
Enhancement of EPR Effect for Passive Tumor Targeting: Current Status and Future Perspectives

 and
and 
Abstract
1. Introduction
2. Distinctive Characteristics of Tumor Microenvironment and Heterogeneity of EPR
2.1. Components of Tumor Microenvironment
2.2. Cell Communication Within Tumor Microenvironment
2.2.1. Direct Intercellular Communication
Gap Junctions
Ligand–Receptor Pairs
Tunnel Nanotubes (TNT) and Tumor Microtubules (TM)
2.2.2. Indirect Cell Communication
Signaling by Extracellular Vesicles
Signaling by Cytokines, Chemokines, and Growth Factors
3. Morphological Characteristics of the Vascular Network
3.1. Vascular Network Morphology and Hypervascularity
3.2. Intravascular Environment
3.3. Types of Blood Vessels
3.4. Functional Characteristics of the Tumor Vascular System
3.4.1. Angiogenesis
Positive Regulators of Angiogenesis
Negative Regulators of Angiogenesis
3.4.2. Extensive Vascular Permeability
3.4.3. Irregular Blood Flow
3.4.4. Decreased Lymphatic Drainage and Increased Interstitial Fluid Pressure (IFP)
Transport of Dissolved Substances Through Vascular Walls
High Interstitial Fluid Pressure (IFP)
Decreased Lymphatic Drainage
3.4.5. Vascular Mediators Related to the EPR Phenomenon
4. The Use of EPR in Passive Tumor Targeting with Nanomedicines
4.1. Active and Passive Tumor Targeting
4.2. Biological Barriers Encountered by Nanoparticles
4.2.1. Nanocarrier Specifications
Utilized Materials
Dependence of the Phenomenon on Nanoparticles’ Size and Shape
Hardness Dependence
Nanoparticles’ Biocompatibility and Stability
Nanoparticles’ Surface Charge Requirement
Conjugation with Polymers—Example of Polyethylene Glycol (PEG)
Significant Parameters
Cellular Barriers for Nucleic-Acid Therapies
5. Enhancing the EPR Effect
5.1. Enhancing the EPR Effect by Targeting the Tumor Microenvironment (TME)
5.1.1. Targeting Molecular Markers in the TME
5.1.2. Enzyme-Activated Drug Delivery Systems
5.1.3. Hyaluronic Acid (HA)-Based Nanodrugs
5.1.4. Targeting EGFR and CD44 in Combination
5.2. Physical Strategies
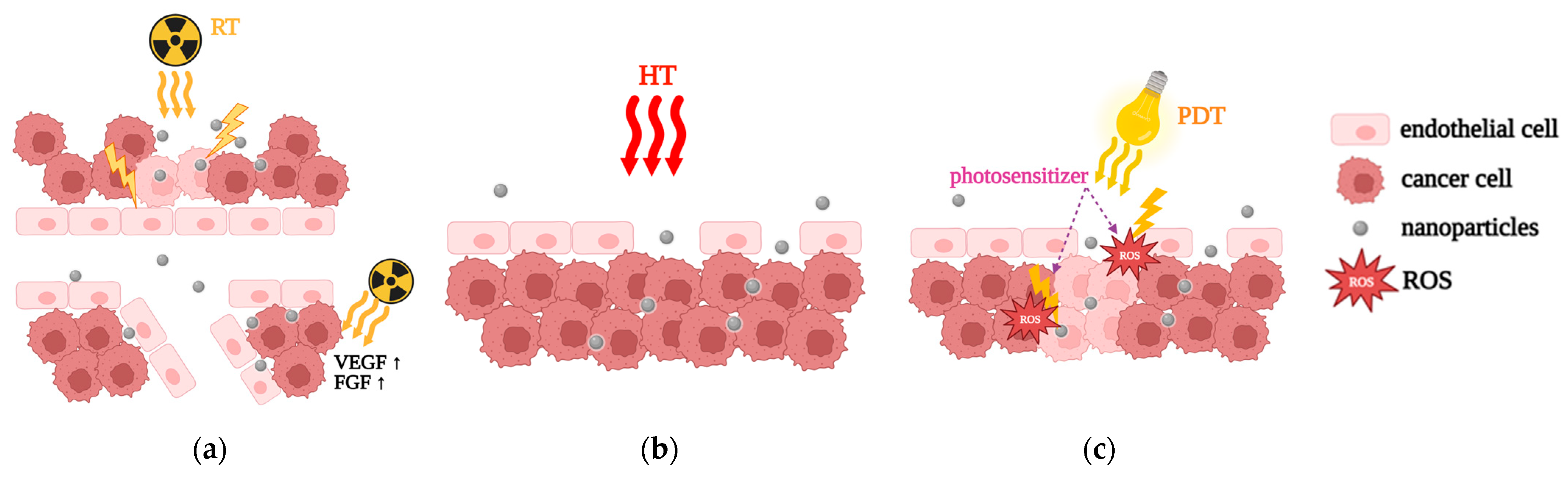
5.2.1. Radiation Therapy (RT)
5.2.2. Hyperthermia (HT)
5.2.3. Photodynamic Therapy (PDT)
5.3. Pharmacological Strategies
5.3.1. Vascular Normalization
5.3.2. Fibrinolytic Co-Therapy
5.3.3. Bradykinin (BK), BK Mediators, Nitric Oxide (NO), and NO Mediators
6. Clinical Trials and Limitations
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Konno, T.; Maeda, H.; Iwai, K.; Maki, S.; Tashiro, S.; Uchida, M.; Miyauchi, Y. Selective targeting of anti-cancer drug and simultaneous image enhancement in solid tumors by arterially administered lipid contrast medium. Cancer 1984, 54, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Lahooti, B.; Akwii, R.G.; Zahra, F.T.; Sajib, M.S.; Lamprou, M.; Alobaida, A.; Lionakis, M.S.; Mattheolabakis, G.; Mikelis, C.M. Targeting endothelial permeability in the EPR effect. J. Control. Release 2023, 361, 212–235. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Xiang, J.; Zhou, Q.; Piao, Y.; Tang, J.; Shao, S.; Zhou, Z.; Bae, Y.H.; Shen, Y. The tumor EPR effect for cancer drug delivery: Current status, limitations, and alternatives. Adv. Drug Deliv. Rev. 2022, 191, 114614. [Google Scholar] [CrossRef]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Ikeda-Imafuku, M.; Wang, L.L.W.; Rodrigues, D.; Shaha, S.; Zhao, Z.; Mitragotri, S. Strategies to improve the EPR effect: A mechanistic perspective and clinical translation. J. Control. Release 2022, 345, 512–536. [Google Scholar] [CrossRef]
- Wu, J. The enhanced permeability and retention (EPR) effect: The significance of the concept and methods to enhance its application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Yhee, J.Y.; Son, S.; Son, S.; Joo, M.K.; Kwon, I.C. The EPR Effect in Cancer Therapy. Cancer Targeted Drug Delivery. In Cancer Targeted Drug Delivery; Bae, Y., Mrsny, R., Park, K., Eds.; Springer: New York, NY, USA, 2013; pp. 621–632. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Youn, Y.S.; Bae, Y.H. Perspectives on the past, present, and future of cancer nanomedicine. Adv. Drug Deliv. Rev. 2018, 130, 3–11. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef]
- Yu, W.; Liu, R.; Zhou, Y.; Gao, H. Size-tunable strategies for a tumor targeted drug delivery system. ACS Cent. Sci. 2020, 6, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Jain, R.K. Design considerations for nanotherapeutics in oncology. Nanomedicine 2015, 10, 1461–1474. [Google Scholar] [CrossRef]
- Kim, T.; Hyeon, T. Applications of inorganic nanoparticles as therapeutic agents. Nanotechnology 2014, 25, 012001. [Google Scholar] [CrossRef]
- Mukherjee, P.; Bhattacharya, R.; Wang, P.; Wang, L.; Basu, S.; Nagy, J.A.; Atala, A.; Mukhopadhyay, D.; Soker, S. Antiangiogenic properties of gold nanoparticles. Clin. Cancer Res. 2005, 11, 3530–3534. [Google Scholar] [CrossRef]
- Barui, A.K.; Nethi, S.K.; Haque, S.; Basuthakur, P.; Patra, C.R. Recent development of metal nanoparticles for angiogenesis study and their therapeutic applications. ACS Appl. Bio Mater. 2019, 2, 5492–5511. [Google Scholar] [CrossRef]
- Kang, H.; Rho, S.; Stiles, W.R.; Hu, S.; Baek, Y.; Hwang, D.W.; Kashiwagi, S.; Kim, M.S.; Choi, H.S. Size-dependent EPR effect of polymeric nanoparticles on tumor targeting. Adv. Healthc. Mater. 2020, 9, e1901223. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Ray, S.; Li, Z.; Hsu, C.H.; Hwang, L.P.; Lin, Y.C.; Chou, P.T.; Lin, Y.Y. Dendrimer- and copolymer-based nanoparticles for magnetic resonance cancer theranostics. Theranostics 2018, 8, 6322–6349. [Google Scholar] [CrossRef] [PubMed]
- Doppalapudi, S.; Jain, A.; Domb, A.J.; Khan, W. Biodegradable polymers for targeted delivery of anti-cancer drugs. Expert Opin. Drug Deliv. 2016, 13, 891–909. [Google Scholar] [CrossRef] [PubMed]
- García-Pinel, B.; Porras-Alcalá, C.; Ortega-Rodríguez, A.; Sarabia, F.; Prados, J.; Melguizo, C.; López-Romero, J.M. Lipid-based nanoparticles: Application and recent advances in cancer treatment. Nanomaterials 2019, 9, 638. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Bendayan, R.; Rauth, A.M.; Li, Y.; Wu, X.Y. Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Adv. Drug Deliv. Rev. 2007, 59, 491–504. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, Q.; Liu, W.; Li, Y.; Tang, H.; Liu, X.; Yang, X. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int. J. Nanomed. 2014, 10, 257–270. [Google Scholar] [CrossRef]
- Lin, X.; Gao, R.; Zhang, Y.; Qi, N.; Zhang, Y.; Zhang, K.; He, H.; Tang, X. Lipid nanoparticles for chemotherapeutic applications: Strategies to improve anticancer efficacy. Expert Opin. Drug Deliv. 2012, 9, 767–781. [Google Scholar] [CrossRef]
- Sailor, M.J.; Park, J.-H. Hybrid nanoparticles for detection and treatment of cancer. Adv. Mater. 2012, 24, 3779–3802. [Google Scholar] [CrossRef]
- Bort, G.; Lux, F.; Dufort, S.; Crémillieux, Y.; Verry, C.; Tillement, O. EPR-mediated tumor targeting using ultrasmall-hybrid nanoparticles: From animal to human with theranostic AGuIX nanoparticles. Theranostics 2020, 10, 1319–1331. [Google Scholar] [CrossRef]
- Upreti, M.; Jyoti, A.; Sethi, P. Tumor microenvironment and nanotherapeutics. Transl. Cancer Res. 2013, 4, 309–319. [Google Scholar] [CrossRef]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2020, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, A.; Xu, J.; Wang, C.; He, F.; Yang, D.; Gai, S.; Yang, P.; Lin, J.; Jin, D.; Xing, B. Tumour microenvironment responsive nanoconstructs for cancer theranostic. Nano Today 2019, 26, 16–56. [Google Scholar] [CrossRef]
- Weber, C.E.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef]
- Arneth, B. Tumor microenvironment. Medicina 2020, 56, 15. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2019, 216, 152729. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulou, P.A.; Koufaki, M.A.; Kostourou, V. The adhesome network: Key components shaping the tumour stroma. Cancers 2021, 13, 525. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Farc, O.; Cristea, V. An overview of the tumor microenvironment, from cells to complex networks. Exp. Ther. Med. 2021, 21, 96. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Atiya, H.; Frisbie, L.; Pressimone, C.; Coffman, L. Mesenchymal stem cells in the tumor microenvironment. In Tumor Microenvironment: Advances in Experimental Medicine and Biology; Birbrair, A., Ed.; Springer: Cham, Switzerland, 2020; Volume 1234, pp. 31–42. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, J.; Li, L.; Liao, S.; He, J.; Zhou, S.; Zhou, Y. Pericytes in the tumor microenvironment. Cancer Lett. 2023, 556, 216074. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A. Chemokines and cancer. Int. J. Cancer 2006, 119, 2026–2029. [Google Scholar] [CrossRef] [PubMed]
- Goustin, A.S.; Leof, E.B.; Shipley, G.D.; Moses, H.L. Growth factors and cancer. Cancer Res. 1986, 46, 1015–1029. [Google Scholar]
- Wang, M.; Rousseau, B.; Qiu, K.; Huang, G.; Zhang, Y.; Su, H.; Le Bihan-Benjamin, C.; Khati, I.; Artz, O.; Foote, M.B.; et al. Killing tumor-associated bacteria with a liposomal antibiotic generates neoantigens that induce anti-tumor immune responses. Nat. Biotechnol. 2024, 42, 1263–1274. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Meşe, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef]
- Schmidt, K.; Windler, R.; De Wit, C. Communication through gap junctions in the endothelium. Adv. Pharmacol. 2016, 77, 209–240. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, A.; Hainz, N.; Tschernig, T.; Meier, C. Facets of communication: Gap junction ultrastructure and function in cancer stem cells and tumor cells. Cancers 2019, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, S.; Song, L.; Wang, B.; Wei, L.; Zhang, F. Applications and analytical tools of cell communication based on ligand-receptor interactions at single cell level. Cell Biosci. 2021, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M. Claudin-based barrier in simple and stratified cellular sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta Biomembr. 2009, 1788, 872–891. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Nature 2008, 453, 1000–1004. [Google Scholar] [CrossRef]
- Mittal, R.; Karhu, E.; Wang, J.S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell. Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef]
- Dagar, S.; Pathak, D.; Oza, H.V.; Mylavarapu, S.V. Tunneling nanotubes and related structures: Molecular mechanisms of formation and function. Biochem. J. 2021, 478, 3977–3998. [Google Scholar] [CrossRef] [PubMed]
- Gurke, S.; Barroso, J.F.; Gerdes, H.H. The art of cellular communication: Tunneling nanotubes bridge the divide. Histochem. Cell Biol. 2008, 129, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling nanotubes and tumor microtubes in cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef]
- Schubert, A.; Boutros, M. Extracellular vesicles and oncogenic signaling. Mol. Oncol. 2021, 15, 3–26. [Google Scholar] [CrossRef]
- Fonseca, P.; Vardaki, I.; Occhionero, A.; Panaretakis, T. Metabolic and signaling functions of cancer cell-derived extracellular vesicles. Int. Rev. Cell Mol. Biol. 2016, 326, 175–199. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Contag, C.H. Signaling by extracellular vesicles advances cancer hallmarks. Trends Cancer 2016, 2, 84–94. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.; Niu, M.; Zhang, H.; Wu, Y.; Wu, K.; Dai, Z. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 176. [Google Scholar] [CrossRef]
- Vicari, A.P.; Caux, C. Chemokines in cancer. Cytokine Growth Factor Rev. 2002, 13, 143–154. [Google Scholar] [CrossRef]
- Lacalle, R.A.; Blanco, R.; Carmona-Rodríguez, L.; Martín-Leal, A.; Mira, E.; Mañes, S. Chemokine receptor signaling and the hallmarks of cancer. Int. Rev. Cell Mol. Biol. 2017, 331, 181–244. [Google Scholar] [CrossRef]
- Subhan, M.A.; Yalamarty, S.S.K.; Filipczak, N.; Parveen, F.; Torchilin, V.P. Recent advances in tumor targeting via EPR effect for cancer treatment. J. Pers. Med. 2021, 11, 571. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular permeability-the essentials. Ups. J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef]
- Nussenbaum, F.; Herman, I.M. Tumor angiogenesis: Insights and innovations. J. Oncol. 2010, 2010, 132641. [Google Scholar] [CrossRef]
- Hiratsuka, S. Vasculogenensis, angiogenesis and special features of tumor blood vessels. Front. Biosci. 2011, 16, 1413–1427. [Google Scholar] [CrossRef]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug delivery: Is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, C.I.; Aschie, M.; Deacu, M.; Bosoteanu, M.; Vamesu, S.; Enciu, M.; Bălţătescu, G.I.; Cozaru, G.C.; Mitroi, A.F.; Voda, R.I. Implications of cellular immaturity in necrosis and microvascularization in glioblastomas IDH-wild-type. Clin. Pract. 2022, 12, 1054–1068. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metastasis 2012, 29, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Tumor blood vessels: Structure, function and classification. In Antiangiogenic Agents in Cancer Therapy. Cancer Drug Discovery and Development; Teicher, B.A., Ellis, L.M., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 205–224. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Cao, Y. Tumor angiogenesis and therapy. Biomed. Pharmacother. 2005, 59, S340–S343. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis. Adv. Cancer Res. 1985, 43, 175–203. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef]
- Detmar, M. Tumor angiogenesis. J. Investig. Dermatol. Symp. Proc. 2000, 5, 20–23. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neurooncol. 2000, 50, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nikitenko, L.L. Vascular endothelium in cancer. Cell Tissue Res. 2009, 335, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Benkheil, M.; Mitola, S.; Struyf, S.; Liekens, S. Tumor angiogenesis revisited: Regulators and clinical implications. Med. Res. Rev. 2017, 37, 1231–1274. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- García-Román, J.; Zentella-Dehesa, A. Vascular permeability changes involved in tumor metastasis. Cancer Lett. 2013, 335, 259–269. [Google Scholar] [CrossRef]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef]
- Kim, J.; Cho, H.; Lim, D.-K.; Joo, M.K.; Kim, K. Perspectives for improving the tumor targeting of nanomedicine via the EPR effect in clinical tumors. Int. J. Mol. Sci. 2023, 24, 10082. [Google Scholar] [CrossRef]
- Fukumura, D.; Jain, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef]
- Netti, P.A.; Roberge, S.; Boucher, Y.; Baxter, L.T.; Jain, R.K. Effect of transvascular fluid exchange on pressure-flow relationship in tumors: A proposed mechanism for tumor blood flow heterogeneity. Microvasc. Res. 1996, 52, 27–46. [Google Scholar] [CrossRef]
- Raghunand, N.; Gatenby, R.A.; Gillies, R.J. Microenvironmental and cellular consequences of altered blood flow in tumours. Br. J. Radiol. 2003, 76, S11–S22. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar] [PubMed]
- Jain, R.K. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987, 6, 559–593. [Google Scholar] [CrossRef]
- Salavati, H.; Debbaut, C.; Pullens, P.; Ceelen, W. Interstitial fluid pressure as an emerging biomarker in solid tumors. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188792. [Google Scholar] [CrossRef] [PubMed]
- Boucher, Y.; Leunig, M.; Jain, R.K. Tumor angiogenesis and interstitial hypertension. Cancer Res. 1996, 56, 4264–4266. [Google Scholar]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor interstitial fluid pressure—A link between tumor hypoxia, microvascular density, and lymph node metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef]
- Zhou, H.; Lei, P.-j.; Padera, T.P. Progression of metastasis through lymphatic system. Cells 2021, 10, 627. [Google Scholar] [CrossRef]
- Nakamura, H.; Jun, F.; Maeda, H. Development of next-generation macromolecular drugs based on the EPR effect: Challenges and pitfalls. Expert Opin. Drug Deliv. 2014, 12, 53–64. [Google Scholar] [CrossRef]
- Karpanen, T.; Alitalo, K. Lymphatic vessels as targets of tumor therapy? J. Exp. Med. 2001, 194, F37–F42. [Google Scholar] [CrossRef]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzym. Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Maeda, H. The link between infection and cancer: Tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci. 2013, 104, 779–789. [Google Scholar] [CrossRef]
- Taurin, S.; Greish, K. Enhanced Vascular Permeability in Solid Tumors: A Promise for Anticancer Nanomedicine. In Tight Junctions in Cancer Metastasis. Cancer Metastasis—Biology and Treatment; Martin, T., Jiang, W., Eds.; Springer: Dordrecht, The Netherlands, 2013; Volume 19, pp. 81–118. [Google Scholar] [CrossRef]
- Maeda, H.; Kabanov, A.; Kataoka, K.; Okano, T. (Eds.) Polymer Drugs in the Clinical Stage: Advantages and Prospects; Springer: New York, NY, USA, 2003; Volume 519, p. 226. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Hussein, W.; Refaat, T. Passive targeting of nanoparticles to cancer: A comprehensive review of the literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Maeda, H.; Fang, J. Factors affecting the dynamics and heterogeneity of the EPR effect: Pathophysiological and pathoanatomic features, drug formulations and physicochemical factors. Expert Opin. Drug Deliv. 2021, 19, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Appiah, E.; Nakamura, H.; Assumang, A.; Etrych, T.; Haratake, M. Chemical modification of bradykinin-polymer conjugates for optimum delivery of nanomedicines to tumors. Nanomed. Nanotechnol. Biol. Med. 2024, 57, 102744. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Kimura, M.; Yamamoto, T.; Maeda, H. Involvement of the kinin-generating cascade in enhanced vascular permeability in tumor tissue. Jpn. J. Cancer Res. 1988, 79, 1327–1334. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y.; Kato, H. Purification and identification of [hydroxyprolyl3] bradykinin in ascitic fluid from a patient with gastric cancer. J. Biol. Chem. 1988, 263, 16051–16054. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Mori, Y.; Feng, H.; Phan, K.Q.; Kishimura, A.; Kang, J.H.; Mori, T.; Katayama, Y. Rapid and continuous accumulation of nitric oxide-releasing liposomes in tumors to augment the enhanced permeability and retention (EPR) effect. Int. J. Pharm. 2019, 565, 481–487. [Google Scholar] [CrossRef]
- Narum, S.M.; Le, T.; Le, D.P.; Lee, J.C.; Donahue, N.D.; Yang, W.; Wilhelm, S. Passive targeting in nanomedicine: Fundamental concepts, body interactions, and clinical potential. In Micro and Nano Technologies, Nanoparticles for Biomedical Applications; Chung, E.J., Leon, L., Rinaldi, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 37–53. [Google Scholar] [CrossRef]
- Parveen, S.; Sahoo, S.K. Nanomedicine: Clinical applications of polyethylene glycol conjugated proteins and drugs. Clin. Pharmacokinet. 2006, 45, 965–988. [Google Scholar] [CrossRef]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- Li, R.; Zheng, K.; Yuan, C.; Chen, Z.; Huang, M. Be active or not: The relative contribution of active and passive tumor targeting of nanomaterials. Nanotheranostics 2017, 1, 346–357. [Google Scholar] [CrossRef]
- Pathak, Y.V. Surface Modification of Nanoparticles for Targeted Drug Delivery; Springer: Cham, Switzerland, 2020; 499p. [Google Scholar] [CrossRef]
- Patel, P.; Hanini, A.; Shah, A.; Patel, D.; Patel, S.; Bhatt, P.; Pathak, Y.V. Surface modification of nanoparticles for targeted drug delivery. In Surface Modification of Nanoparticles for Targeted Drug Delivery; Pathak, Y., Ed.; Springer: Cham, Switzerland, 2019; pp. 19–31. [Google Scholar] [CrossRef]
- Onzi, G.; Guterres, S.S.; Pohlmann, A.R.; Frank, L.A. Passive Targeting and the Enhanced Permeability and Retention (EPR) Effect. In The ADME Encyclopedia; Talevi, A., Ed.; Springer: Cham, Switzerland, 2020; pp. 1–13. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Halwani, A.A. Development of pharmaceutical nanomedicines: From the bench to the market. Pharmaceutics 2022, 14, 106. [Google Scholar] [CrossRef]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Allela, O.Q.B.; Pecho, R.D.C.; Jayasankar, N.; Rao, D.P.; Thamaraikani, T.; Vasanthan, M.; Viktor, P.; Lakshmaiya, N.; et al. Progressing nanotechnology to improve targeted cancer treatment: Overcoming hurdles in its clinical implementation. Mol. Cancer 2023, 22, 169. [Google Scholar] [CrossRef]
- Parveen, S.; Sahoo, S.K. Polymeric nanoparticles for cancer therapy. J. Drug Target. 2008, 16, 108–123. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Q.; Peng, F.; Liu, L.; Gong, C. Strategies of polymeric nanoparticles for enhanced internalization in cancer therapy. Colloids Surf. B 2015, 135, 56–72. [Google Scholar] [CrossRef]
- Goodall, S.; Jones, M.L.; Mahler, S. Monoclonal antibody-targeted polymeric nanoparticles for cancer therapy—Future pro-spects. J. Chem. Technol. Biotechnol. 2014, 90, 1169–1176. [Google Scholar] [CrossRef]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef]
- Black, K.C.; Wang, Y.; Luehmann, H.P.; Cai, X.; Xing, W.; Pang, B.; Zhao, Y.; Cutler, C.S.; Wang, L.V.; Liu, Y.; et al. Radio-active 198Au-doped nanostructures with different shapes for in vivo analyses of their biodistribution, tumor uptake, and intratumoral distribution. ACS Nano 2014, 8, 4385–4394. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Xu, M.; Liu, S. Single-particle hyperspectral imaging for monitoring of gold nanoparticle aggregates in macrophages. J. Phys. Chem. B 2023, 127, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Baeza, A.; Manzano, M.; Colilla, M.; Vallet-Regí, M. Recent advances in mesoporous silica nanoparticles for antitumor therapy: Our contribution. Biomater. Sci. 2016, 4, 803–813. [Google Scholar] [CrossRef]
- He, C.; Lu, J.; Lin, W. Hybrid nanoparticles for combination therapy of cancer. J. Control. Release 2015, 219, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Taurin, S.; Nehoff, H.; Greish, K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J. Control. Release 2012, 164, 265–275. [Google Scholar] [CrossRef]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. 2008, 26, 57–64. [Google Scholar] [CrossRef]
- Loverde, S.M.; Klein, M.L.; Discher, D.E. Nanoparticle shape improves delivery: Rational coarse grain molecular dynamics (rCG-MD) of taxol in worm-like PEG-PCL micelles. Adv. Mater. 2012, 24, 3823–3830. [Google Scholar] [CrossRef]
- Hui, Y.; Yi, X.; Hou, F.; Wibowo, D.; Zhang, F.; Zhao, D.; Gao, H.; Zhao, C.X. Role of nanoparticle mechanical properties in cancer drug delivery. ACS Nano 2019, 13, 7410–7424. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Assani, K.; Neidhard-Doll, A.; Goswami, T. Mechanical properties of nanoparticles in the drug delivery kinetics. J. Pharm. Biopharm. Res. 2022, 4, 248–255. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zang, F.; Wu, H.; Li, J.; Xie, J.; Ma, M.; Gu, N.; Zhang, Y. Using PEGylated magnetic nanoparticles to describe the EPR effect in tumor for predicting therapeutic efficacy of micelle drugs. Nanoscale 2018, 10, 1788–1797. [Google Scholar] [CrossRef]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly(ethylene glycol)-prodrug conjugates: Concept, design, and applications. J. Drug Deliv. 2012, 2012, 103973. [Google Scholar] [CrossRef]
- Hong, M.; Zhu, S.; Jiang, Y.; Tang, G.; Pei, Y. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin. J. Control. Release 2009, 133, 96–102. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nano-materials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef]
- Dutta, S.; Ganguly, B.N. Characterization of ZnO nanoparticles grown in presence of folic acid template. J. Nanobiotechnol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar] [CrossRef]
- Linsley, C.S.; Wu, B.M. Recent advances in light-responsive on-demand drug-delivery systems. Ther. Deliv. 2017, 8, 89–107. [Google Scholar] [CrossRef]
- Arruebo, M.; Fernandez-Pacheco, R.; Ibarra, M.R.; Santamaría, J. Magnetic nanoparticles for drug delivery. Nano Today 2007, 2, 22–32. [Google Scholar] [CrossRef]
- Jung, H.N.; Lee, S.Y.; Lee, S.; Youn, H.; Im, H.J. Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics 2022, 12, 7509–7531. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Xia, F.; Zhang, J.; Shi, Q.; Meng, Y.; Wang, C.; Pang, H.; Gu, L.; Xu, C.; Guo, Q.; et al. Advanced strategies for overcoming endosomal/lysosomal barrier in nanodrug delivery. Research 2023, 6, 0148. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR effect: Combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef]
- Fu, S.; Xu, X.; Ma, Y.; Zhang, S.; Zhang, S. RGD peptide-based non-viral gene delivery vectors targeting integrin αvβ3 for cancer therapy. J. Drug Target. 2019, 27, 1–11. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Jiang, K.; Zhang, Y.; Zhan, C.; Ying, M.; Zhang, M.; Lu, L.; Wang, R.; Wang, S.; et al. Liposomes with cyclic RGD peptide motif triggers acute immune response in mice. J. Control. Release 2019, 293, 201–214. [Google Scholar] [CrossRef]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.H.; Kang, S.W.; Seo, J.W.; Hyun, S.W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xu, C.; Zhao, X.; Lin, C.; Yang, X.; Xin, X.; Zhang, L.; Qin, C.; Han, X.; Yang, L.; et al. Nanoplatform assembled from a CD44-targeted prodrug and smart liposomes for dual targeting of tumor microenvironment and cancer cells. ACS Nano 2018, 12, 1519–1536. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; Contreras, A.M.; Merino, M.; Navarro, I.; de Ilarduya, C.T.; Trocóniz, I.F.; Koning, G.; Garrido, M.J. EGF-liposomes promote efficient EGFR targeting in xenograft colocarcinoma model. Nanomedicine 2016, 11, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Korn, R.L.; Rosen, L.S.; LoRusso, P.; Dychter, S.S.; Zhu, J.; Maneval, D.C.; Jiang, P.; Shepard, H.M.; Frost, G.; et al. Phase 1 trials of PEGylated recombinant human hyaluronidase PH20 in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 153–161. [Google Scholar] [CrossRef]
- Park, J.S.; Qiao, L.; Su, Z.Z.; Hinman, D.; Willoughby, K.; McKinstry, R.; Yacoub, A.; Duigou, G.J.; Young, C.S.; Grant, S.; et al. Ionizing radiation modulates vascular endothelial growth factor (VEGF) expression through multiple mitogen activated protein kinase dependent pathways. Oncogene 2001, 20, 3266–3280. [Google Scholar] [CrossRef]
- Dhaliwal, A.; Zheng, G. Improving accessibility of EPR-insensitive tumor phenotypes using EPR-adaptive strategies: Designing a new perspective in nanomedicine delivery. Theranostics 2019, 9, 8091–8108. [Google Scholar] [CrossRef]
- Davies Cde, L.; Lundstrøm, L.M.; Frengen, J.; Eikenes, L.; Bruland, S.Ø.S.; Kaalhus, O.; Hjelstuen, M.H.; Brekken, C. Radiation improves the distribution and uptake of liposomal doxorubicin (caelyx) in human osteosarcoma xenografts. Cancer Res. 2004, 64, 547–553. [Google Scholar] [CrossRef]
- Lammers, T.; Peschke, P.; Kühnlein, R.; Subr, V.; Ulbrich, K.; Debus, J.; Huber, P.; Hennink, W.; Storm, G. Effect of radiotherapy and hyperthermia on the tumor accumulation of HPMA copolymer-based drug delivery systems. J. Control. Release 2007, 117, 333–341. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P.; Li, F.; Jin, X.; Li, J.; Chen, W.; Li, Q. Metal-based nanoenhancers for future radiotherapy: Radiosensitizing and synergistic effects on tumor cells. Theranostics 2018, 8, 1824–1849. [Google Scholar] [CrossRef]
- Ashton, J.R.; Castle, K.D.; Qi, Y.; Kirsch, D.G.; West, J.L.; Badea, C.T. Dual-energy CT imaging of tumor liposome delivery after gold nanoparticle-augmented radiation therapy. Theranostics 2018, 8, 1782–1797. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Regenold, M.; Allen, C. Hyperthermia can alter tumor physiology and improve chemo- and radio-therapy efficacy. Adv. Drug Deliv. Rev. 2020, 163–164, 98–124. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, T.; Yang, Y.; Jiang, L.; Wang, W.; Fu, L.; Zhu, Y.; Hao, Y. Activatable nanomedicine for overcoming hypoxia-induced resistance to chemotherapy and inhibiting tumor growth by inducing collaborative apoptosis and ferroptosis in solid tumors. Biomaterials 2021, 268, 120537. [Google Scholar] [CrossRef]
- Vujaskovic, Z.; Kim, D.W.; Jones, E.; Lan, L.; McCall, L.; Dewhirst, M.W.; Craciunescu, O.; Stauffer, P.; Liotcheva, V.; Betof, A.; et al. A phase I/II study of neoadjuvant liposomal doxorubicin, paclitaxel, and hyperthermia in locally advanced breast cancer. Int. J. Hyperth. 2010, 26, 514–521. [Google Scholar] [CrossRef]
- Zagar, T.M.; Vujaskovic, Z.; Formenti, S.; Rugo, H.; Muggia, F.; O’Connor, B.; Myerson, R.; Stauffer, P.; Hsu, I.C.; Diederich, C.; et al. Two phase I dose-escalation/pharmacokinetics studies of low temperature liposomal doxorubicin (LTLD) and mild local hyperthermia in heavily pretreated patients with local regionally recurrent breast cancer. Int. J. Hyperth. 2014, 30, 285–294. [Google Scholar] [CrossRef]
- Kim, Y.; Uthaman, S.; Pillarisetti, S.; Noh, K.; Huh, K.M.; Park, I.K. Bioactivatable reactive oxygen species-sensitive nanopar-ticulate system for chemo-photodynamic therapy. Acta Biomater. 2020, 108, 273–284. [Google Scholar] [CrossRef]
- Huang, T.; Zhao, M.; Yu, Q.; Feng, Z.; Xie, M.; Liu, S.; Zhang, K.Y.; Zhao, Q.; Huang, W. De novo design of polymeric carrier to photothermally release singlet oxygen for hypoxic tumor treatment. Research 2019, 2019, 9269081. [Google Scholar] [CrossRef]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Chen, J.; Yang, Z.; Liang, C.; Yang, Y.; Liu, Z. Tumor vasculature normalization by orally fed erlotinib to modulate the tumor microenvironment for enhanced cancer nanomedicine and immunotherapy. Biomaterials 2017, 148, 69–80. [Google Scholar] [CrossRef]
- Caine, G.J.; Stonelake, P.S.; Lip, G.Y.; Kehoe, S.T. The hypercoagulable state of malignancy: Pathogenesis and current debate. Neoplasia 2002, 4, 465–473. [Google Scholar] [CrossRef]
- Navi, B.B.; Reiner, A.S.; Kamel, H.; Iadecola, C.; Okin, P.M.; Tagawa, S.T.; Panageas, K.S.; DeAngelis, L.M. Arterial throm-boembolic events preceding the diagnosis of cancer in older persons. Blood 2019, 133, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jiang, T.; She, X.; Shen, S.; Wang, S.; Deng, J.; Shi, W.; Mei, H.; Hu, Y.; Pang, Z.; et al. Fibrin degradation by rtPA enhances the delivery of nanotherapeutics to A549 tumors in nude mice. Biomaterials 2016, 96, 63–71. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Sadhukha, T.; Kim, H.; Khanna, V.; Koniar, B.; Panyam, J. Fibrinolytic enzyme cotherapy improves tumor perfusion and therapeutic efficacy of anticancer nanomedicine. Cancer Res. 2017, 77, 1465–1475. [Google Scholar] [CrossRef]
- Zuraw, B.L.; Christiansen, S.C. HAE pathophysiology and underlying mechanisms. Clin. Rev. Allergy Immunol. 2016, 51, 216–229. [Google Scholar] [CrossRef]
- Fang, J.; Liao, L.; Yin, H.; Nakamura, H.; Shin, T.; Maeda, H. Enhanced bacterial tumor delivery by modulating the EPR effect and therapeutic potential of Lactobacillus casei. J. Pharm. Sci. 2014, 103, 3235–3243. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, T.; Tuo, Y.; Jin, K.; Luo, Z.; Shi, W.; Mei, H.; Hu, Y.; Pang, Z.; Jiang, X. Captopril improves tumor nanomedicine delivery by increasing tumor blood perfusion and enlarging endothelial gaps in tumor blood vessels. Cancer Lett. 2017, 410, 12–19. [Google Scholar] [CrossRef]
- Yang, B.; Cai, B.; Deng, P.; Wu, X.; Guan, Y.; Zhang, B.; Cai, W.; Schaper, J.; Schaper, W. Nitric oxide increases arterial endotheial permeability through mediating VE-cadherin expression during arteriogenesis. PLoS ONE 2015, 10, e0127931. [Google Scholar] [CrossRef] [PubMed]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention effect with nitric oxide-generating agents improves the therapeutic effects of nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef]
- Andrew, Z.; Wang, A.Z. EPR or no EPR? The billion-dollar question. Sci. Transl. Med. 2015, 7, 294ec112. [Google Scholar] [CrossRef]
- Schiering, C.; Guarnerio, J.; Basso, V.; Muzio, L.; Mondino, A. Antigen-experienced CD4(+) T cells limit naïve T-cell priming in response to therapeutic vaccination in vivo. Cancer Res. 2010, 70, 6161–6170. [Google Scholar] [CrossRef]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: Clinical implications. Cancer Res. 1999, 59, 3776–3782. [Google Scholar] [PubMed]
- Almutairi, S.; Kalloush, H.M.; Manoon, N.A.; Bardaweel, S.K. Matrix Metalloproteinases Inhibitors in Cancer Treatment: An Updated Review (2013–2023). Molecules 2023, 28, 5567. [Google Scholar] [CrossRef] [PubMed]
- Look, J.; Wilhelm, N.; von Briesen, H.; Noske, N.; Günther, C.; Langer, K.; Gorjup, E. Ligand-modified human serum albumin nanoparticles for enhanced gene delivery. Mol. Pharm. 2015, 12, 3202–3213. [Google Scholar] [CrossRef]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef]
- Phase 3 Study of ThermoDox with Radiofrequency Ablation (RFA) in Treatment of Hepatocellular Carcinoma (HCC). Available online: https://clinicaltrials.gov/search?term=NCT00617981 (accessed on 13 September 2024).
- COVID-OUT: Early Outpatient Treatment for SARS-CoV-2 Infection (COVID-19). Available online: https://clinicaltrials.gov/search?term=NCT04510194 (accessed on 13 September 2024).
- Magnetic Nanoparticle Thermoablation-Retention and Maintenance in the Prostate:A Phase 0 Study in Men (MAGNABLATE I). Available online: https://clinicaltrials.gov/search?term=NCT02033447 (accessed on 13 September 2024).
- Disulfiram in Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/search?term=NCT02678975 (accessed on 13 September 2024).
- Study of MM-398 with or Without 5-FU/LV, Versus 5-FU/LV in Patients with Metastatic Pancreatic Cancer (NAPOLI-1). Available online: https://clinicaltrials.gov/search?term=NCT01494506 (accessed on 13 September 2024).



{kind=link}
{kind=link}
{kind=link}
| Components of the Tumor Microenvironment | |
|---|---|
| Cellular Components | |
| Component | Characteristics |
| Cancerous [31] |
|
| Fibroblasts [31,45] |
|
| Endothelial cells [31,45] |
|
| Pericytes [31] |
|
| Immune cells [31,46] |
|
| Adipocytes [31] |
|
| Mesenchymal stem cells (MSCs) [47] |
|
| Non-cellular components | |
| Component | Characteristics |
| Extracellular matrix (ECM) [31] |
|
| Extracellular vesicles [48] |
|
| Chemokines [49] |
|
| Growth factors [50] |
|
| Section | Details | References |
|---|---|---|
| Vascular Network Morphology | Tumor vasculature is disorganized with structural and functional abnormalities due to overexpression of angiogenic factors. | [72,73] |
| Vascular Network Hypervascularity | Tumors exhibit hypervascularity with regions of increased microvascular density (hot spots), increasing the likelihood of metastasis. | [8,84,85] |
| Intravascular Environment | Endothelial cells (ECs) are poorly connected in tumors and may form multilayered structures with irregular basement membranes. | [74,75,79,80,81] |
| Tumor Vascularity vs. Normal Vascularity | Tumor vessels are more variable in size and structure, larger than normal, and show branching and convoluted pathways. | [74,76,77,78] |
| Types of Blood Vessels in Tumors | Tumors include arteries, arterioles, capillaries, venules, and veins, with varying structures based on tissue function. | [74,75,76,77,78,79,80,81] |
| Factors Influencing Vascularization | Cancer and stromal cells secrete angiogenic and growth factors, which stimulate abnormal blood vessel formation. | [74,75] |
| Vascular Smooth Muscle and Pericytes | Vascular smooth muscle cells support arteries, and pericytes stabilize smaller vessels, contributing to endothelial cell (EC) proliferation. | [81,88] |
| Blood Flow | Tumor blood flow follows a similar pattern as in normal tissue, with greater structural abnormalities in blood vessels. | [74] |
| Irregular Blood Flow | Increased microvascular pressures, vessel abnormalities, and blood flow resistance contribute to tumor hypoxia and reduced perfusion despite high vascular density. | [85,100,102,104] |
| Blood Flow Regions in Tumors | Vascular regions in tumors: necrotic (no blood flow), semi-necrotic (some blood flow), stabilized microcirculation, and advanced fronts. | [104] |
| Tumor Vascular System Functional Features | Unique features like angiogenesis, hyperangiogenesis, irregular blood flow, vascular permeability, and abnormal lymphatic drainage describe the EPR phenomenon. | [72,85] |
| Angiogenesis | Essential for tumor growth, triggered by hypoxia, enables migration, nutrient supply, and waste disposal. Process involves sprouting, de novo angiogenesis, and division of parent vessels. | [74,77,80,89,93] |
| Angiogenesis Mechanism | A 4-stage process involving cytokine release, endothelial cell proliferation, and pericyte fixation. Endothelial cells form filopodia, proliferate, and recruit pericytes. | [77,92,93,96] |
| Angiogenesis Regulation | Balance between pro-angiogenic and anti-angiogenic factors. Overexpression of pro-angiogenic factors leads to cancer growth. | [87,94,95] |
| Positive Regulators of Angiogenesis | VEGF family (VEGF-A, B, C, D), PlGF, angiopoietins, CXC chemokines, TGF-alpha, PDGF-BB, and others stimulate endothelial proliferation and migration. | [77,94,98] |
| Negative Regulators of Angiogenesis | Inhibitors like angiostatin, interleukins, and other cytokines suppress angiogenesis and prevent tumor growth. | [77,87,94] |
| Permeability and Fluid Diffusion | Tumor permeability is altered due to ECM changes and is regulated by EC junctions (TJs, AJs, GJs), whose disruption in cancer leads to endothelial barrier breakdown. This increases vascular permeability and facilitates molecular diffusion and transport. | [72,73,75,85,99,100] |
| Lymphatic Drainage and Interstitial Fluid Pressure | Decreased lymphatic drainage leads to increased interstitial fluid pressure (IFP) and cancer tissue stiffness. IFP ranges from 5 to 40 mm Hg in tumors. | [85,106] |
| Clinical Trial | Therapeutic Approach | Cancer Type | Mechanism to Enhance EPR Effect | Reference |
|---|---|---|---|---|
| ThermoDox® + RFA | Thermosensitive liposomal doxorubicin | Hepatocellular carcinoma (HCC) | Heat-induced vascular permeability increase | [204] |
| Sonoporation (ultrasound + microbubbles) | Enhanced NP delivery | Pancreatic cancer | Temporary pore formation in tumor vasculature | [205] |
| Magnetic NPs | Magnetic field-directed targeting | Glioblastoma multiforme | Improved tumor accumulation | [206] |
| Doxorubicin-loaded NPs and vascular-modulating agents | Chemotherapy enhancement | Breast cancer | Improved nanoparticle retention | [207] |
| Liposomal irinotecan(Onivyde®) | Nanoparticle-based chemotherapy | Metastatic pancreatic cancer | Enhanced drug retention via EPR | [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagena, I.-A.; Malapani, C.; Gatou, M.-A.; Lagopati, N.; Pavlatou, E.A. Enhancement of EPR Effect for Passive Tumor Targeting: Current Status and Future Perspectives. Appl. Sci. 2025, 15, 3189. https://doi.org/10.3390/app15063189
Vagena I-A, Malapani C, Gatou M-A, Lagopati N, Pavlatou EA. Enhancement of EPR Effect for Passive Tumor Targeting: Current Status and Future Perspectives. Applied Sciences. 2025; 15(6):3189. https://doi.org/10.3390/app15063189
Chicago/Turabian StyleVagena, Ioanna-Aglaia, Christina Malapani, Maria-Anna Gatou, Nefeli Lagopati, and Evangelia A. Pavlatou. 2025. "Enhancement of EPR Effect for Passive Tumor Targeting: Current Status and Future Perspectives" Applied Sciences 15, no. 6: 3189. https://doi.org/10.3390/app15063189
APA StyleVagena, I.-A., Malapani, C., Gatou, M.-A., Lagopati, N., & Pavlatou, E. A. (2025). Enhancement of EPR Effect for Passive Tumor Targeting: Current Status and Future Perspectives. Applied Sciences, 15(6), 3189. https://doi.org/10.3390/app15063189