The Accuracy of Determining Cluster Size by Analyzing Ripley’s K Function in Single Molecule Localization Microscopy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Methods for SMLM Image and Nanoclusters Analysis
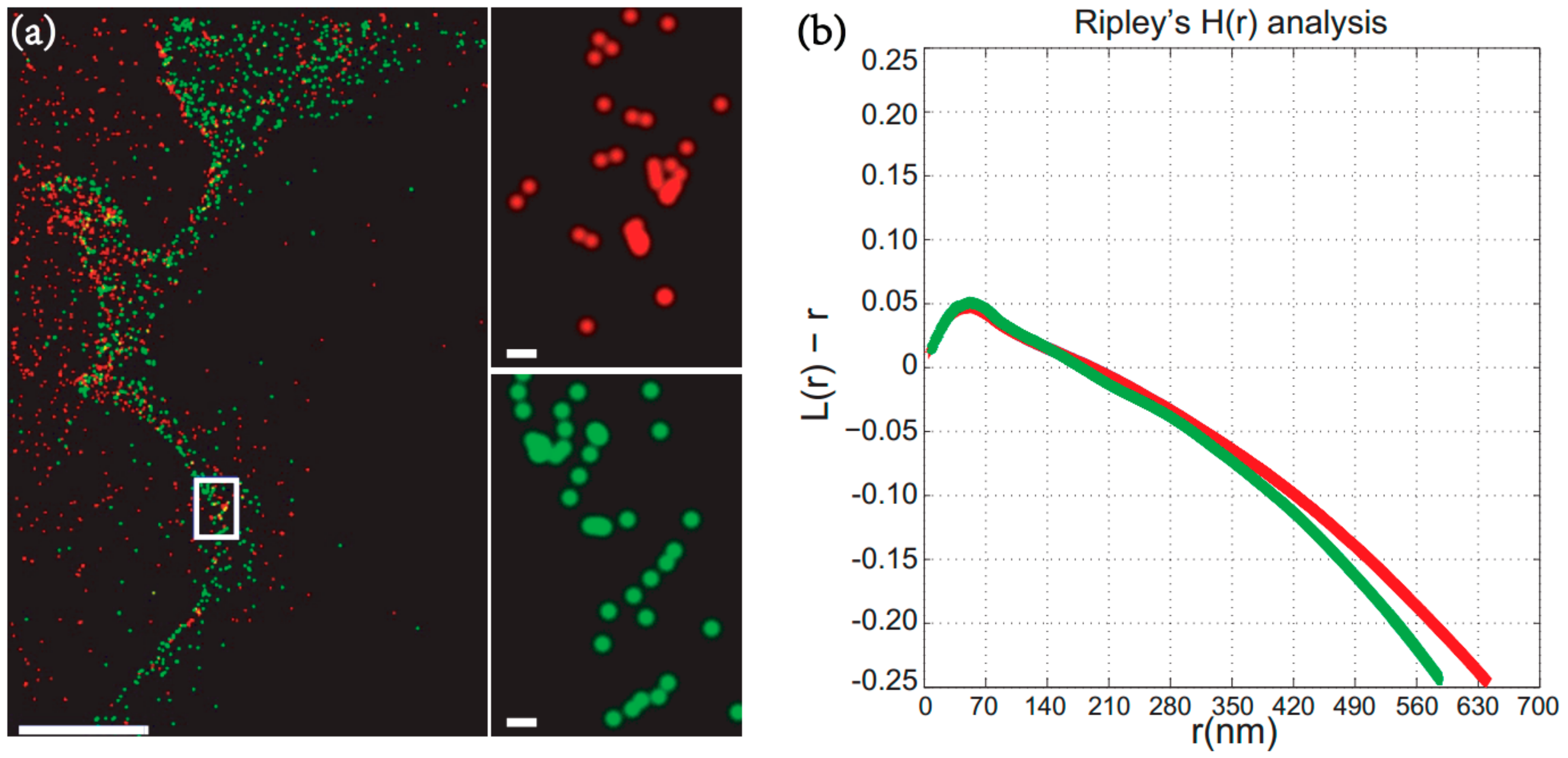
2.2. Material for EGFR (Epidermal Growth Factor Receptor) Measurements at Fibroblast-Like Cell Lines Derived from Monkey Kidney Tissue—COS7 Cell Surface
3. Results and Discussion
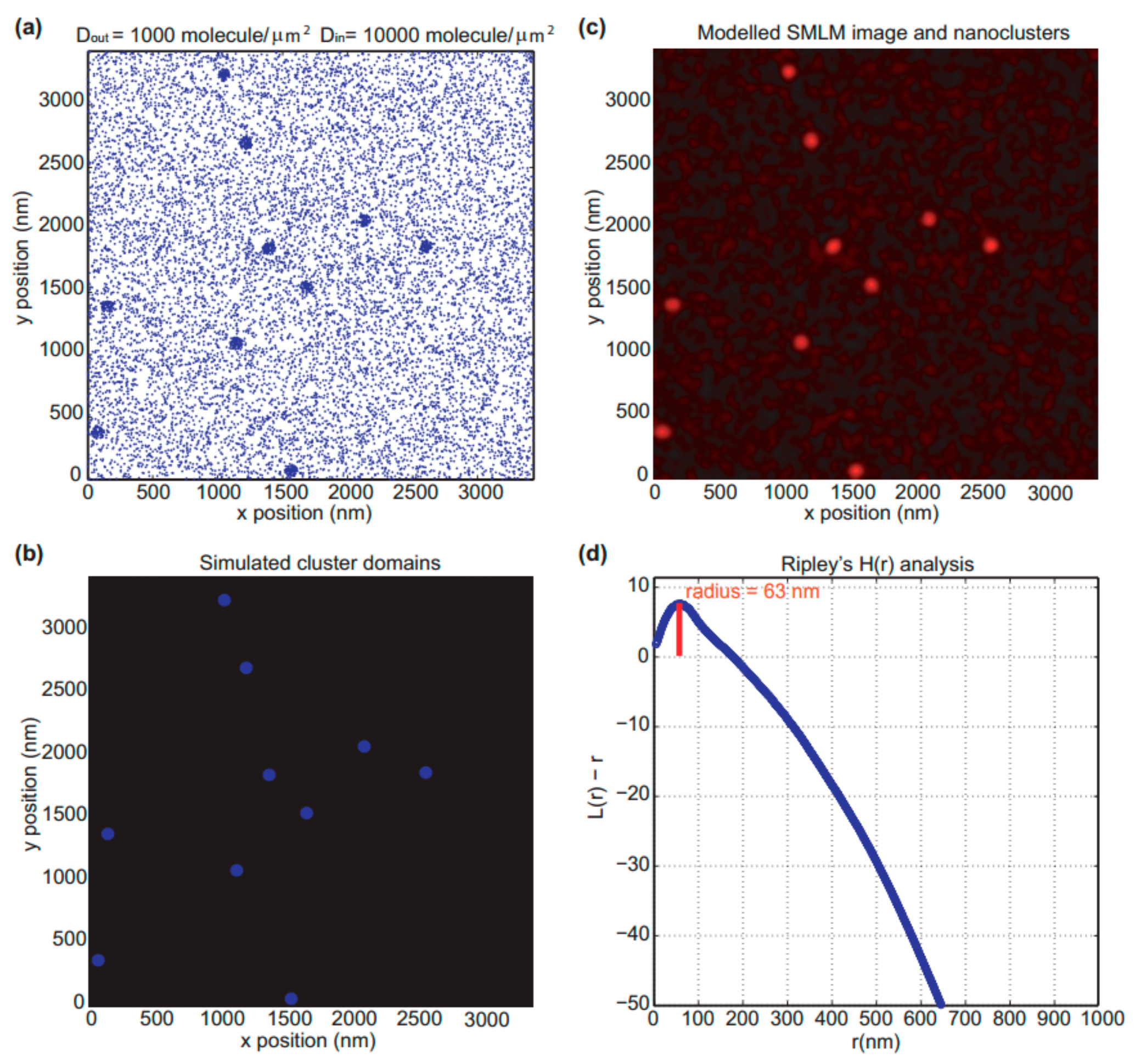
3.1. The Modeled SMLM Image and Nanoclusters
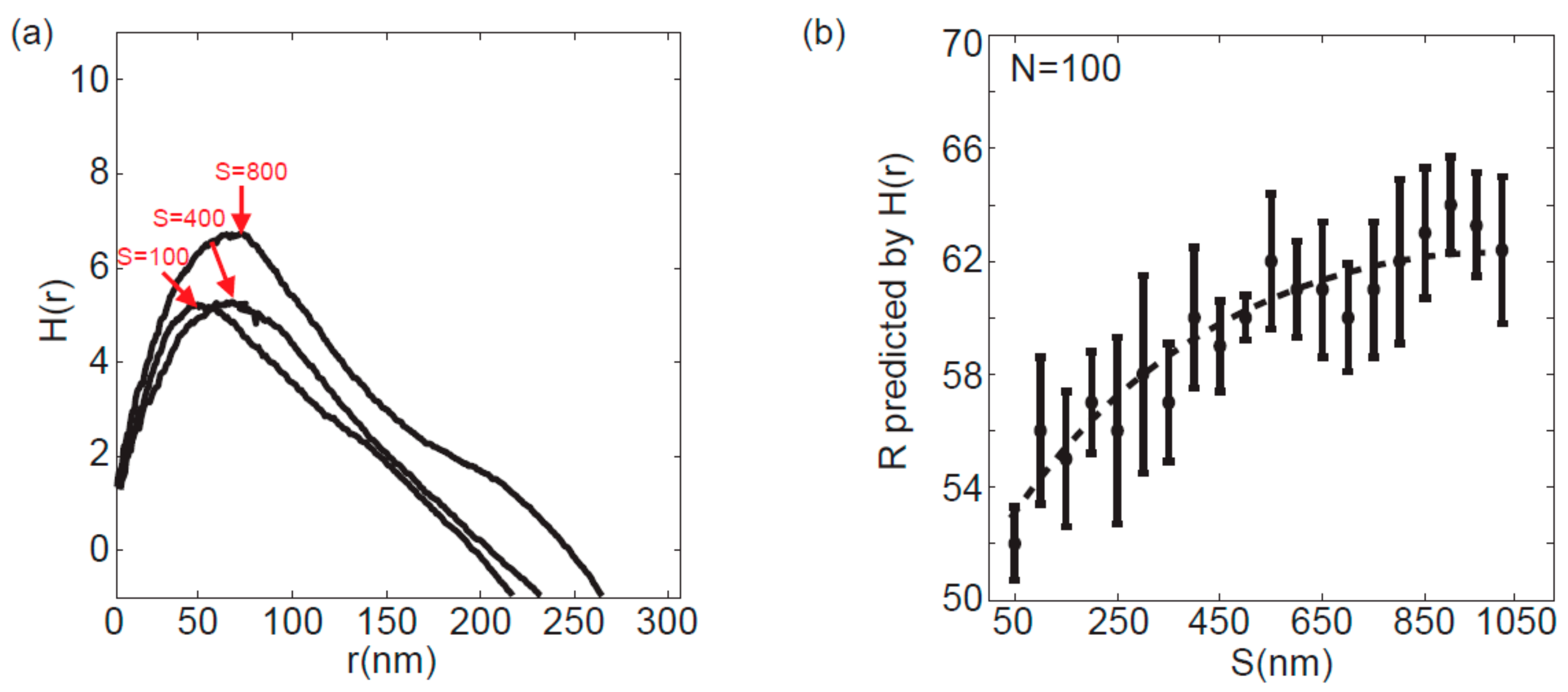
3.2. Effect of the Clusters Separation Distance on the Accuracy of Peak Analysis Method
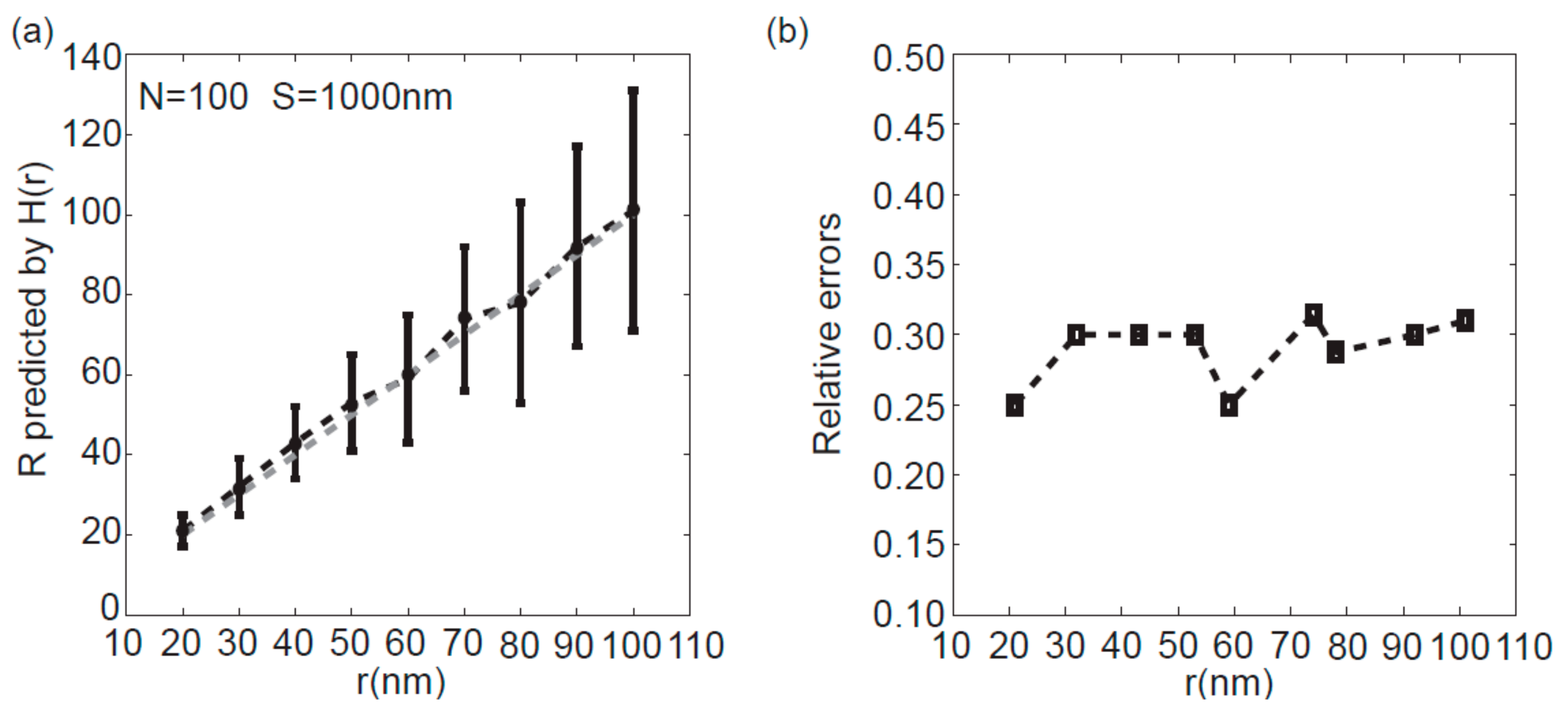
3.3. Effect of the Cluster Diameter on the Accuracy of Peak Analysis Method
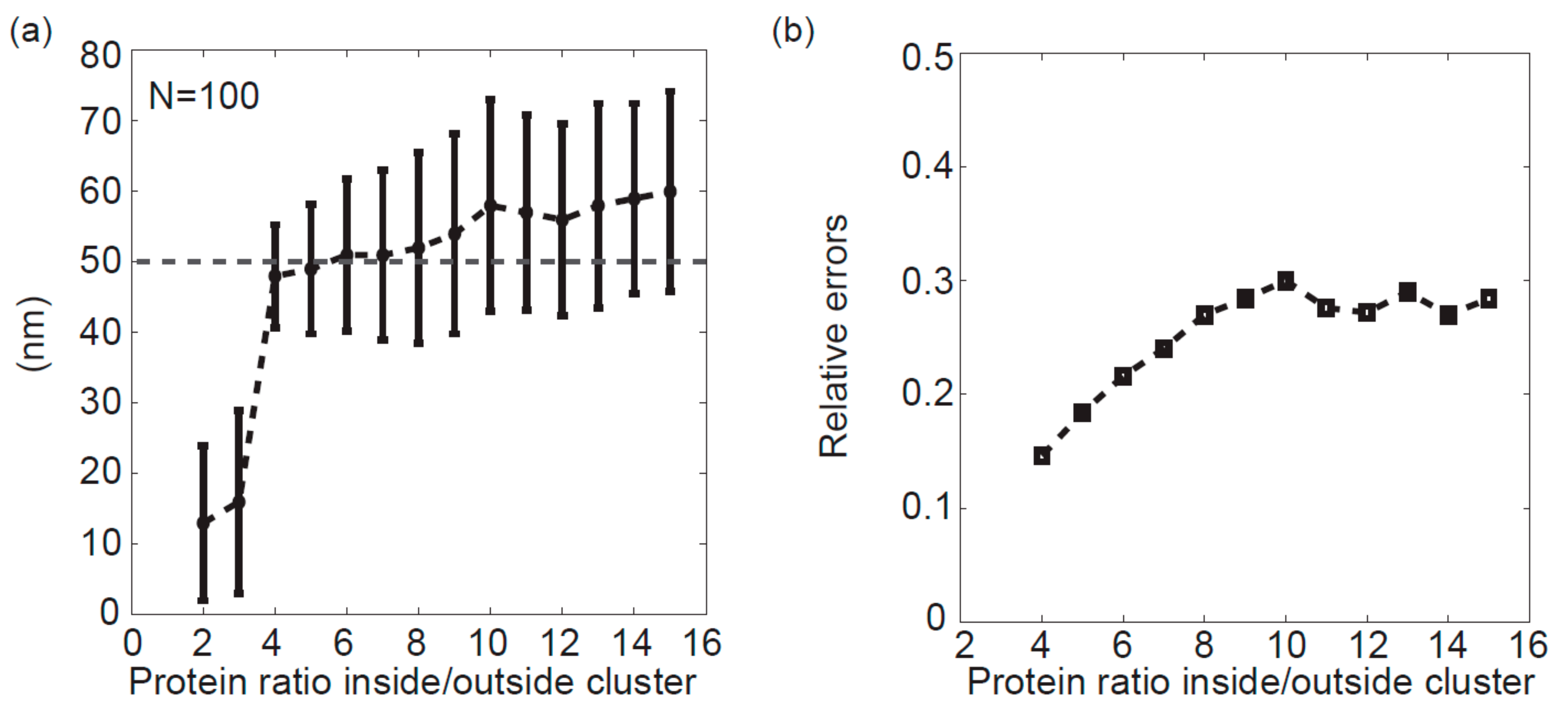
3.4. Effects of the Protein Density Ratio Inside/Outside Cluster on the Accuracy of Peak Analysis Method
3.5. A Quantitative Analysis of EGFR (Epidermal Growth Factor Receptor) Cluster at COS7 Cell Surface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Gould, T.J.; Gunewardene, M.; Bewersdorf, J.; Mason, M.D. Ultrahigh Resolution Imaging of Biomolecules by Fluorescence Photoactivation Localization Microscopy; Humana Press, Springer: Totowa, NJ, USA, 2009; pp. 483–522. [Google Scholar]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2010, 3, 793. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, J.; Guo, X.; Tong, T.; Shi, X.; Li, L.; Qi, M.; Wang, Y.; Cai, M.; Jiang, J.; et al. Regulation of EGFR nanocluster formation by ionic protein-lipid interaction. Cell Res. 2014, 24, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhai, J.; Gao, X.Y.; Zhao, H.K.; Su, W.Y.; Zhao, L.N. Targeted peptide-Au cluster binds to epidermal growth factor receptor (EGFR) in both active and inactive states: A clue for cancer inhibition through dual pathways. Sci. Bull. 2018, 63, 349–355. [Google Scholar] [CrossRef]
- Park, K.; Han, S.; Shin, E.; Kim, H.J.; Kim, J.Y. EGFR gene and protein expression in breast cancers. Eur. J. Surg. Oncol. 2007, 33, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Ramarao, M.K.; Cohen, J.B. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc. Natl. Acad. Sci. USA 1998, 95, 4007–4012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lukas, R.J. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem. Pharm. 2011, 82, 800–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, N.C.; Groves, J.T. Signaling clusters in the cell membrane. Curr. Opin. Cell Biol. 2011, 23, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Armson, P.F.; Cha, J.; Phillips, W.D. Clustering of GABAA receptors by rapsyn/43kD protein in vitro. Mol. Cell. Neurosci. 1997, 8, 430–438. [Google Scholar] [CrossRef]
- Waard, M.D.; Pragnell, M.; Campbell, K.P. Ca2+ channel regulation by a conserved β subunit domain. Neuron 1994, 13, 495–503. [Google Scholar] [CrossRef]
- Kazachenko, V.N.; Geletiuk, V.I. Blocking of the counter-K+ fluxes through the single Ca2+-activated K+-channel. Cluster organization of ion channels. Dokl. Akad. Nauk SSSR 1987, 293, 990. [Google Scholar] [PubMed]
- Ripley, B.D. Modelling spatial patterns. J. R. Stat. Soc. Ser. B Stat. Methodol. 1977, 39, 172–212. [Google Scholar] [CrossRef]
- Owen, D.M.; Rentero, C.; Rossy, J.; Magenau, A.; Williamson, D.; Rodriguez, M.; Gaus, K. Palm imaging and cluster analysis of protein heterogeneity at the cell surface. J. Biophotonics 2010, 3, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Shivanandan, A.; Unnikrishnan, J.; Radenovic, A. Accounting for limited detection efficiency and localization precision in cluster analysis in single molecule localization microscopy. PLoS ONE 2015, 10, e0118767. [Google Scholar] [CrossRef] [PubMed]
- Kiskowski, M.A.; Hancock, J.F.; Kenworthy, A.K. On the Use of Ripley’s K-Function and Its Derivatives to Analyze Domain Size. Biophys. J. 2009, 97, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Haase, P. Spatial pattern analysis in ecology based on Ripley’s K-function: Introduction and methods of edge correction. J. Veg. Sci. 1995, 6, 575–582. [Google Scholar] [CrossRef]
- Perry, G.L.W. SpPack: Spatial point pattern analysis in Excel using Visual Basic for Applications (VBA). Environ. Model. Softw. 2004, 19, 559–569. [Google Scholar] [CrossRef]
- Malkusch, S.; Heilemann, M. Extracting quantitative information from single-molecule super-resolution imaging data with LAMA—LocAlization Microscopy Analyzer. Sci. Rep. 2016, 6, 34486. [Google Scholar] [CrossRef]
- Malkusch, S.; Muranyi, W.; Müller, B.; Kräusslich, H.-G.; Heilemann, M. Single-molecule coordinate-based analysis of the morphology of HIV-1 assembly sites with near-molecular spatial resolution. Histochem. Cell Biol. 2013, 139, 173–179. [Google Scholar] [CrossRef]
- Winckler, P.; Lartigue, L.; Giannone, G.; De Giorgi, F.; Ichas, F.; Sibarita, J.B.; Lounis, B.; Cognet, L. Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Sci. Rep. 2013, 3, 2387. [Google Scholar] [CrossRef]
- Giannone, G.; Hosy, E.; Levet, F.; Constals, A.; Schulze, K.; Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E.; Tampe, R.; Choquet, D.; et al. Dynamic Superresolution Imaging of Endogenous Proteins on Living Cells at Ultra-High Density. Biophys. J. 2010, 99, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, R.; Lelek, M.; Fornasiero, E.F.; Valtorta, F.; Zimmer, C.; Mhlanga, M.M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 2010, 7, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Levet, F.; Hosy, E.; Kechkar, A.; Butler, C.; Beghin, A.; Choquet, D.; Sibarita, J.-B. SR-Tesseler: A method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 2015, 12, 1065. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A density-based algorithm for discovering clusters a density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996; pp. 226–231. [Google Scholar]
- Zessin, P.J.; Krüger, C.L.; Malkusch, S.; Endesfelder, U.; Heilemann, M. A hydrophilic gel matrix for single-molecule super-resolution microscopy. Opt. Nanoscopy 2013, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Endesfelder, U.; Malkusch, S.; Fricke, F.; Heilemann, M. A simple method to estimate the average localization precision of a single-molecule localization microscopy experiment. Histochem. Cell Biol. 2014, 141, 629–638. [Google Scholar] [CrossRef] [PubMed]
- De, D.C.; Schmidt, T.H.; Schloetel, J.G.; Lang, T. Packing Density of the Amyloid Precursor Protein in the Cell Membrane. Biophys. J. 2018, 114, 1128–1141. [Google Scholar]
- Amgad, M.; Itoh, A.; Tsui, M.M. Extending Ripley’s K-Function to Quantify Aggregation in 2-D Grayscale Images. PLoS ONE 2015, 10, e0144404. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ncluster | Sarea (μm2) | Scluster (μm2) | Average Rcluster (nm) | Average Separation of Cluster (μm) | % of Molecule inside Cluster | Din (Molecule/μm2) | Dout (Molecule/μm2) | |
|---|---|---|---|---|---|---|---|---|
| EGFR (Panitumumab + Atto647) | 3 | 3.75 | 0.0079 (0.21%) | 49.8279 ± 3.1652 | 0.9462 ± 0.1128 | 20.79% ± 0.05% | 8410 ± 208 | 836 ± 16 |
| EGFR (Panitumumab + Atto532) | 4 | 3.75 | 0.0082 (0.22%) | 50.7771 ± 3.2657 | 1.1321 ± 0.0952 | 18.11% ± 0.04% | 8172 ± 172 | 971 ± 27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, Y.; Yin, P.; Li, F.; Li, D.; Lin, Q.; Li, K. The Accuracy of Determining Cluster Size by Analyzing Ripley’s K Function in Single Molecule Localization Microscopy. Appl. Sci. 2019, 9, 3271. https://doi.org/10.3390/app9163271
Ruan Y, Yin P, Li F, Li D, Lin Q, Li K. The Accuracy of Determining Cluster Size by Analyzing Ripley’s K Function in Single Molecule Localization Microscopy. Applied Sciences. 2019; 9(16):3271. https://doi.org/10.3390/app9163271
Chicago/Turabian StyleRuan, Yi, Ping Yin, Fei Li, Dongmei Li, Qiang Lin, and Kan Li. 2019. "The Accuracy of Determining Cluster Size by Analyzing Ripley’s K Function in Single Molecule Localization Microscopy" Applied Sciences 9, no. 16: 3271. https://doi.org/10.3390/app9163271
APA StyleRuan, Y., Yin, P., Li, F., Li, D., Lin, Q., & Li, K. (2019). The Accuracy of Determining Cluster Size by Analyzing Ripley’s K Function in Single Molecule Localization Microscopy. Applied Sciences, 9(16), 3271. https://doi.org/10.3390/app9163271