

Computational Insights into Aluminum and Cation Placement in Clinoptilolite: Optimizing DFT Parameters for Natural Zeolites, Part I


Abstract
1. Introduction and State of the Research
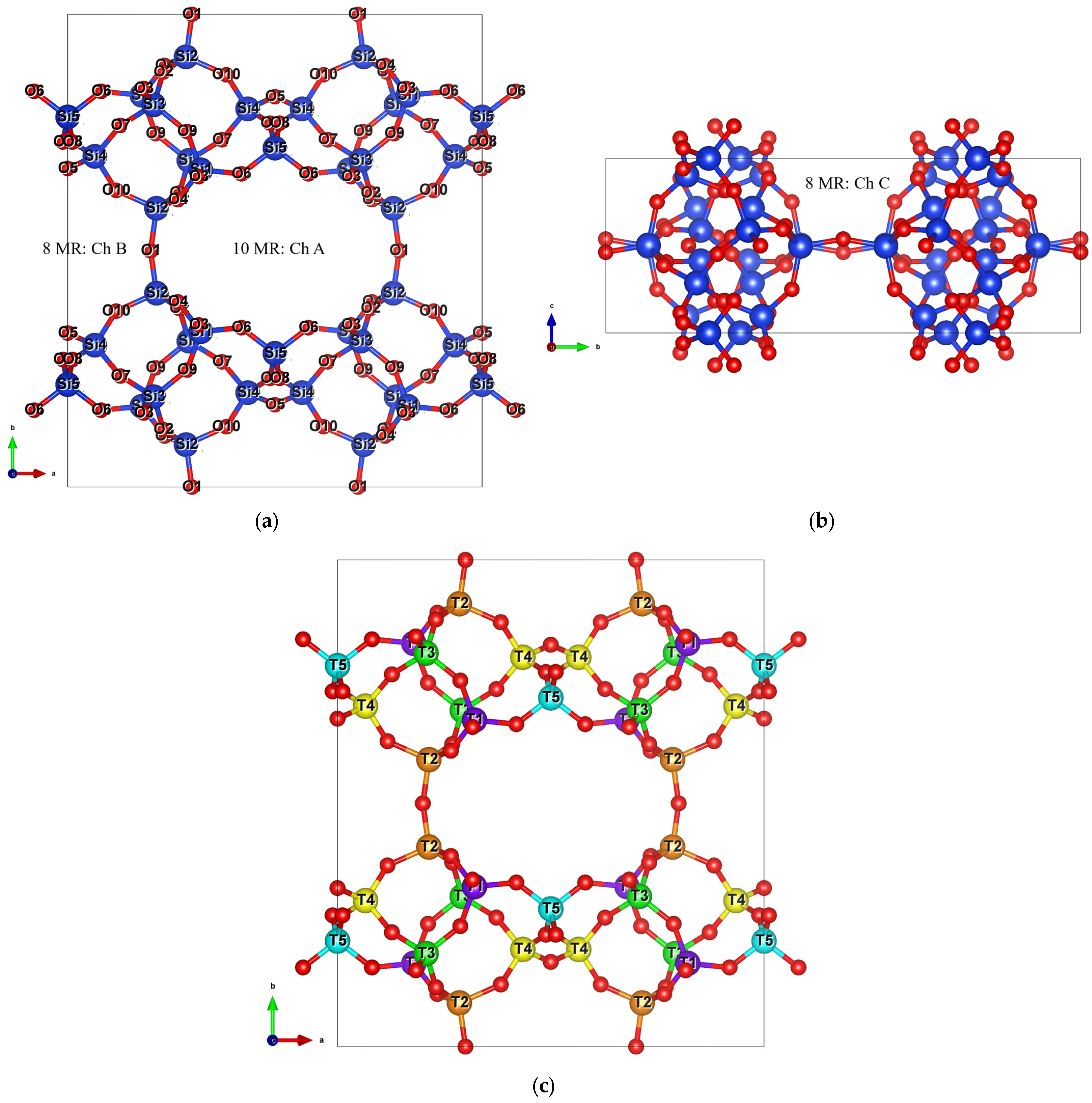
2. Structural Features and Methodology
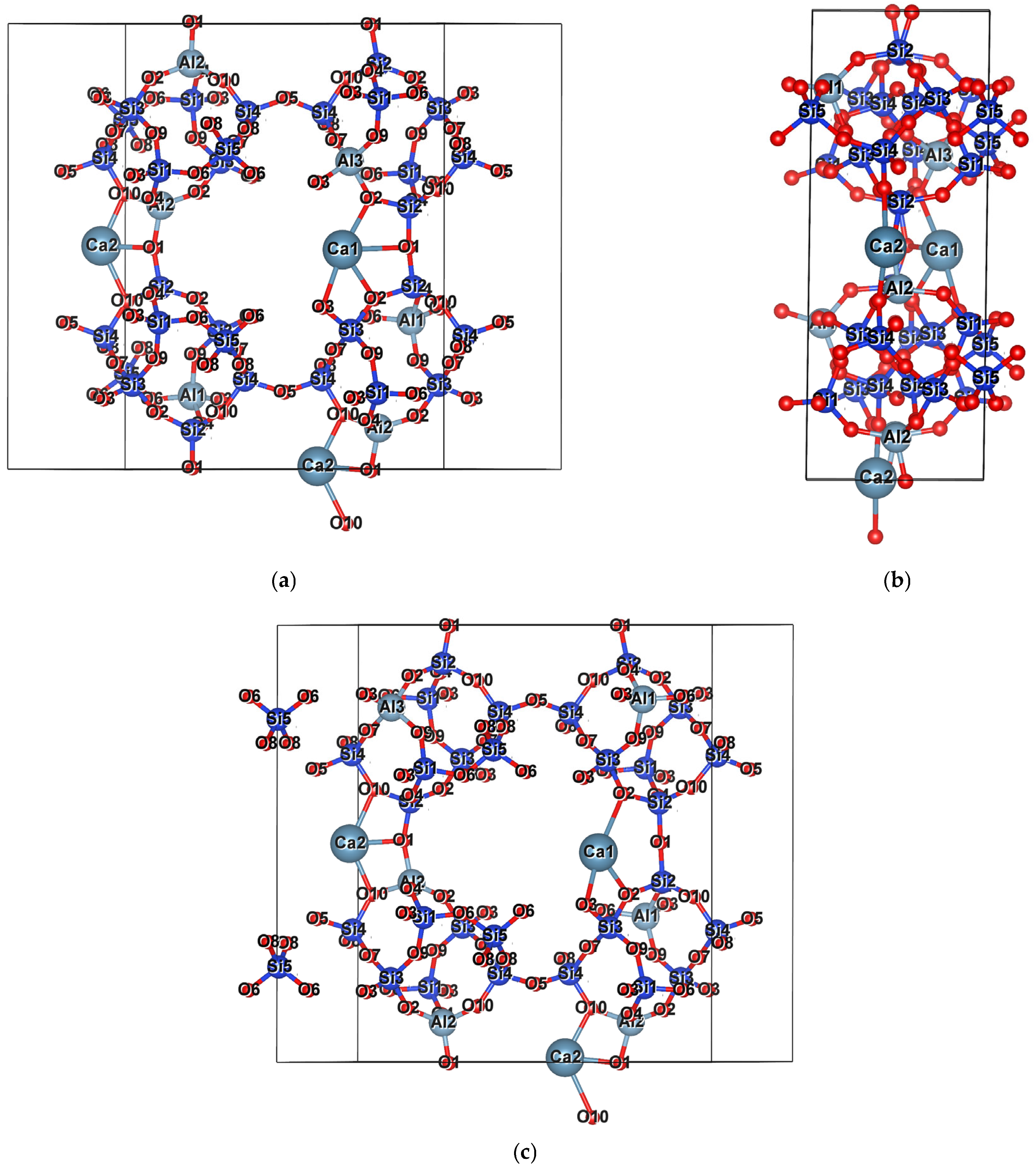
2.1. Aluminum Distribution
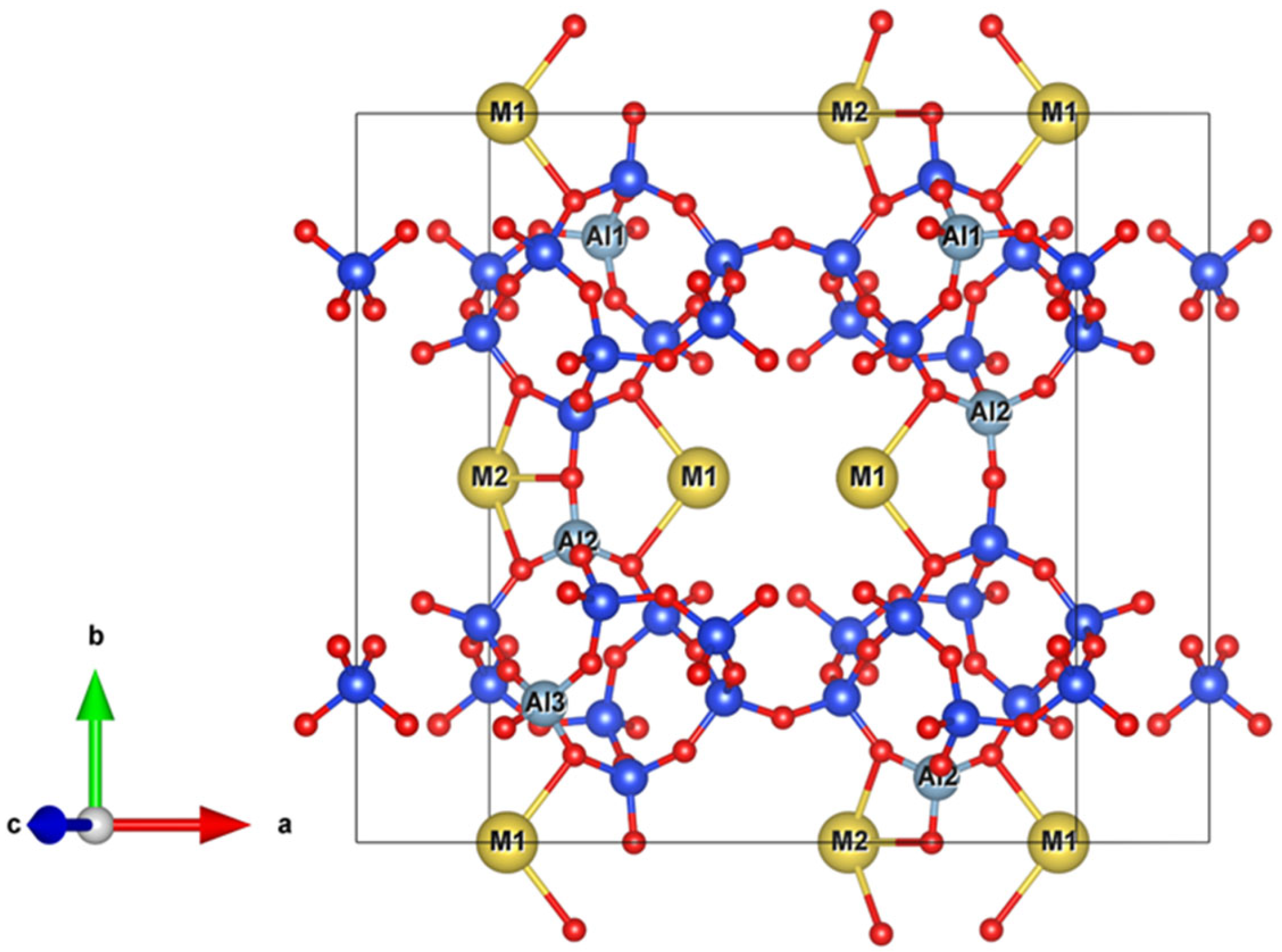
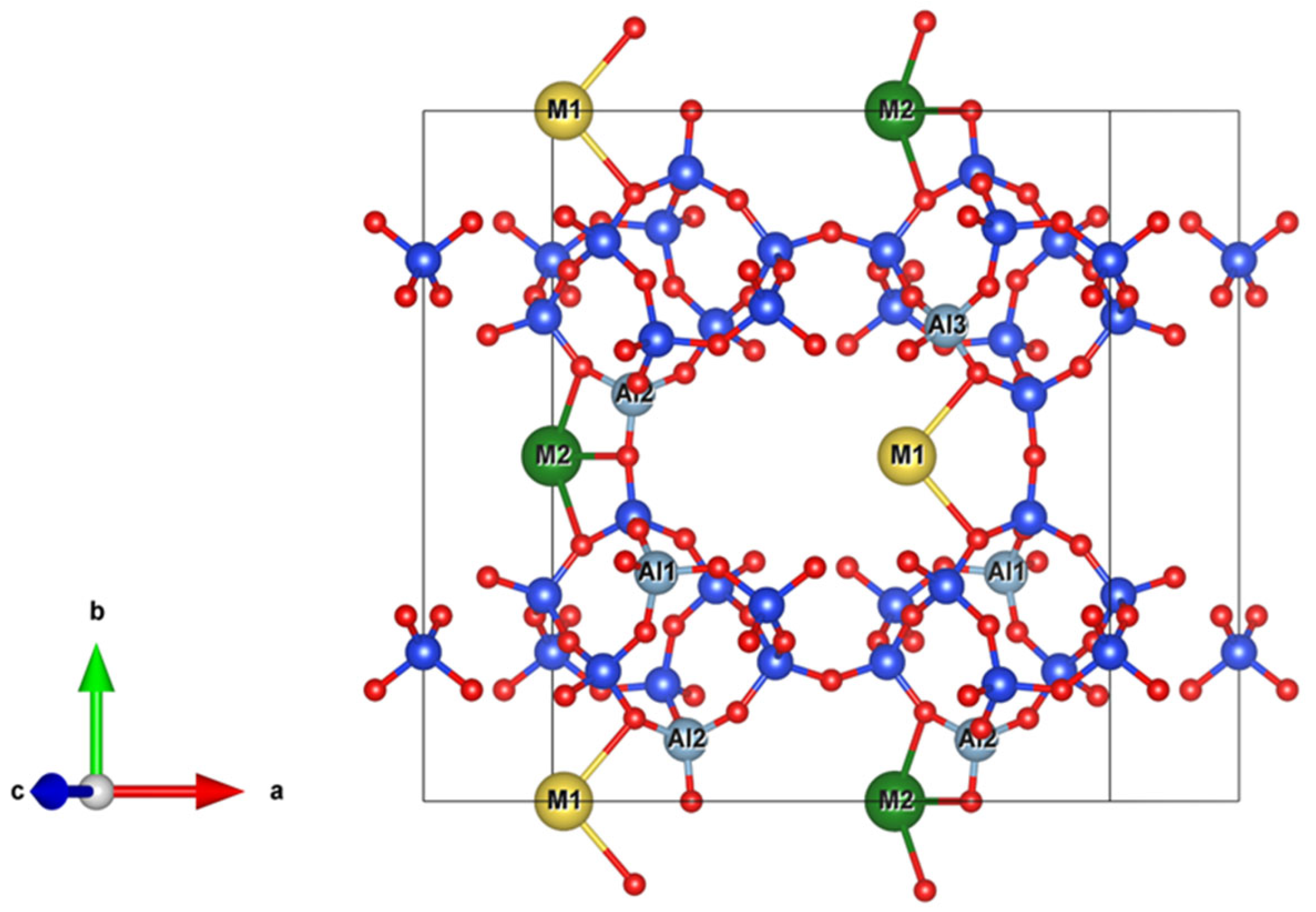
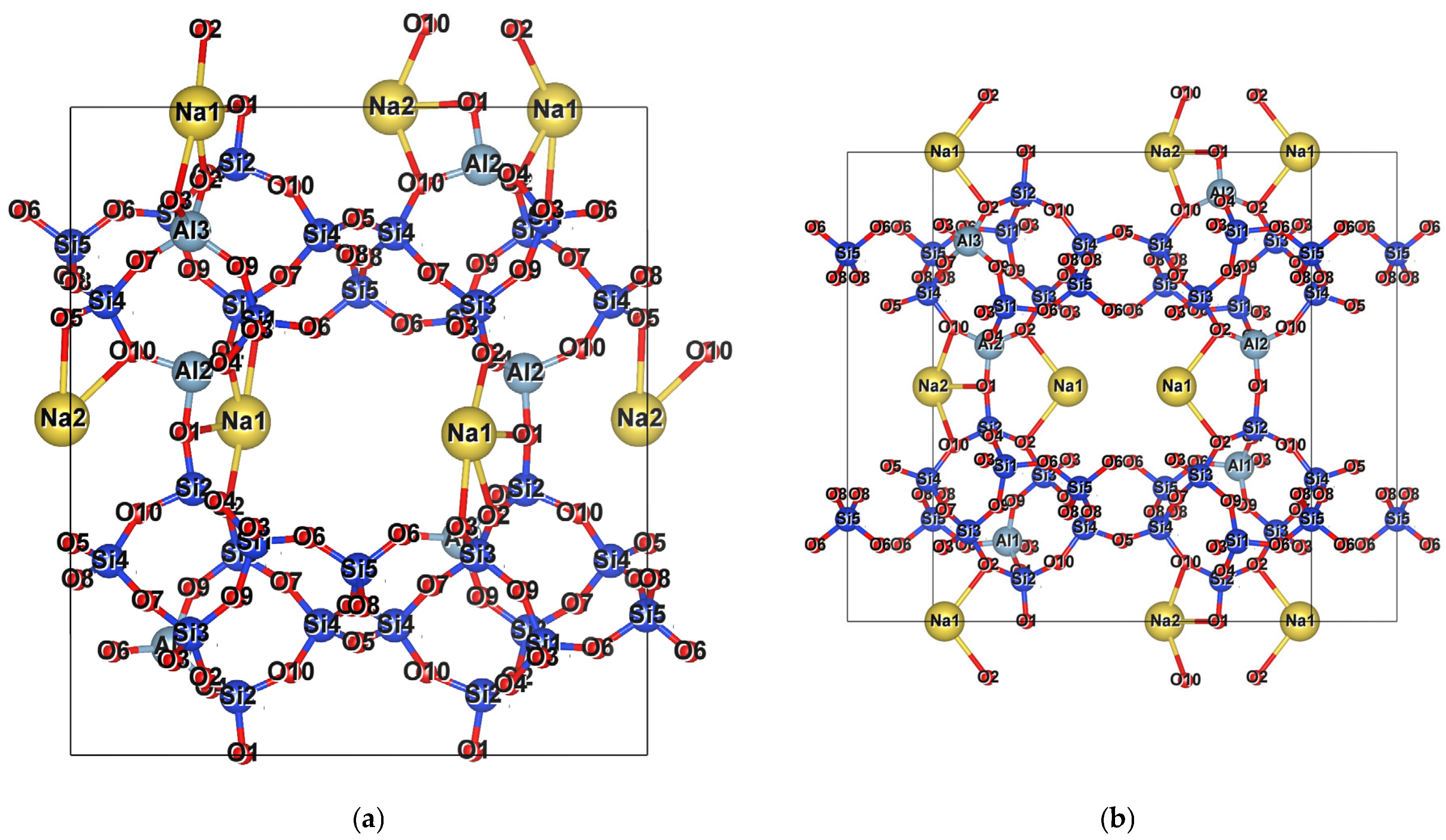
2.2. Cations
Na-Dominated, Ca-Dominated, Na-Ca, and Na-Ca-K Clinoptilolites
2.3. Computational Details
2.3.1. All-Silica Clinoptilolites
2.3.2. Basis Sets
2.3.3. Density Functionals
3. Results and Discussion
3.1. Na-Dominant Clinoptilolite
3.1.1. XC Functionals
3.1.2. Bond Lengths and Interatomic Distances
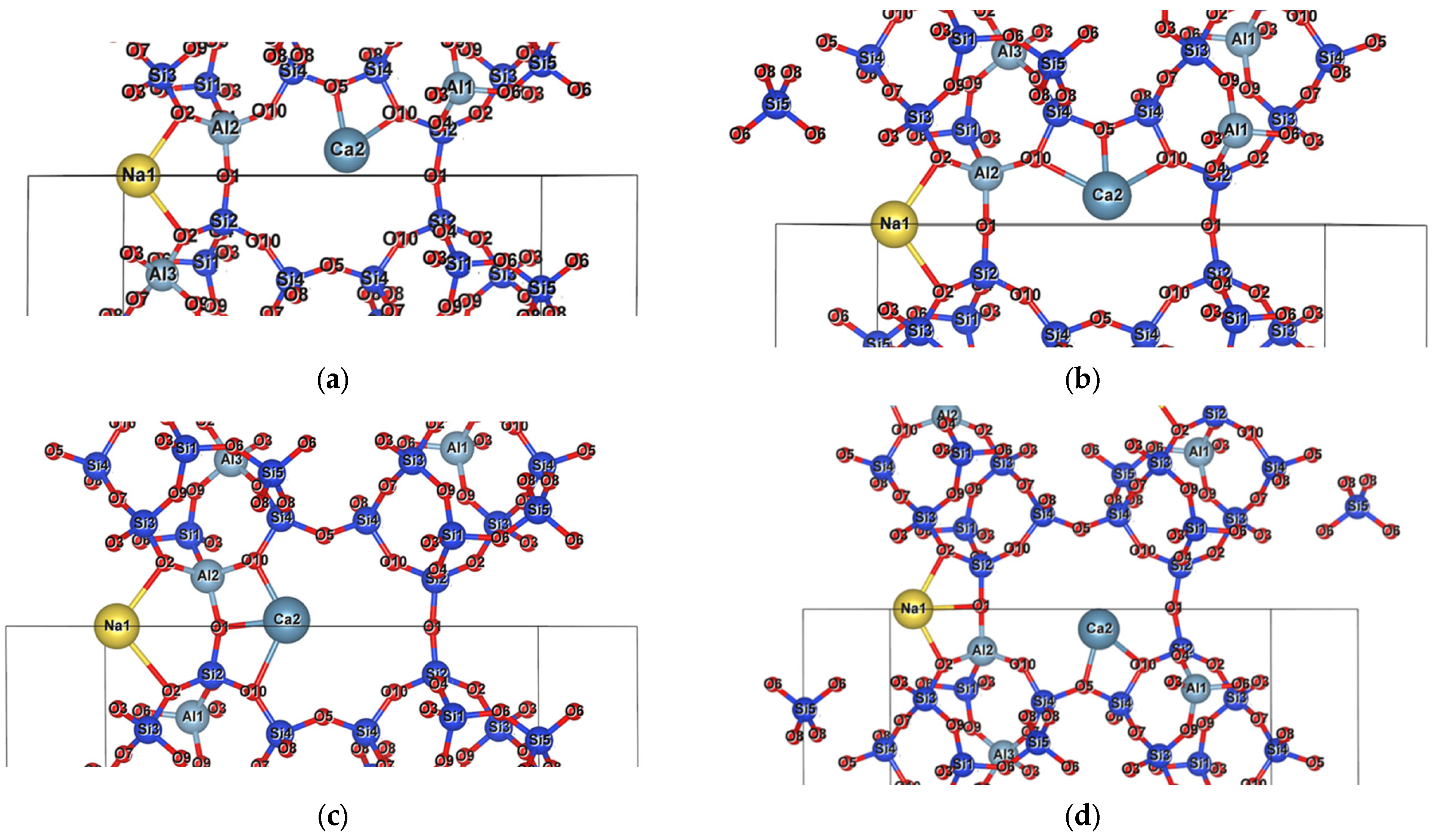
3.1.3. Interaction of Na Cations with the Framework
3.2. Ca-Dominant Clinoptilolite
3.3. Migration of Ca Cations
3.4. Na-Ca Clinoptilolite
3.5. Na-Ca-K Clinoptilolite
4. Conclusions and Outlook
- 1.
- Surface models of the different clinoptilolite models studied here.
- 2.
- An exploration of the interaction of the emerging contaminants, such as PPCPS, with those surface models.
‘We must now note a confusing, but important, notion that while DFT is exact in principle, in practice it is implemented with approximations. As Becke wrote, we should thus keep in mind that “The failures we report at meetings and in papers are not failures of DFT, but failures of DFAs” (whereby DFAs stand for Density Functional Approximations)’ …
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, M. Simulation-Based Evaluation of Zeolite Adsorbents for the Removal of Emerging Contaminants. Mater. Adv. 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Yang, Y.; Ok, Y.S.; Kim, K.-H.; Kwon, E.E.; Tsang, Y.F. Occurrences and Removal of Pharmaceuticals and Personal Care Products (PPCPs) in Drinking Water and Water/Sewage Treatment Plants: A Review. Sci. Total Environ. 2017, 596–597, 303–320. [Google Scholar] [CrossRef]
- Tondar, A.; Mirzaei-Parsa, M.; Yousefpoor, Y.; Sharifi, A.M.; Shetab-Boushehri, S. Feasibility of Clinoptilolite Application as a Microporous Carrier for pH-Controlled Oral Delivery of Aspirin. Acta Chim. Slov. 2014, 61, 688–693. [Google Scholar] [PubMed]
- Cerri, G.; Degennaro, M.; Bonferoni, M.; Caramella, C. Zeolites in Biomedical Application: Zn-Exchanged Clinoptilolite-Rich Rock as Active Carrier for Antibiotics in Topical Therapy. Appl. Clay Sci. 2004, 27, 141–150. [Google Scholar] [CrossRef]
- De Gennaro, B.; Catalanotti, L.; Cappelletti, P.; Langella, A.; Mercurio, M.; Serri, C.; Biondi, M.; Mayol, L. Surface Modified Natural Zeolite as a Carrier for Sustained Diclofenac Release: A Preliminary Feasibility Study. Colloids Surf. B Biointerfaces 2015, 130, 101–109. [Google Scholar] [CrossRef]
- Ochiuz, L.; Ghiciuc, C.; Ignat, M.; Popa, M.; Peptu, C.A.; Vasile, A. Development of a Modified—Release Drug Delivery System with Bexarotene Loaded in Clinoptilolite. Mater. Plast. 2017, 54, 581–585. [Google Scholar] [CrossRef]
- Bacakova, L.; Vandrovcova, M.; Kopova, I.; Jirka, I. Applications of Zeolites in Biotechnology and Medicine—A Review. Biomater. Sci. 2018, 6, 974–989. [Google Scholar] [CrossRef]
- Kraljević Pavelić, S.; Simović Medica, J.; Gumbarević, D.; Filošević, A.; Pržulj, N.; Pavelić, K. Critical Review on Zeolite Clinoptilolite Safety and Medical Applications in Vivo. Front. Pharmacol. 2018, 9, 1350. [Google Scholar] [CrossRef]
- Natural Zeolites: Occurrence, Properties, Applications; Bish, D.L., Ming, D.W., Eds.; Reviews in mineralogy and geochemistry; Mineralogical Society of America: Washington, DC, USA, 2001; ISBN 978-0-939950-57-7. [Google Scholar]
- Uzunova, E.L.; Mikosch, H. Cation Site Preference in Zeolite Clinoptilolite: A Density Functional Study. Microporous Mesoporous Mater. 2013, 177, 113–119. [Google Scholar] [CrossRef]
- Ruiz-Salvador, R.A.; Gómez, A.; Lewis, D.W.; Richard, A.; Catlow, C.; Marleny Rodríguez-Albelo, L.; Montero, L.; Rodríguez-Fuentes, G. Clinoptilolite–Heulandite Polymorphism: Structural Features from Computer Simulation. Phys. Chem. Chem. Phys. 2000, 2, 1803–1813. [Google Scholar] [CrossRef]
- Lewis, D.W.; Ruiz-Salvador, A.R.; Almora-Barrios, N.; Gómez, A.; Mistry, M. Modelling of Hydrated Ca-Rich Zeolites. Mol. Simul. 2002, 28, 649–661. [Google Scholar] [CrossRef]
- Ruiz-Salvador, A.R.; Lewis, D.W.; Rubayo-Soneira, J.; Rodríguez-Fuentes, G.; Sierra, L.R.; Catlow, C.R.A. Aluminum Distribution in Low Si/Al Zeolites: Dehydrated Na−Clinoptilolite. J. Phys. Chem. B 1998, 102, 8417–8425. [Google Scholar] [CrossRef]
- Ruiz-Salvador, A.R.; Gómez, A.; Lewis, D.W.; Rodríguez-Fuentes, G.; Montero, L. Silicon–Aluminium Distribution in Dehydrated Calcium Heulandite. Phys. Chem. Chem. Phys. 1999, 1, 1679–1685. [Google Scholar] [CrossRef]
- Valdiviés Cruz, K.; Lam, A.; Zicovich-Wilson, C.M. Periodic Quantum Chemical Studies on Anhydrous and Hydrated Acid Clinoptilolite. J. Phys. Chem. A 2014, 118, 5779–5789. [Google Scholar] [CrossRef]
- Valdiviés-Cruz, K.; Lam, A.; Zicovich-Wilson, C.M. Chemical Interaction of Water Molecules with Framework Al in Acid Zeolites: A Periodic Ab Initio Study on H-Clinoptilolite. Phys. Chem. Chem. Phys. 2015, 17, 23657–23666. [Google Scholar] [CrossRef]
- Awuah, J.B.; Dzade, N.Y.; Tia, R.; Adei, E.; Kwakye-Awuah, B.; Richard, A.; Catlow, C.; de Leeuw, N.H. A Density Functional Theory Study of Arsenic Immobilization by the Al(III)-Modified Zeolite Clinoptilolite. Phys. Chem. Chem. Phys. 2016, 18, 11297–11305. [Google Scholar] [CrossRef]
- Uzunova, E.L.; Mikosch, H. Adsorption of Phosphates and Phosphoric Acid in Zeolite Clinoptilolite: Electronic Structure Study. Microporous Mesoporous Mater. 2016, 232, 119–125. [Google Scholar] [CrossRef]
- Abatal, M.; Ruiz-Salvador, A.R.; Cruz Hernández, N. A DFT-Based Simulated Annealing Method for the Optimization of Global Energy in Zeolite Framework Systems: Application to Natrolite, Chabazite and Clinoptilolite. Microporous Mesoporous Mater. 2020, 294, 8. [Google Scholar] [CrossRef]
- Diógenes, T.S.; Santiago, R.G.; Maia, D.A.S.; Gonçalves, D.V.; Azevedo, D.C.S.; Lucena, S.M.P.; Bastos-Neto, M. Experimental and Theoretical Assessment of CO2 Capture by Adsorption on Clinoptilolite. Chem. Eng. Res. Des. 2022, 177, 640–652. [Google Scholar] [CrossRef]
- Alberti, A. The Crystal Structure of Two Clinoptilolites. TMPM Tschermaks Mineral. Petrogr. Mitteilungen 1975, 22, 25–37. [Google Scholar] [CrossRef]
- Clinoptilolite: The Distribution of Potassium Atoms and Its Role in Thermal Stability. Z. Für Krist.—Cryst. Mater. 1977, 145, 216–239. [CrossRef]
- Okhotnikov, K.; Charpentier, T.; Cadars, S. Supercell Program: A Combinatorial Structure-Generation Approach for the Local-Level Modeling of Atomic Substitutions and Partial Occupancies in Crystals. J. Cheminform. 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Supercell. Available online: https://orex.github.io/supercell/ (accessed on 14 February 2024).
- Dempsey, E.; Kuehl, G.H.; Olson, D.H. Variation of the Lattice Parameter with Aluminum Content in Synthetic Sodium Faujasites. Evidence for Ordering of the Framework Ions. J. Phys. Chem. 1969, 73, 387–390. [Google Scholar] [CrossRef]
- Den Ouden, C.J.J.; Jackson, R.A.; Catlow, C.R.A.; Post, M.F.M. Location of Nickel(2+) Ions in Siliceous Mordenite: A Computational Approach. J. Phys. Chem. 1990, 94, 5286–5290. [Google Scholar] [CrossRef]
- Ackley, M.W.; Yang, R.T. Diffusion in Ion-Exchanged Clinoptilolites. AIChE J. 1991, 37, 1645–1656. [Google Scholar] [CrossRef]
- Davarpanah, E.; Armandi, M.; Hernández, S.; Fino, D.; Arletti, R.; Bensaid, S.; Piumetti, M. CO2 Capture on Natural Zeolite Clinoptilolite: Effect of Temperature and Role of the Adsorption Sites. J. Environ. Manag. 2020, 275, 111229. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package—Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and Augmented-Plane-Wave Density Functional Method for Ab Initio Molecular Dynamics Simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Shanno, D.F. Conditioning of Quasi-Newton Methods for Function Minimization. Math. Comput. 1970, 24, 647–656. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. An Efficient Orbital Transformation Method for Electronic Structure Calculations. J. Chem. Phys. 2003, 118, 4365–4369. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple-PBE. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Fischer, M.; Kim, W.J.; Badawi, M.; Lebègue, S. Benchmarking the Performance of Approximate van Der Waals Methods for the Structural and Energetic Properties of SiO2 and AlPO4 Frameworks. J. Chem. Phys. 2019, 150, 094102. [Google Scholar] [CrossRef]
- Fischer, M.; Evers, F.O.; Formalik, F.; Olejniczak, A. Benchmarking DFT-GGA Calculations for the Structure Optimisation of Neutral-Framework Zeotypes. Theor. Chem. Acc. 2016, 135, 257. [Google Scholar] [CrossRef]
- Fischer, M.; Angel, R.J. Accurate Structures and Energetics of Neutral-Framework Zeotypes from Dispersion-Corrected DFT Calculations. J. Chem. Phys. 2017, 146, 174111. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, D.; Crawford, N.R.M.; Furche, F.; Burke, K. Which Functional Should I Choose? Comput. Inorg. Bioinorg. Chem. 2008, 594, 1–25. [Google Scholar]
- Zhang, Y.; Yang, W. Comment on ``Generalized Gradient Approximation Made Simple’’. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van Der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van Der Waals Density Functionals Applied to Solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef]
- Lee, K.; Murray, É.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-Accuracy van Der Waals Density Functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef]
- Hamada, I. Van Der Waals Density Functional Made Accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Becke, A.D. On the Large-Gradient Behavior of the Density Functional Exchange Energy. J. Chem. Phys. 1986, 85, 7184–7187. [Google Scholar] [CrossRef]
- Wellendorff, J.; Lundgaard, K.T.; Møgelhøj, A.; Petzold, V.; Landis, D.D.; Nørskov, J.K.; Bligaard, T.; Jacobsen, K.W. Density Functionals for Surface Science: Exchange-Correlation Model Development with Bayesian Error Estimation. Phys. Rev. B 2012, 85, 235149. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Wojdyr, M. GEMMI: A Library for Structural Biology. J. Open Source Softw. 2022, 7, 4200. [Google Scholar] [CrossRef]
- Jackson, R.A.; Catlow, C.R.A. Computer Simulation Studies of Zeolite Structure. Mol. Simul. 1988, 1, 207–224. [Google Scholar] [CrossRef]
- Armbruster, T. Dehydration Mechanism of Clinoptilolite and Heulandite: Single-Crystal X-ray Study of Na-Poor, Ca-, K-, Mg-Rich Clinoptilolite at 100 K. Am. Mineral. 1993, 78, 260–264. [Google Scholar]
- Goh, T. Exchange-Correlation Functionals: GGA vs. Meta-GGA; University of Chicago: Chicago, IL, USA, 2020. [Google Scholar]
- Becke, A.D. Perspective: Fifty Years of Density-Functional Theory in Chemical Physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef]
- Kudin, K.N.; Scuseria, G.E. Linear-Scaling Density-Functional Theory with Gaussian Orbitals and Periodic Boundary Conditions: Efficient Evaluation of Energy and Forces via the Fast Multipole Method. Phys. Rev. B 2000, 61, 16440–16453. [Google Scholar] [CrossRef]
- CCCBDB Compare Calculated Bond Lengths. Available online: https://cccbdb.nist.gov/calcbondcomp1x.asp (accessed on 16 April 2024).
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Exercises:2015_cecam_tutorial:Basis_set_optimisation_using_optimize_basis [CP2K Open Source Molecular Dynamics]. Available online: https://www.cp2k.org/exercises:2015_cecam_tutorial:basis_set_optimisation_using_optimize_basis (accessed on 16 April 2024).
- Ling, S. Optimisation of Basis Sets and Pseudopotentials; University College London: London, UK, 2007. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell | a (Å) | b (Å) | c (Å) | β (°) |
|---|---|---|---|---|
| Experimental Agoura | 17.646 | 17.898 | 7.397 | 116.22 |
| Calculated Agoura with PBE | 17.696 | 17.746 | 7.458 | 116.67 |
| Calculated Agoura with PBE-D3 | 17.711 | 17.781 | 7.436 | 116.59 |
| Experimental Siusi | 17.637 or 17.641 | 18.024 or 18.031 | 7.399 or 7.402 | 116.23 or 116.26 |
| Calculated Siusi with PBE | 17.688 | 17.877 | 7.416 | 116.09 |
| Calculated Siusi with PBE-D3 | 17.652 | 17.861 | 7.401 | 116.11 |
| Structure Type | a [Å] | b [Å] | c [Å] | β [°] |
|---|---|---|---|---|
| HEU: clinoptilolites and heulandites | 17.62–17.74 | 17.81–18.05 | 7.39–7.53 | 116.13–116.90 |
| Na-Clinoptilolite | a [Å] | b [Å] | c [Å] | β [°] | Volume [Å3] | ||
|---|---|---|---|---|---|---|---|
| Before optimization | 17.653 | 17.862 | 7.401 | 90 | 116.11 | 90 | 2095.51 |
| 1. PBE-D3 | 17.778 | 17.727 | 7.426 | 89.65 | 116.77 | 89.94 | 2089.70 |
| 2. PBE-D3(BJ) | 17.775 | 17.721 | 7.421 | 89.76 | 116.68 | 90.06 | 2088.69 |
| 3. revPBE-D3 | 17.735 | 17.828 | 7.407 | 89.66 | 116.76 | 90 | 2090.99 |
| 4. PBEsol-D3 | 17.697 | 17.719 | 7.402 | 89.74 | 117.01 | 90.02 | 2068.05 |
| 5. B97-D3 | 17.735 | 17.847 | 7.416 | 89.71 | 116.33 | 90.20 | 2103.89 |
| 6. TPSS-D3 | 17.747 | 17.790 | 7.408 | 89.89 | 116.13 | 90.10 | 2099.94 |
| 7. optB86b-vdW | No convergence | ||||||
| 8. rev-vdW-DF2 | 17.737 | 17.663 | 7.420 | 89.54 | 117.21 | 90.31 | 2067.65 |
| 9. BEEF-vdW | 17.836 | 17.816 | 7.438 | 89.83 | 116.47 | 90.13 | 2115.84 |
| Structure | a (Å) | b (Å) | c (Å) | β (°) |
|---|---|---|---|---|
| Agoura_revPBE-D3 | 17.63–17.73 | 17.71–17.76 | 7.37–7.39 | 116.84–117.03 |
| Siusi_revPBE-D3 | 17.65–17.71 | 17.72–17.76 | 7.37–7.39 | 116.73–117.04 |
| Agoura_B97-D3 | 17.72–17.77 | 17.68–17.81 | 7.41–7.42 | 116.25–116.52 |
| Siusi_B97-D3 | 17.70–17.74 | 17.82–17.87 | 7.39–7.41 | 116.12–116.35 |
| Optimized Agoura Na-Clinoptilolite (Si-O) | |||||
|---|---|---|---|---|---|
| T-Sites (Å) | Avg. Exp. | revPBE-D3 (Avg. Distances) | B97-D3 (Avg. Distances) | FF [13] | ONIOM/PBC [10] |
| Si (1)—O3 —O4 —O6 —O9 | 1.625 | 1.632, 1.626, 1.628, 1.629, 1.629, 1.631 | 1.628, 1.617, 1.611, 1.620, 1.616, 1.616 | 1.599, 1.603, 1.602, 1.615 | 1.643/1.656 |
| Si (2)—O1 —O10 —O2 —O4 | 1.645 | 1.622, 1.620, 1.623, 1.627, 1.620 | 1.612, 1.618, 1.622, 1.618, 1.612 | 1.589, 1.602, 1.613, 1.599 | 1.638/1.675 |
| Si (3)—O2 —O3 —O7 —O9 | 1.61 | 1.620, 1.626, 1.629, 1.620,1.625, 1.626, 1.627 | 1.618, 1.611, 1.626, 1.613, 1.619, 1.612, 1.614 | 1.591, 1.600, 1.599, 1.609 | 1.657/1.653 |
| Si (4)—O7 —O5 —O8 —O10 | 1.61 | 1.620, 1.624, 1.622 (3×), 1.625, 1.627, 1.623 | 1.620, 1.615, 1.610, 1.611, 1.613, 1.614, 1.613, 1.617 | 1.599, 1.6060, 1.594, 1.598 | Not reported |
| Si (5)—O6 —O8 | 1.615 | 1.622, 1.626, 1.630, 1.623 | 1.617, 1.611, 1.615, 1.621 | 1.606 (2×), 1.597(2×) | Not reported |
| Optimized Siusi Na-Clinoptilolite | |||
|---|---|---|---|
| T-Sites (Å) | Exp. | revPBE-D3 (Avg. Distances) | B97-D3 (Avg. Distances) |
| Si (1)—O3 —O4 —O6 —O9 | 1.61 | 1.634, 1.635, 1.623, 1.629, 1.633, 1.622 | 1.616, 1.617, 1.624, 1.609, 1.617, 1.624 |
| Si (2)—O1 —O10 —O2 —O4 | 1.67 | 1.622, 1.624, 1.626, 1.627, 1.627 | 1.616, 1.613, 1.619, 1.622, 1.612 |
| Si (3)—O2 —O3 —O7 —O9 | 1.615 | 1.623, 1.630, 1.621, 1.615, 1.627, 1.621, 1.621 | 1.617, 1.614, 1.622, 1.602, 1.616, 1.622, 1.613 |
| Si (4)—O7 —O5 —O8 —O10 | 1.62 | 1.624, 1.622, 1.622, 1.622, 1.625, 1.620, 1.623, 1.627 | 1.614, 1.611, 1.615, 1.614, 1.609, 1.616, 1.613, 1.611 |
| Si (5)—O6 —O8 | 1.615 | 1.628, 1.630, 1.632, 1.629 | 1.612, 1.618, 1.611, 1.619 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, L.; Fischer, M. Computational Insights into Aluminum and Cation Placement in Clinoptilolite: Optimizing DFT Parameters for Natural Zeolites, Part I. Appl. Sci. 2025, 15, 3955. https://doi.org/10.3390/app15073955
Saeed L, Fischer M. Computational Insights into Aluminum and Cation Placement in Clinoptilolite: Optimizing DFT Parameters for Natural Zeolites, Part I. Applied Sciences. 2025; 15(7):3955. https://doi.org/10.3390/app15073955
Chicago/Turabian StyleSaeed, Lobna, and Michael Fischer. 2025. "Computational Insights into Aluminum and Cation Placement in Clinoptilolite: Optimizing DFT Parameters for Natural Zeolites, Part I" Applied Sciences 15, no. 7: 3955. https://doi.org/10.3390/app15073955
APA StyleSaeed, L., & Fischer, M. (2025). Computational Insights into Aluminum and Cation Placement in Clinoptilolite: Optimizing DFT Parameters for Natural Zeolites, Part I. Applied Sciences, 15(7), 3955. https://doi.org/10.3390/app15073955