
Bioinformatics-Based Management of Vitellogenin-like Protein’s Role in Pathogen Defense in Nicotiana tabacum L.


Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of Pathogen Defense Protein Sequence
2.2. Structural and Functional Annotation Using InterPro
2.3. Physiochemical Properties
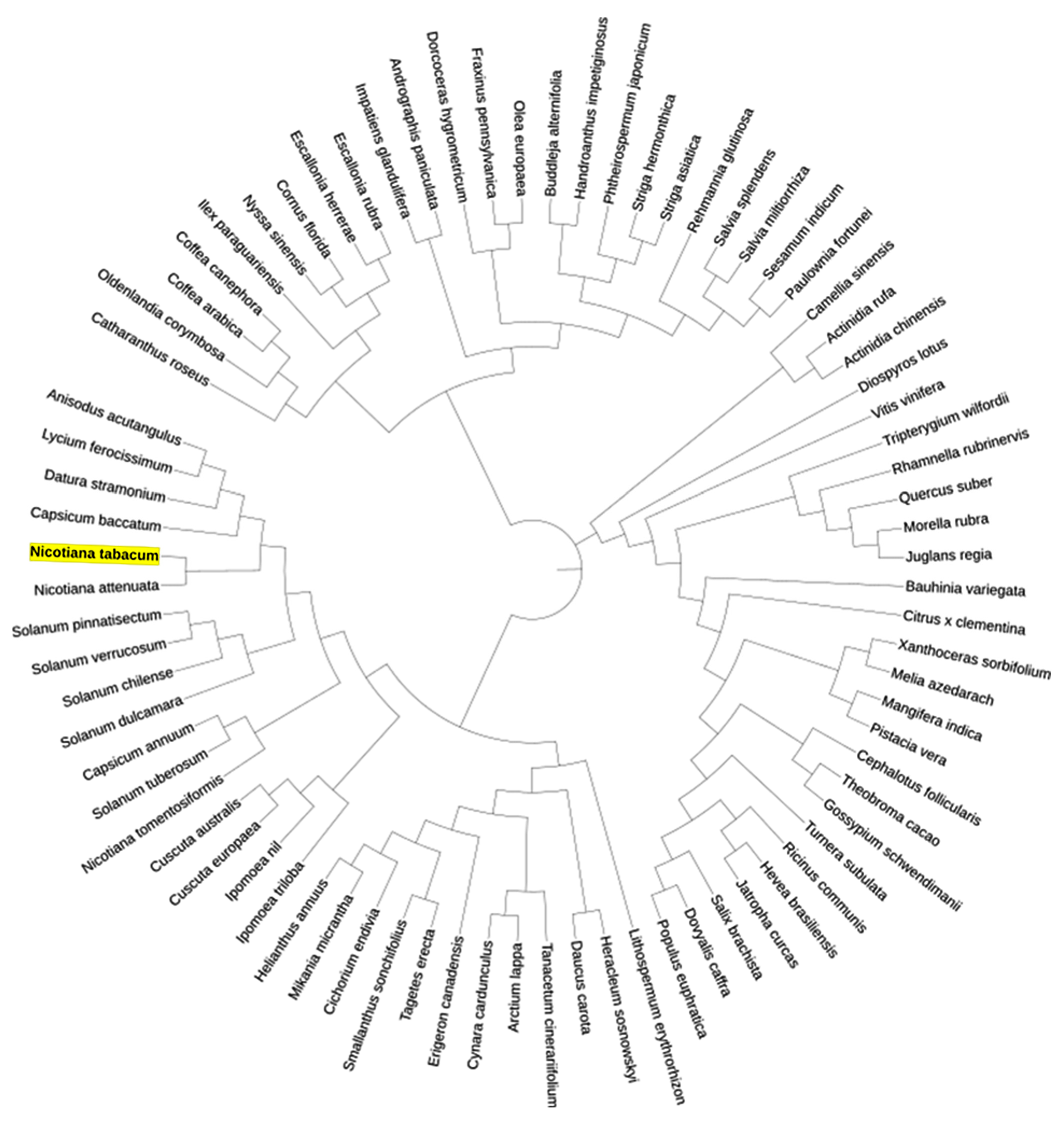
2.4. Phylogenetic Analysis
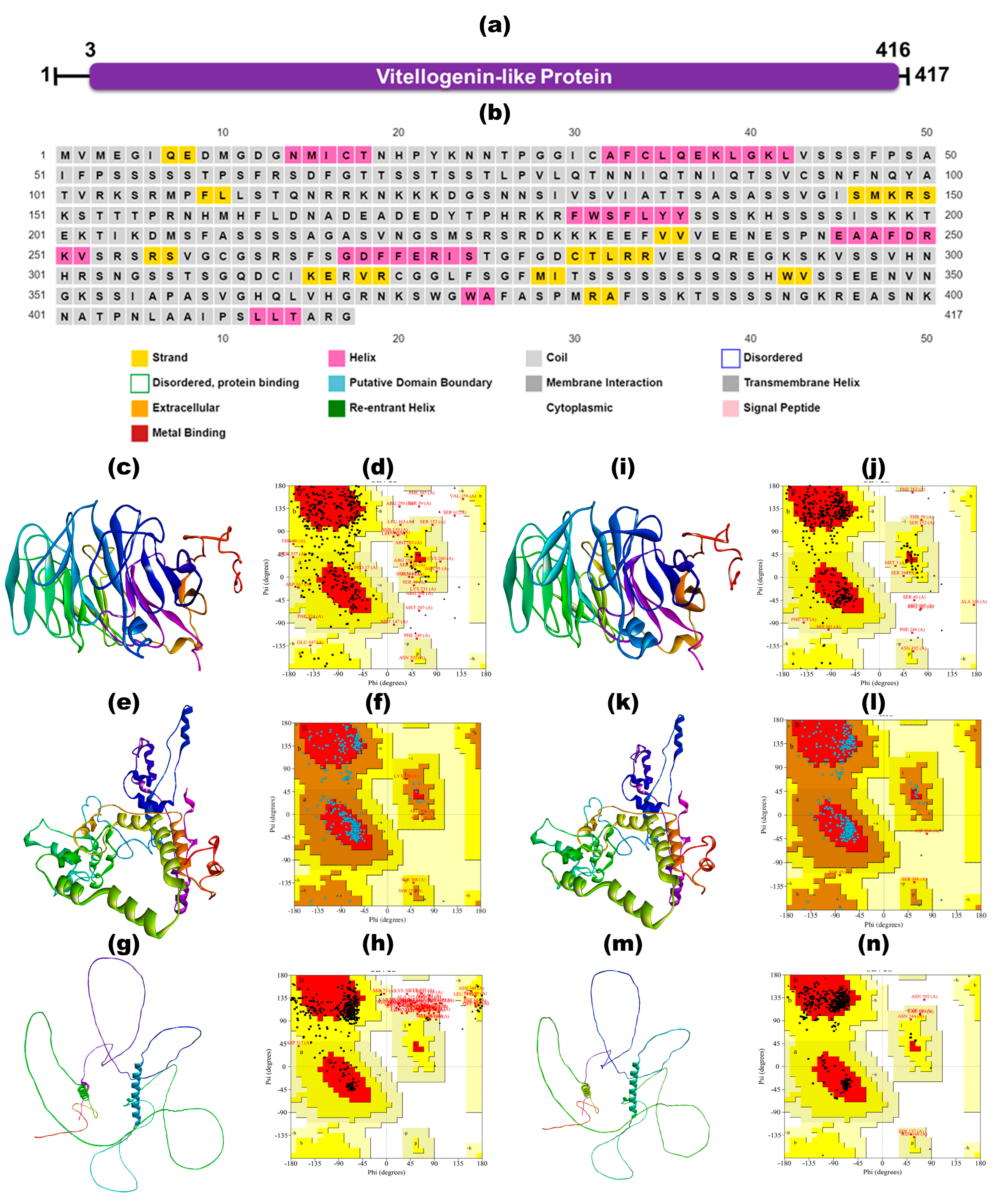
2.5. Structure Prediction, Refinement, and Validation
2.5.1. Secondary Structure Prediction
2.5.2. 3D Structure Modeling
2.5.3. Ab Initio Structure Prediction Using I-TASSER, RoseTTAFold, and AlphaFold
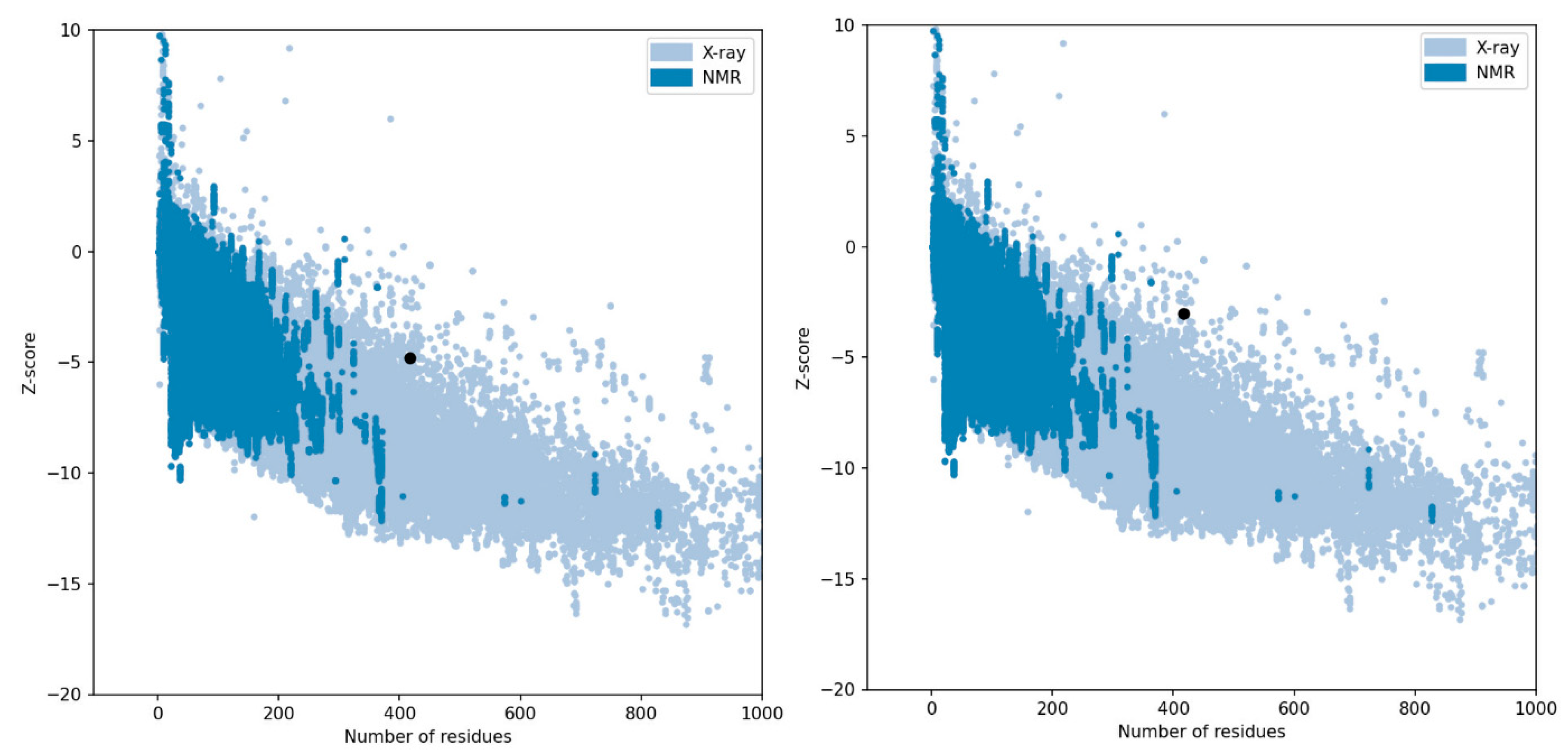
2.5.4. Structure Validation, Refinement, and Quality Assessment
2.6. Molecular Docking
2.6.1. Ligands Selection and Preparation
2.6.2. Protein Preparation
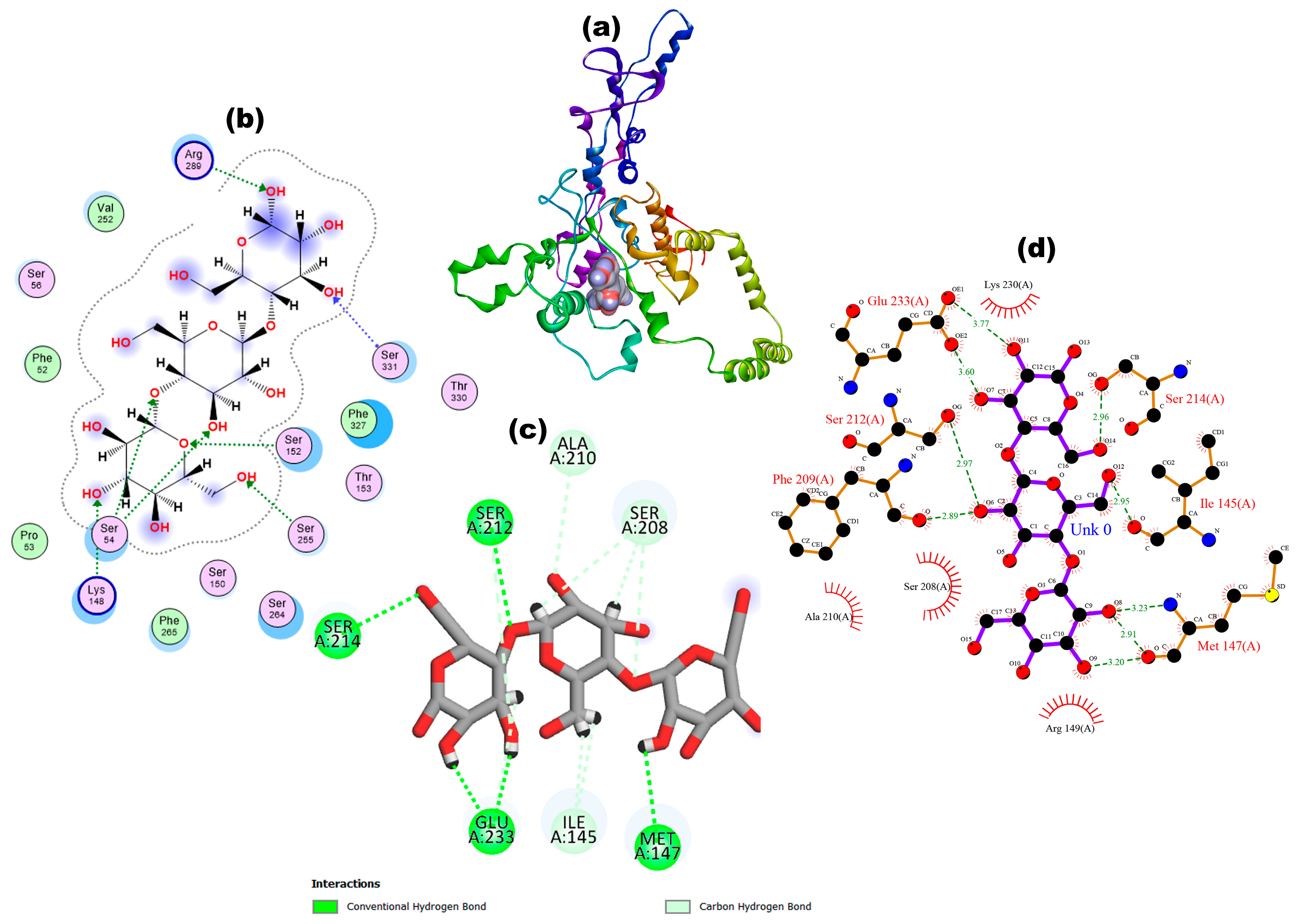
2.6.3. Functional Interaction Site Prediction
2.6.4. Molecular Docking
2.7. Internal Coordinates Normal Mode Analysis
2.8. Molecular Dynamic Simulation
2.9. Post-Simulation Analysis
2.9.1. Principal Components and Dynamic Cross-Correlation Matrix Analysis
2.9.2. Binding Affinity Calculations Using MM-GBSA
2.10. Tools Used in This Study
3. Results
3.1. Retrieval of Pathogen Defense Protein Sequence
3.2. Structural and Functional Annotation Using InterPro
3.3. Physiochemical Properties
3.4. Phylogenetic Analysis
3.5. Structure Prediction, Refinement and Validation
3.5.1. Secondary Structure Prediction
3.5.2. Ab Initio Structure Prediction Using I-TASSER, RoseTTAFold, and AlphaFold
3.5.3. Structure Validation, Refinement, Quality Assessment, and Model Selection
3.6. Functional Interaction Site Prediction
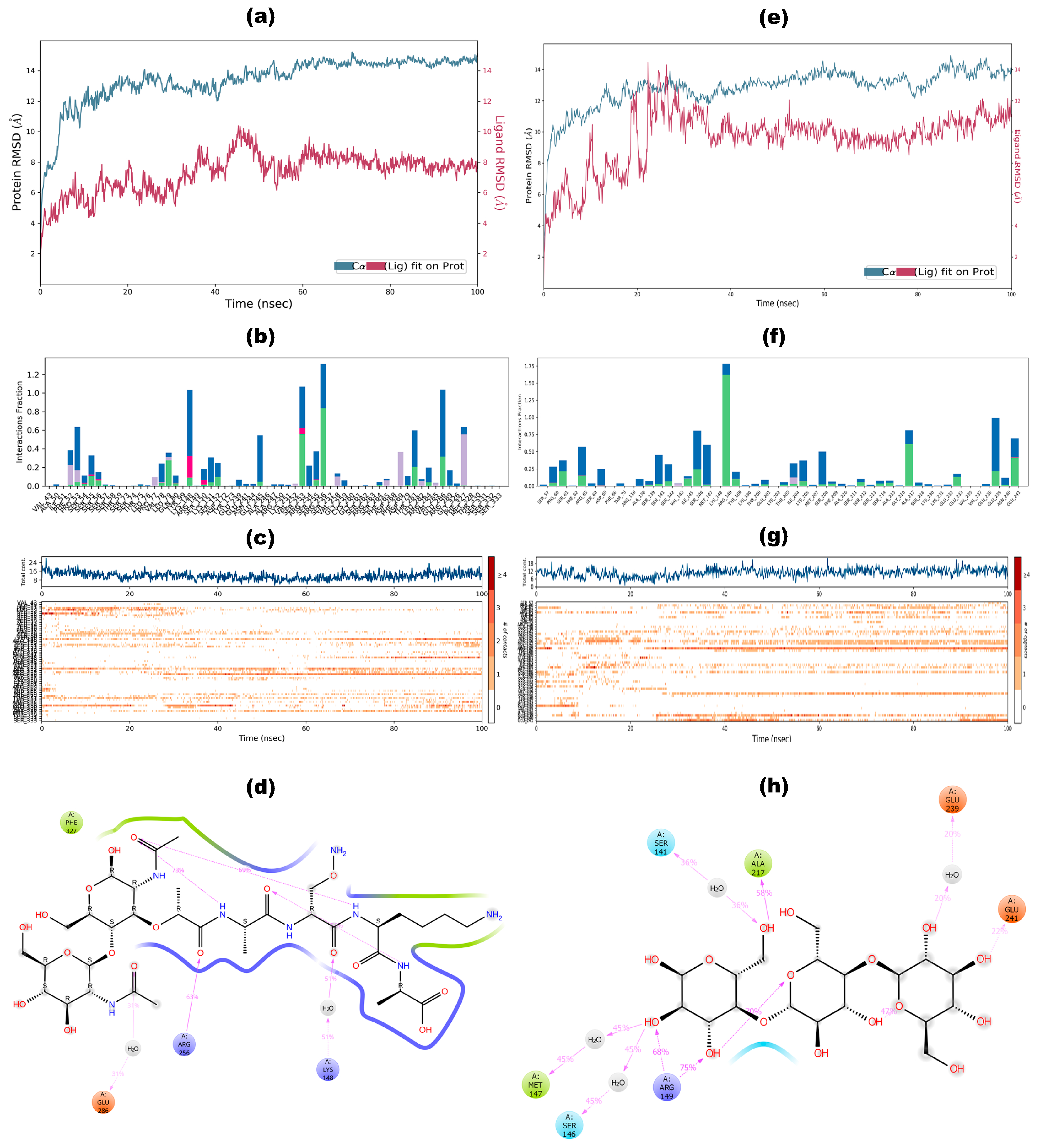
3.7. Molecular Docking
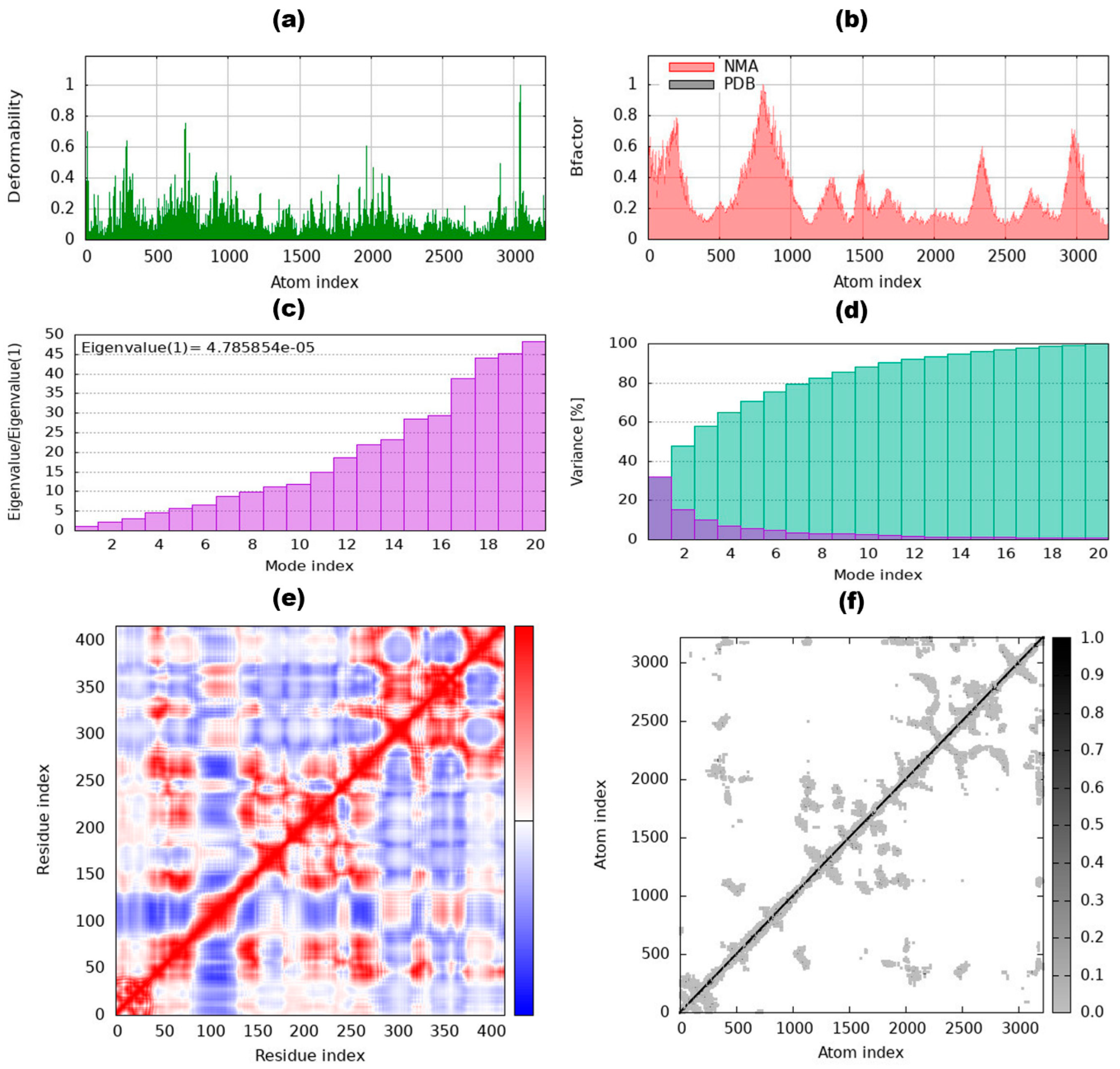
3.8. Internal Coordinates Normal Mode Analysis
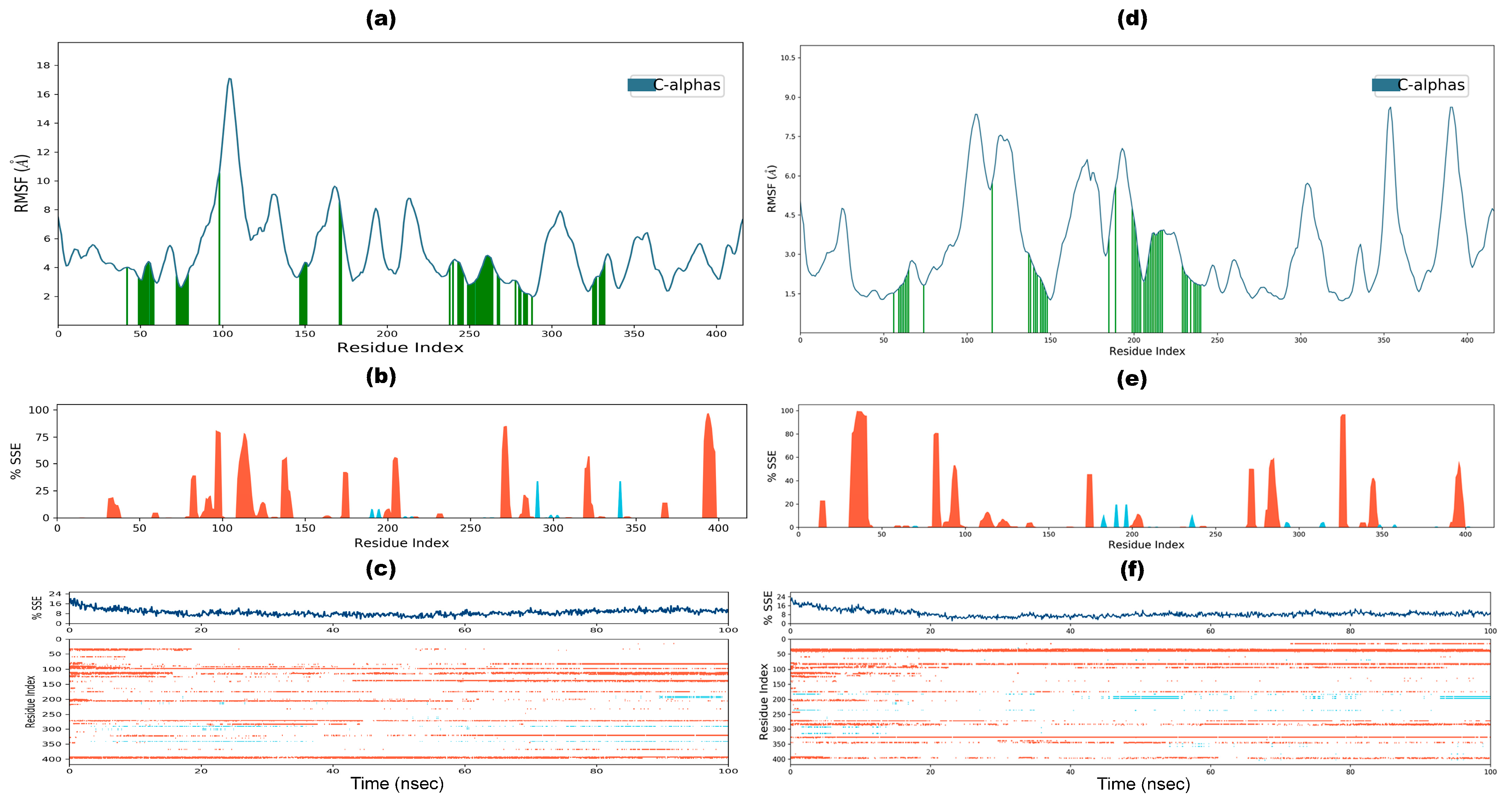
3.9. Molecular Dynamic Simulation
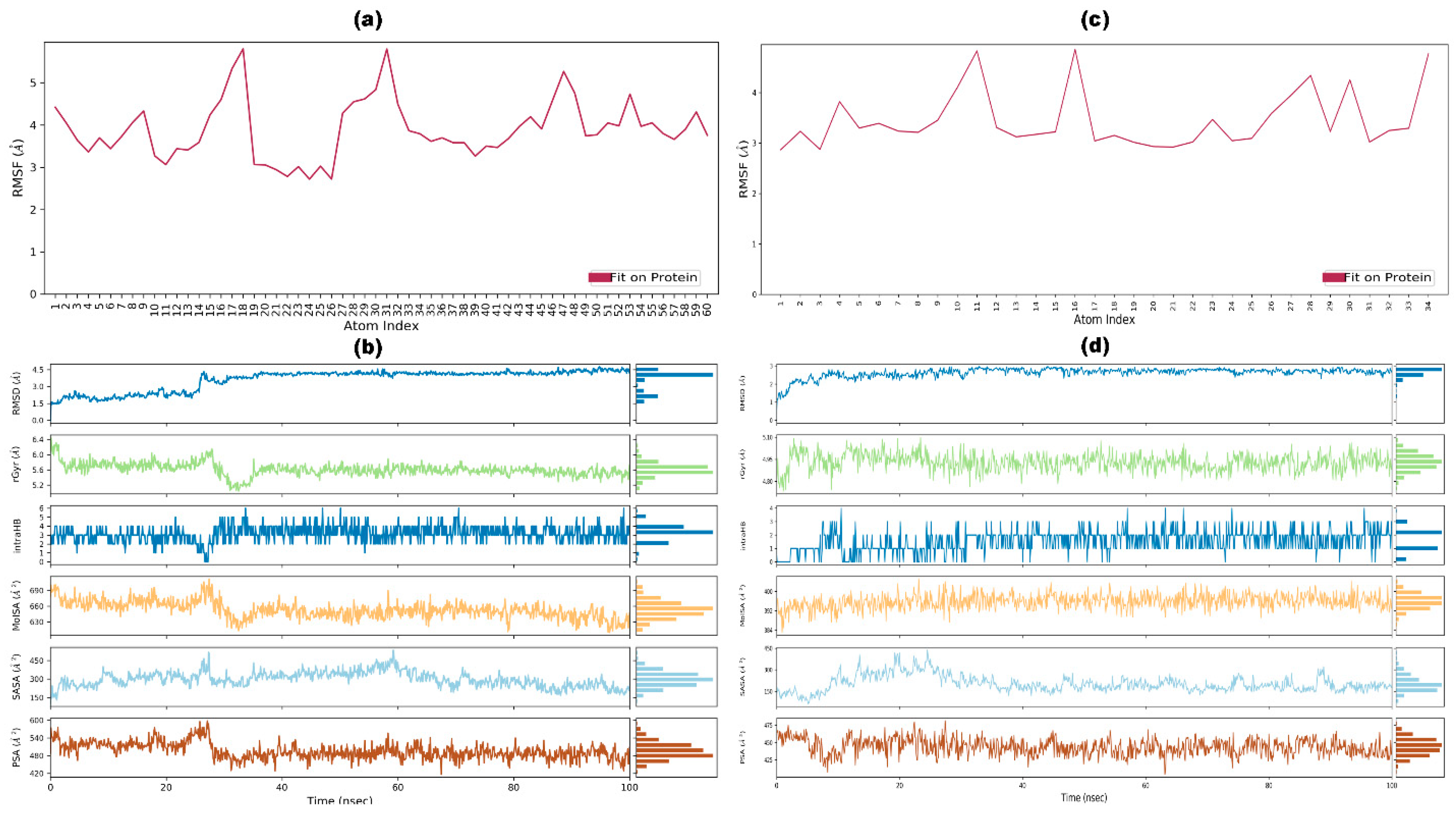
3.10. Post-Simulation Analysis
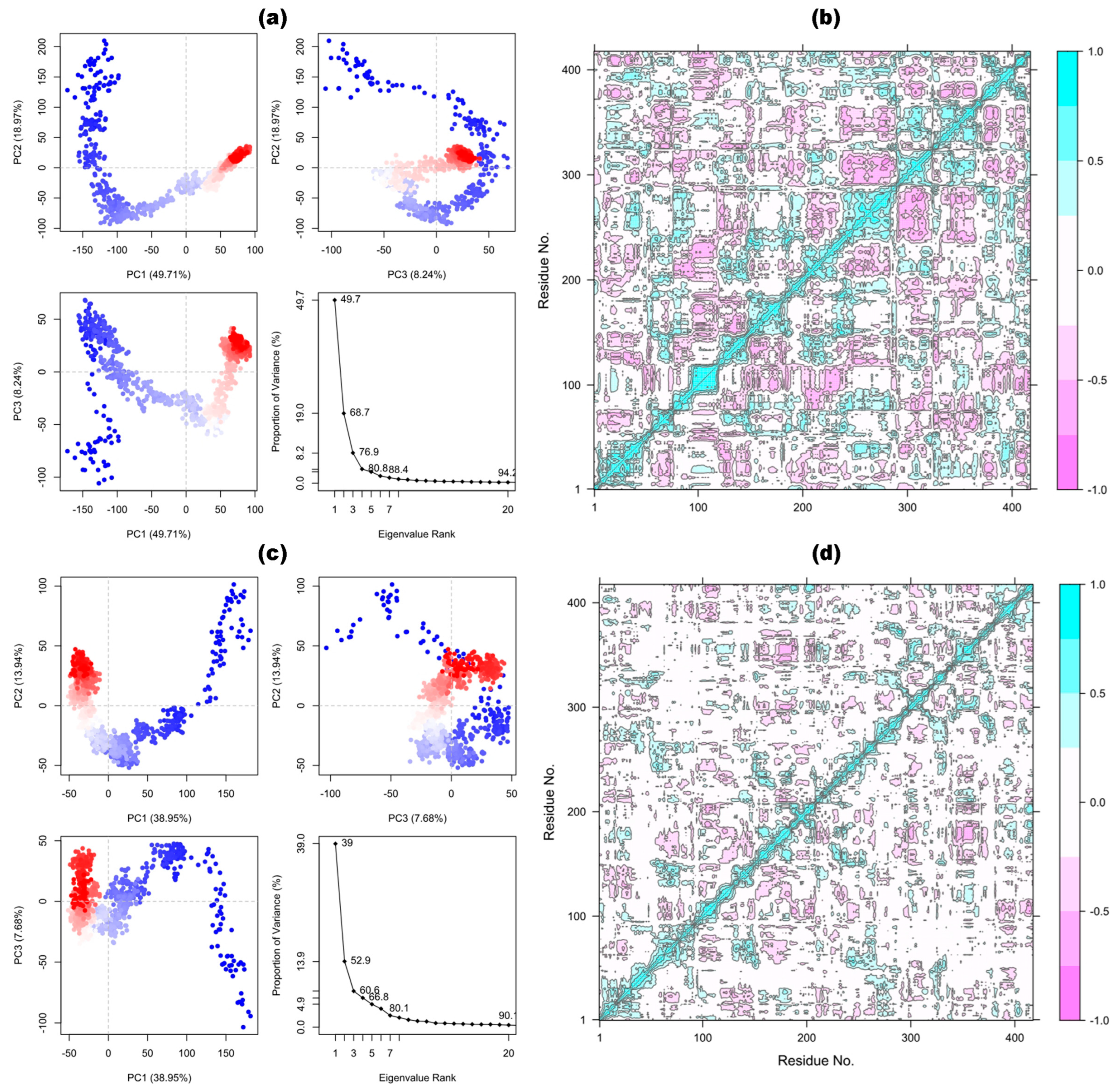
3.10.1. Principal Components and Dynamic Cross-Correlation Matrix Analysis
3.10.2. Binding Affinity Calculations Using MM-GBSA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Otávio, O.A.; dos Santos, M.S.; Antunes Filho, S.; Backx, B.P. Nanotechnology for the Control of Plant Pathogens and Pests. Plant Nano Biol. 2024, 8, 100080. [Google Scholar] [CrossRef]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of Plant Defense against Insect Herbivores. Plant Signal. Behav. 2012, 7, 1306. [Google Scholar] [CrossRef] [PubMed]
- Hönig, M.; Roeber, V.M.; Schmülling, T.; Cortleven, A. Chemical Priming of Plant Defense Responses to Pathogen Attacks. Front. Plant Sci. 2023, 14, 1146577. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Feng, H.; Zhang, T.; Wang, Q. Editorial: Plant Defense Mechanisms in Plant-Pathogen Interactions; Frontiers Media SA: Lausanne, Switzerland, 2023; Volume 14, p. 1292294. [Google Scholar]
- Saeed, A. Plant Defense Mechanisms Against Bacterial Pathogens. Genet. Genom. Sci. 2019, 4, 1–6. [Google Scholar] [CrossRef]
- Mithöfer, A.; Maffei, M.E. General Mechanisms of Plant Defense and Plant Toxins. In Plant Toxins; Springer: Dordrecht, The Netherlands, 2017; pp. 3–24. ISBN 978-94-007-6464-4. [Google Scholar]
- Kaur, S.; Samota, M.K.; Choudhary, M.; Choudhary, M.; Pandey, A.K.; Sharma, A.; Thakur, J. How Do Plants Defend Themselves against Pathogens-Biochemical Mechanisms and Genetic Interventions. Physiol. Mol. Biol. Plants 2022, 28, 485–504. [Google Scholar] [CrossRef]
- Drapal, M.; Enfissi, E.M.A.; Fraser, P.D. The Chemotype Core Collection of Genus Nicotiana. Plant J. 2022, 110, 1516–1528. [Google Scholar] [CrossRef]
- Zou, X.; Bk, A.; Rauf, A.; Saeed, M.; Al-Awthan, Y.S.; Al-Duais, M.A.; Bahattab, O.; Hamayoon Khan, M.; Suleria, H.A.R. Screening of Polyphenols in Tobacco (Nicotiana tabacum) and Determination of Their Antioxidant Activity in Different Tobacco Varieties. ACS Omega 2021, 6, 25361–25371. [Google Scholar] [CrossRef]
- Zhang, W.; Pan, X.; Fu, J.; Cheng, W.; Lin, H.; Zhang, W.; Huang, Z. Phytochemicals Derived from Nicotiana tabacum L. Plant Contribute to Pharmaceutical Development. Front. Pharmacol. 2024, 15, 1372456. [Google Scholar] [CrossRef]
- Stewart, G.G. A History of the Medicinal Use of Tobacco 1492–1860. Med. Hist 1967, 11, 228–268. [Google Scholar] [CrossRef]
- Zou, X.; BK, A.; Abu-Izneid, T.; Aziz, A.; Devnath, P.; Rauf, A.; Mitra, S.; Emran, T.B.; Mujawah, A.A.H.; Lorenzo, J.M.; et al. Current Advances of Functional Phytochemicals in Nicotiana Plant and Related Potential Value of Tobacco Processing Waste: A Review. Biomed. Pharmacother. 2021, 143, 112191. [Google Scholar] [CrossRef]
- Sharma, Y.; Srivastava, N.; Bala, K. Antioxidant and Antimicrobial Activity of Protein-AgNPs from the Stem of Nicotiana tabacum. Curr. Bioact. Compd. 2021, 17, 37–46. [Google Scholar] [CrossRef]
- Akinpelu, D.A.; Obuotor, E.M. Antibacterial Activity of Nicotiana tabacum Leaves. Fitoterapia 2000, 71, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K. Antiparasitic Potential of Plant Natural Products against Protozoans, Nematodes and Cestodes: A Review. Gen. Med. Clin. Pract. 2024, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Prommaban, A.; Kheawfu, K.; Chittasupho, C.; Sirilun, S.; Hemsuwimon, K.; Chaiyana, W. Phytochemical, Antioxidant, Antihyaluronidase, Antityrosinase, and Antimicrobial Properties of Nicotiana tabacum L. Leaf Extracts. Evid. Based Complement. Altern. Med. 2022, 2022, 5761764. [Google Scholar] [CrossRef]
- Tariq, M.; Ahmad, Z.; Shah, S.A.; Gul, Z.; Khan, S.A. Phytochemical Analysis and Antibacterial Activity of Nicotiana tabacum and Nicotiana Rustica. RADS J. Biol. Res. Appl. Sci. 2021, 12, 60–65. [Google Scholar] [CrossRef]
- Ganapathi, T.R.; Suprasanna, P.; Rao, P.S.; Bapat, V.A. Tobacco (Nicotiana tabacum L.)—A Model System for Tissue Culture Interventions and Genetic Engineering. Indian J. Biotechnol. 2004, 3, 171–184. [Google Scholar]
- Al-Snafi, A.E. Pharmacological and Toxicological Effects of Nicotiana tabacum. World J. Adv. Pharm. Med. Res. 2022, 3, 006–018. [Google Scholar] [CrossRef]
- Manzoor, S.; Nabi, S.U.; Rather, T.R.; Gani, G.; Mir, Z.A.; Wani, A.W.; Ali, S.; Tyagi, A.; Manzar, N. Advancing Crop Disease Resistance through Genome Editing: A Promising Approach for Enhancing Agricultural Production. Front. Genome Ed. 2024, 6, 1399051. [Google Scholar] [CrossRef]
- Yin, Q.; Ren, Z.; Wu, D.; Feng, Z.; Zhu, Z.; Jaisi, A.; Wang, H.; Yang, M. Comparative Effects of Biocontrol Agent and Pathogen on Nicotiana tabacum: Insights into Fungal-Plant Interactions. Plant Signal Behav. 2025, 20, 2453562. [Google Scholar] [CrossRef]
- Ahmad, Z.; Niyazi, S.; Firdoos, A.; Wang, C.; Manzoor, M.A.; Ramakrishnan, M.; Upadhyay, A.; Ding, Y. Enhancing Plant Resilience: Nanotech Solutions for Sustainable Agriculture. Heliyon 2024, 10, e40735. [Google Scholar] [CrossRef]
- Kęska, P.; Gustaw, W.; Stadnik, J. Trends in in Silico Approaches to the Prediction of Biologically Active Peptides in Meat and Meat Products as an Important Factor for Preventing Food-Related Chronic Diseases. Appl. Sci. 2021, 11, 11236. [Google Scholar] [CrossRef]
- Bhat, G.R.; Sethi, I.; Rah, B.; Kumar, R.; Afroze, D. Innovative in Silico Approaches for Characterization of Genes and Proteins. Front. Genet. 2022, 13, 865182. [Google Scholar] [CrossRef]
- Kingdon, A.D.H.; Alderwick, L.J. Structure-Based in Silico Approaches for Drug Discovery against Mycobacterium Tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3708–3719. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef]
- Dhiman, V.; Biswas, S.; Shekhawat, R.S.; Sadhukhan, A.; Yadav, P. In Silico Characterization of Five Novel Disease-Resistance Proteins in Oryza sativa Sp. Japonica against Bacterial Leaf Blight and Rice Blast Diseases. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hasan, R.; Rony, M.N.H.; Ahmed, R. In Silico Characterization and Structural Modeling of Bacterial Metalloprotease of Family M4. J. Genet. Eng. Biotechnol. 2021, 19, 25. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Protein Structure Prediction. In Bioinformatics: Methods and Applications; Academic Press: Cambridge, MA, USA, 2021; pp. 177–188. ISBN 9780323897754. [Google Scholar]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. ISBN 978-1-59259-890-8. [Google Scholar]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher Sequence Analysis Tools Framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y.K. Protein Database Searches Using Compositionally Adjusted Substitution Matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.B.; Sajib, E.H.; Hoque, S.F.; Bappy, M.N.I.; Elahi, F.; Ghosh, A.; Muhit, S.; Hassan, M.M.; Hasan, M.; Chelliah, R.; et al. Genomic Diversity and Molecular Dynamics Interaction on Mutational Variances among RB Domains of SARS-CoV-2 Interplay Drug Inactivation. Infect. Genet. Evol. 2022, 97, 105128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Yuan, Y.; Pang, Y.X.; Yu, F.L.; Yuan, C.; Hu, X.; Wang, D. Phylogenetic Reconstruction and Divergence Time Estimation of Blumea Dc. (Asteraceae: Inuleae) in China Based on Nrdna Its and Cpdna Trnl-f Sequences. Plants 2019, 8, 210. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; McInerney, J.O. Assessment of Methods for Amino Acid Matrix Selection and Their Use on Empirical Data Shows That Ad Hoc Assumptions for Choice of Matrix Are Not Justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. Sopma: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A Deep-Learning-Based Platform for Multi-Domain Protein Structure and Function Prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding Non-Homologous Proteins by Coupling Deep-Learning Contact Maps with I-TASSER Assembly Simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Chinea, G.; Lopez, N.; Pons, T.; Vriend, G. Homology Modeling, Model and Software Evaluation: Three Related Resources. Bioinformatics 1998, 14, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB Server for Protein Structure Prediction and Refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Ménard, R.; Alban, S.; De Ruffray, P.; Jamois, F.; Franz, G.; Fritig, B.; Yvin, J.C.; Kauffmann, S. β-1,3 Glucan Sulfate, but Not β-1,3 Glucan, Induces the Salicylic Acid Signaling Pathway in Tobacco and Arabidopsis. Plant Cell 2004, 16, 3020. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. Beta-Glucan Recognition by the Innate Immune System. Immunol. Rev. 2009, 230, 38–50. [Google Scholar] [CrossRef]
- Fesel, P.H.; Zuccaro, A. β-Glucan: Crucial Component of the Fungal Cell Wall and Elusive MAMP in Plants. Fungal Genet. Biol. 2016, 90, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.Z.; Hameed, S.; Ali, M.; Zaheer, A. In Silico Molecular Docking Analysis of Limonene with The Fat Mass and Obesity-Associated Protein by Using Autodock Vina. Sci. J. Inform. 2021, 8, 154–160. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Katuwal, S.; Upadhyaya, S.R.; Marahatha, R.; Shrestha, A.; Regmi, B.P.; Khadayat, K.; Basnet, S.; Basnyat, R.C.; Parajuli, N. In Silico Study of Coumarins: Wedelolactone as a Potential Inhibitor of the Spike Protein of the SARS-CoV-2 Variants. J. Trop. Med. 2023, 2023, 4771745. [Google Scholar] [CrossRef]
- Shah, M.; Yamin, R.; Ahmad, I.; Wu, G.; Jahangir, Z.; Shamim, A.; Nawaz, H.; Nishan, U.; Ullah, R.; Ali, E.A.; et al. In-Silico Evaluation of Natural Alkaloids against the Main Protease and Spike Glycoprotein as Potential Therapeutic Agents for SARS-CoV-2. PLoS ONE 2024, 19, e0294769. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. IMODS: Internal Coordinates Normal Mode Analysis Server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Hossain, A. Molecular Docking, Drug-Likeness and ADMET Analysis, Application of Density Functional Theory (DFT) and Molecular Dynamics (MD) Simulation to the Phytochemicals from Withania Somnifera as Potential Antagonists of Estrogen Receptor Alpha (ER-α). Curr. Comput. Aided Drug Des. 2020, 17, 797–805. [Google Scholar] [CrossRef]
- Singh, S.; Singh, V.K. Molecular Dynamics Simulation: Methods and Application. In Frontiers in Protein Structure, Function, and Dynamics; Springer: Singapore, 2020; pp. 213–238. [Google Scholar]
- Fujii, S.; Kono, H.; Takenaka, S.; Go, N.; Sarai, A. Sequence-Dependent DNA Deformability Studied Using Molecular Dynamics Simulations. Nucleic Acids Res. 2007, 35, 6063–6074. [Google Scholar] [CrossRef]
- Petrov, D.; Margreitter, C.; Grandits, M.; Oostenbrink, C.; Zagrovic, B. A Systematic Framework for Molecular Dynamics Simulations of Protein Post-Translational Modifications. PLoS Comput. Biol. 2013, 9, e1003154. [Google Scholar] [CrossRef]
- Lopéz-Blanco, J.R.; Garzón, J.I.; Chacón, P. IMod: Multipurpose Normal Mode Analysis in Internal Coordinates. Bioinformatics 2011, 27, 2843–2850. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC’06, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Ferreira, L.G.; Oliva, G.; Andricopulo, A.D. Target-Based Molecular Modeling Strategies for Schistosomiasis Drug Discovery. Future Med. Chem. 2015, 7, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384. [Google Scholar] [CrossRef] [PubMed]
- Fateen, A.; Zeeshan Ahmed, M.; Mahmood Ashraf, N.; Khurshid, M.; Ali, M.; Mutahir, Z. Computational analysis of action mechanism and evolutionary insight of chitin degrading enzymes in Bacillus cereus. Xi’an Shiyou Daxue Xuebao (Ziran Kexue Ban)/J. Xi’an Shiyou Univ. Nat. Sci. Ed. 2024, 67, 55–101. [Google Scholar]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing Molecular Dynamics Simulation Data into View; Elsevier Current Trends: Amsterdam, The Netherlands, 2019; Volume 44, pp. 902–913. [Google Scholar]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of Lead Compounds against Scm (Fms10) in Enterococcus Faecium Using Computer Aided Drug Designing. Life 2021, 11, 77. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the Opls Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef]
- Bahuguna, A.; Khaket, T.P.; Bajpai, V.K.; Shukla, S.; Park, I.W.; Na, M.K.; Huh, Y.S.; Han, Y.K.; Kang, S.C.; Kim, M. N-Acetyldopamine Dimers from Oxya Chinensis Sinuosa Attenuates Lipopolysaccharides Induced Inflammation and Inhibits Cathepsin C Activity. Comput. Struct. Biotechnol. J. 2022, 20, 1177–1188. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Azhar, E.I.; Kamal, M.A.; Bajrai, L.H.; Dubey, A.; Jha, K.; Yadava, U.; Kang, S.G.; Dwivedi, V.D. SARS-CoV-2 Mpro Inhibitors: Identification of Anti-SARS-CoV-2 Mpro Compounds from FDA Approved Drugs. J. Biomol. Struct. Dyn. 2022, 40, 2769–2784. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Bolaji, O.Q.; Ojo, T.O.; Adegun, I.P.; Adebodun, S. Molecular Mechanics with Generalized Born Surface Area (MMGBSA) Calculations and Docking Studies Unravel Some Antimalarial Compounds Using Heme O Synthase as Therapeutic Target. ChemistrySelect 2023, 8, e202303686. [Google Scholar] [CrossRef]
- Ghosh, P.; Bhakta, S.; Bhattacharya, M.; Sharma, A.R.; Sharma, G.; Lee, S.S.; Chakraborty, C. A Novel Multi-Epitopic Peptide Vaccine Candidate Against Helicobacter Pylori: In-Silico Identification, Design, Cloning and Validation Through Molecular Dynamics. Int. J. Pept. Res. Ther. 2021, 27, 1149–1166. [Google Scholar] [CrossRef]
- Mirzadeh, A.; Kobakhidze, G.; Vuillemot, R.; Jonic, S.; Rouiller, I. In Silico Prediction, Characterization, Docking Studies and Molecular Dynamics Simulation of Human P97 in Complex with P37 Cofactor. BMC Mol. Cell Biol. 2022, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the HACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef] [PubMed]
- El Khatabi, K.; Aanouz, I.; Alaqarbeh, M.; Ajana, M.A.; Lakhlifi, T.; Bouachrine, M. Molecular Docking, Molecular Dynamics Simulation, and ADMET Analysis of Levamisole Derivatives against the SARS-CoV-2 Main Protease (MPro). BioImpacts 2022, 12, 107–113. [Google Scholar] [CrossRef]
- Shivanika, C.; Deepak Kumar, S.; Ragunathan, V.; Tiwari, P.; Sumitha, A.; Brindha Devi, P. Molecular Docking, Validation, Dynamics Simulations, and Pharmacokinetic Prediction of Natural Compounds against the SARS-CoV-2 Main-Protease. J. Biomol. Struct. Dyn. 2022, 40, 585–611. [Google Scholar] [CrossRef]
- Sparling, D.W. Bioindicators of Contaminant Exposure. In Ecotoxicology Essentials; Academic Press: Cambridge, MA, USA, 2016; pp. 45–66. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, S.; Li, H.; Li, L. Vitellogenin, a Multivalent Sensor and an Antimicrobial Effector. Int. J. Biochem. Cell Biol. 2011, 43, 303–305. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, S. Immune-Relevant and Antioxidant Activities of Vitellogenin and Yolk Proteins in Fish. Nutrients 2015, 7, 8818. [Google Scholar] [CrossRef]
- Sheng, Y.; Chen, J.; Jiang, H.; Lu, Y.; Dong, Z.; Pang, L.; Zhang, J.; Wang, Y.; Chen, X.; Huang, J. The Vitellogenin Receptor Gene Contributes to Mating and Host-Searching Behaviors in Parasitoid Wasps. iScience 2023, 26, 106298. [Google Scholar] [CrossRef]
- Li, L.; Li, X.J.; Wu, Y.M.; Yang, L.; Li, W.; Wang, Q. Vitellogenin Regulates Antimicrobial Responses in Chinese Mitten Crab, Eriocheir Sinensis. Fish Shellfish Immunol. 2017, 69, 6–14. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Singh, H.; Dixit, S.; Mendu, V.; Verma, P.C. Molecular Characterization of Vitellogenin and Vitellogenin Receptor of Bemisia Tabaci. PLoS ONE 2016, 11, e0155306. [Google Scholar] [CrossRef]
- Huo, Y.; Yu, Y.; Chen, L.; Li, Q.; Zhang, M.; Song, Z.; Chen, X.; Fang, R.; Zhang, L. Insect Tissue-Specific Vitellogenin Facilitates Transmission of Plant Virus. PLoS Pathog. 2018, 14, e1006909. [Google Scholar] [CrossRef]
- He, Y.-Z.; Wang, Y.-M.; Yin, T.-Y.; Cuellar, W.J.; Liu, S.-S.; Wang, X.-W. Gut-Expressed Vitellogenin Facilitates the Movement of a Plant Virus across the Midgut Wall in Its Insect Vector. mSystems 2021, 6, e0058121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiochemical Properties | VLP |
|---|---|
| Number of amino acids | 417 |
| Molecular weight (kDa) | 45.145 |
| Average Residue Weight (EMBOSS-PEPSTATS) | 108.264 |
| Theoretical pI (ExPASy-ProtParam) | 9.81 |
| Theoretical pI (EMBOSS-PEPSTATS) | 10.43 |
| Negatively charged residues (Asp + Glu) | 33 |
| Positively charged residues (Arg + Lys) | 54 |
| Formula | C1916H3048N584O642S19 |
| Total number of atoms | 6209 |
| Ext. coefficient (ExPASy-ProtParam) | 29,450, Abs 0.1% (=1 g/L) 0.652, assuming all Cys residues are reduced, and 29,950, Abs 0.1% (=1 g/L) 0.663, assuming all pairs of Cys residues form cystines |
| Molar ext. coefficients (EMBOSS-PEPSTATS) | 0.652 (reduced), 0.663 (cystine bridges) |
| Estimated Half-life (mammalian reticulocytes, in vitro) (hours) | 30 h |
| Estimated Half-life (yeast, in vivo) (hours) | >20 h |
| Estimated Half-life (E. coli, in vivo) (hours) | >10 h |
| Aliphatic index | 49.81 |
| Grand average of hydropathicity (GRAVY) | −0.658 |
| Solubility | 0.345 |
| Improbability of expression in inclusion bodies | 0.979 |
| Category | Metric | Refined RoseTTAFold | Refined AlphaFold | |
|---|---|---|---|---|
| All-Atom Contacts | Clashscore (number of serious steric overlaps (>0.4 Å) per 1000 atoms), all atoms | 1.93 (99th percentile) | 18.88 (36th percentile) | |
| Protein Geometry | Poor rotamers | 1 (0.27%) | 2 (0.55%) | Goal: <0.3% |
| Favored rotamers | 362 (98.91%) | 357 (97.54%) | Goal: >98% | |
| Ramachandran outliers | 1 (0.24%) | 3 (0.72%) | Goal: <0.05% | |
| Ramachandran favored | 406 (97.83%) | 400 (96.39%) | Goal: >98% | |
| Rama distribution Z-score | −0.25 ± 0.38 | −2.65 ± 0.27 | Goal: abs (Z score) < 2 | |
| MolProbity score | 1.00 (100th percentile) | 2.01 (75th percentile) | ||
| Cβ deviations >0.25Å | 2 (0.51%) | 0 (0.00%) | Goal: 0 | |
| Bad bonds | 17/3218 (0.53%) | 21/3218 (0.65%) | Goal: 0% | |
| Bad angles | 25/4324 (0.58%) | 22/4324 (0.51%) | Goal: <0.1% | |
| Peptide Omegas | Cis Prolines | 0/14 (0.00%) | 0/14 (0.00%) | Expected: ≤1 per chain, or ≤5% |
| Low-resolution Criteria | CaBLAM outliers | 8 (1.9%) | 10 (2.4%) | Goal: <1.0% |
| CA Geometry outliers | 2 (0.48%) | 3 (0.73%) | Goal: <0.5% |
| Functional Interaction Site | Area (SA) Å2 | Volume (SA) Å3 | Interaction Amino Acids |
|---|---|---|---|
 | 2461.98 | 2893.06 | Met1, Met3, Phe33, Gln36, Glu37, Leu39, Gly40, Lys41, Val43, Ser44, Ser49, Ile51, Phe52, Pro53, Ser54, Ser55, Ser56, Ser57, Ser58, Thr59, Ser61, Phe62, Arg63, Ser64, Ser74, Thr75, Leu76, Pro77, Val78, Leu79, Thr81, Asn82, Gln85, Thr86, Ser91, Ser139, Ser142, Val143, Ile145, Ser146, Met147, Lys148, Arg149, Ser150, Lys151, Ser152, Thr153, Thr154, Pro156, Arg157, Phe183, Tyr185, Glu201, Ile204, Lys205, Met207, Ser208, Phe209, Ala210, Ser212, Ala215, Lys230, Glu233, Phe234, Val235, Glu245, Ala246, Ala247, Phe248, Arg250, Val252, Ser253, Arg254, Ser255, Arg256, Gly259, Cys260, Gly261, Ser262, Arg263, Ser264, Phe265, Ser266, Gly267, Phe269, Glu271, Ile273, Asp279, Thr281, Leu282, Arg283, Val285, Glu286, Arg289, Leu323, Phe324, Met328, Thr330, Ser331, His341, Leu364, and His366. |
| Protein | Ligand | Binding Affinity (kcal/mol) | RMSD | Amino Acid Residues | Number and Types of Bond Interaction | Bond Length (A°) |
|---|---|---|---|---|---|---|
| VLP | PGN | −10.16 | 2.3 | Ser150 | 1 Unfavourable donor–donor | 2.38 |
| Ser152 | 1 Conventional hydrogen bond, 2 Carbon hydrogen bond | 2.99, 3.50, 3.38 | ||||
| Ser262 | 1 Conventional hydrogen bond | 2.69 | ||||
| Ser264 | 1 Carbon hydrogen bond | 3.26 | ||||
| Phe265 | 1 Pi Alkyl | 5.41 | ||||
| Arg289 | 2 Unfavourable donor–donor | 2.23, 2.02 | ||||
| Ser331 | 1 Carbon hydrogen bond | 3.71 | ||||
| β-glucan | −7.19 | 1.45 | Ile145 | 2 Carbon hydrogen bond | 2.51, 2.57 | |
| Met147 | 1 Conventional hydrogen bond | 2.03 | ||||
| Ser208 | 3 Carbon hydrogen bond | 2.35, 2.86, 3.48 | ||||
| Ala210 | 1 Carbon hydrogen bond | 3.42 | ||||
| Ser212 | 1 Conventional hydrogen bonds, 1 Carbon hydrogen bond | 2.36, 3.3 | ||||
| Ser214 | 1 Conventional hydrogen bonds | 2.08 | ||||
| Glu233 | 2 Conventional hydrogen bonds, 1 Carbon hydrogen bond | 2.00, 2.25, 2.44 |
| Free Energies (kcal/mol) | ΔGbind | ΔGbindLipo | ΔGbindCovalent | ΔGbindvdW | ΔGbindCoulomb | ΔGbindHbond | ΔGbindSolv GB | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | VLP-PGN | VLP β-glucan | |
| 0 ns | −85.4146 | −66.0888 | −19.9471 | −12.8334 | 10.93931 | 3.71073 | −84.2806 | −37.3172 | −44.2088 | −50.9057 | −3.3445 | −7.44361 | 55.42714 | 38.70038 |
| 25 ns | −48.0244 | −28.0909 | −15.5065 | −5.35445 | 4.143636 | 0.789766 | −49.1873 | −26.3458 | −32.4691 | −25.0725 | −3.03638 | −2.51674 | 48.03121 | 30.40881 |
| 50 ns | −65.3214 | −49.0764 | −18.1111 | −11.9729 | 7.669821 | 5.886676 | −61.9296 | −38.4276 | −32.3607 | −44.9816 | −2.56722 | −4.76744 | 41.97732 | 45.18656 |
| 75 ns | −76.5874 | −48.3983 | −20.4385 | −12.4229 | 12.45735 | 6.199441 | −62.9388 | −41.6997 | −39.7965 | −47.324 | −3.70663 | −4.4175 | 37.83573 | 51.26634 |
| 100 ns | −64.6475 | −43.8721 | −19.869 | −11.294 | −1.8028 | 2.74656 | −77.6791 | −34.9507 | −27.0734 | −35.1664 | −2.51416 | −4.05589 | 64.29094 | 38.84835 |
| Mean ± SD | −68.00 | −47.11 | −18.77 | −10.78 | 6.68 | 3.87 | −67.20 | −35.75 | −35.18 | −40.69 | −3.03 | −4.64 | 49.51 | 40.88 |
| SD | 14.09 | 13.58 | 2.03 | 3.08 | 5.72 | 2.25 | 13.89 | 5.79 | 6.78 | 10.50 | 0.51 | 1.79 | 10.59 | 7.83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maoz, H.; Elalouf, A.; Rosenfeld, A.Y. Bioinformatics-Based Management of Vitellogenin-like Protein’s Role in Pathogen Defense in Nicotiana tabacum L. Appl. Sci. 2025, 15, 4463. https://doi.org/10.3390/app15084463
Maoz H, Elalouf A, Rosenfeld AY. Bioinformatics-Based Management of Vitellogenin-like Protein’s Role in Pathogen Defense in Nicotiana tabacum L. Applied Sciences. 2025; 15(8):4463. https://doi.org/10.3390/app15084463
Chicago/Turabian StyleMaoz, Hanan, Amir Elalouf, and Amit Yaniv Rosenfeld. 2025. "Bioinformatics-Based Management of Vitellogenin-like Protein’s Role in Pathogen Defense in Nicotiana tabacum L." Applied Sciences 15, no. 8: 4463. https://doi.org/10.3390/app15084463
APA StyleMaoz, H., Elalouf, A., & Rosenfeld, A. Y. (2025). Bioinformatics-Based Management of Vitellogenin-like Protein’s Role in Pathogen Defense in Nicotiana tabacum L. Applied Sciences, 15(8), 4463. https://doi.org/10.3390/app15084463