fabry Disease: Towards New Prognostic Markers. Retina 2017, 37, 592–603. [Google Scholar] [CrossRef]
- Wagner, S.K.; Fu, D.J.; Faes, L.; Liu, X.; Huemer, J.; Khalid, H.; Ferraz, D.; Korot, E.; Kelly, C.; Balaskas, K.; et al. Insights into Systemic Disease through Retinal Imaging-Based Oculomics. Transl. Vis. Sci. Technol. 2020, 9, 6. [Google Scholar] [CrossRef]
- Honavar, S.G. Oculomics—The Eyes Talk a Great Deal. Indian. J. Ophthalmol. 2022, 70, 713. [Google Scholar] [CrossRef]
- Rozhyna, A.; Somfai, G.M.; Atzori, M.; DeBuc, D.C.; Saad, A.; Zoellin, J.; Müller, H. Exploring Publicly Accessible Optical Coherence Tomography Datasets: A Comprehensive Overview. Diagnostics 2024, 14, 1668. [Google Scholar] [CrossRef]
- Arnould, L.; Meriaudeau, F.; Guenancia, C.; Germanese, C.; Delcourt, C.; Kawasaki, R.; Cheung, C.Y.; Creuzot-Garcher, C.; Grzybowski, A. Using Artificial Intelligence to Analyse the Retinal Vascular Network: The Future of Cardiovascular Risk Assessment Based on Oculomics? A Narrative Review. Ophthalmol. Ther. 2023, 12, 657–674. [Google Scholar] [CrossRef]
- Litjens, G.; Kooi, T.; Bejnordi, B.E.; Setio, A.A.A.; Ciompi, F.; Ghafoorian, M.; van der Laak, J.A.W.M.; van Ginneken, B.; Sánchez, C.I. A Survey on Deep Learning in Medical Image Analysis. Med. Image Anal. 2017, 42, 60–88. [Google Scholar] [CrossRef]
- Kaizu, Y.; Nakao, S.; Wada, I.; Yamaguchi, M.; Fujiwara, K.; Yoshida, S.; Hisatomi, T.; Ikeda, Y.; Hayami, T.; Ishibashi, T.; et al. Imaging of Retinal Vascular Layers: Adaptive Optics Scanning Laser Ophthalmoscopy Versus Optical Coherence Tomography Angiography. Transl. Vis. Sci. Technol. 2017, 6, 2. [Google Scholar] [CrossRef]
- Li, R.; Du, Z.; Qian, X.; Li, Y.; Martinez-Camarillo, J.-C.; Jiang, L.; Humayun, M.S.; Chen, Z.; Zhou, Q. High Resolution Optical Coherence Elastography of Retina under Prosthetic Electrode. Quant. Imaging Med. Surg. 2021, 11, 918–927. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).