A Multi-Scale Modeling of CH4 and H2O Adsorption on Coal Molecules and the Water Blocking Effect in Coalbed Methane Extraction


Abstract
:
1. Introduction
2. Materials and Methods
2.1. DFT Calculations
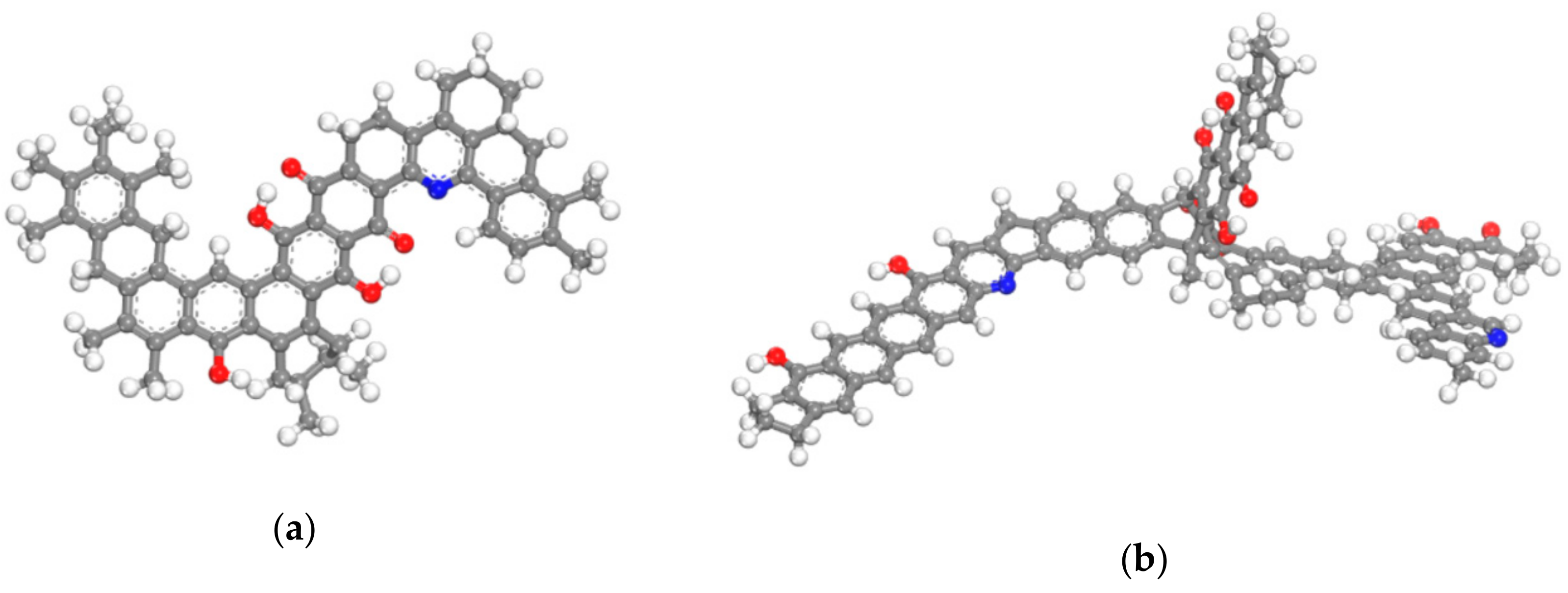
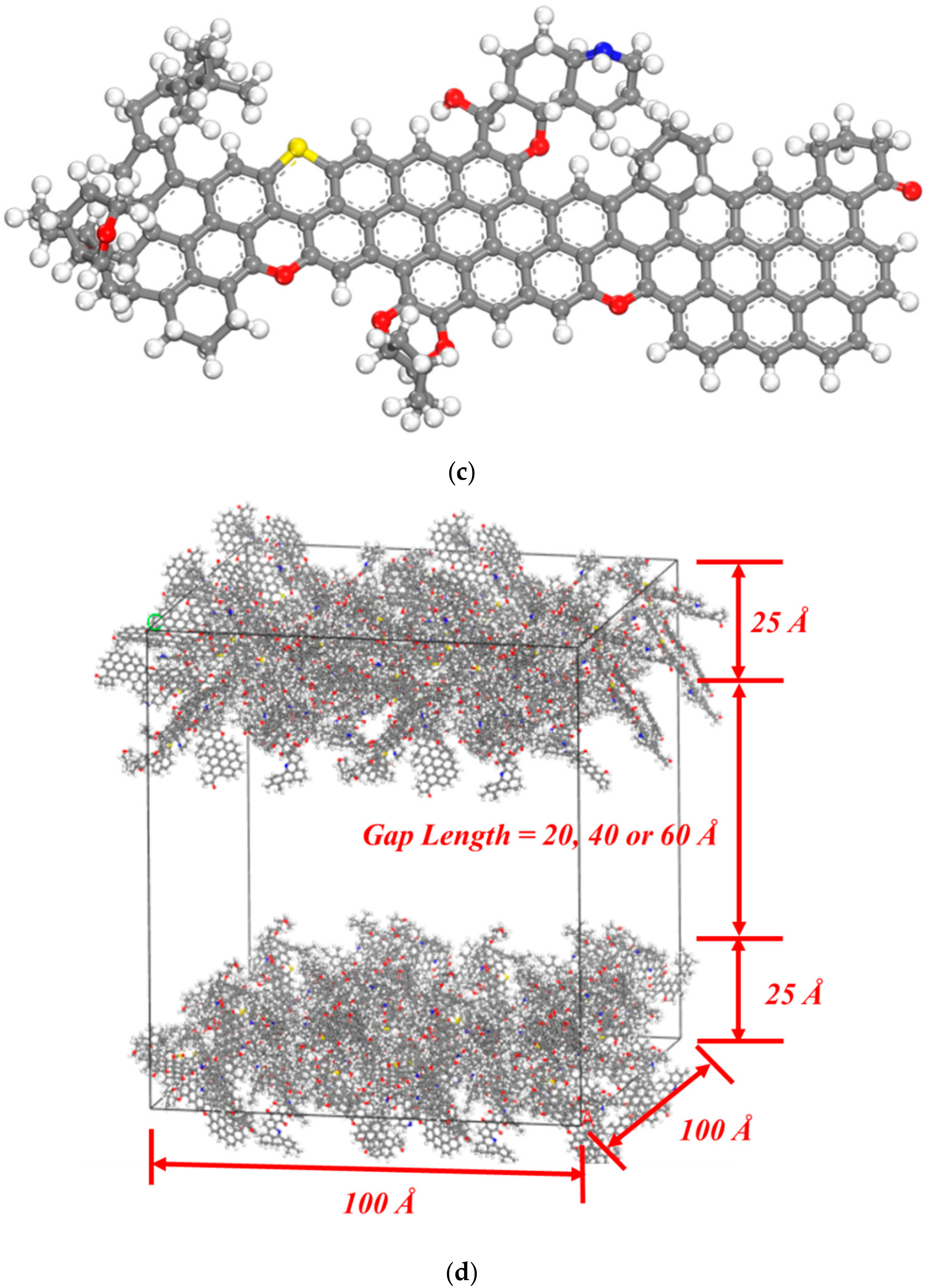
2.2. Molecular Simulation
3. Results and Discussion
3.1. DFT Calculated CH4 and H2O Adsorption on Coal
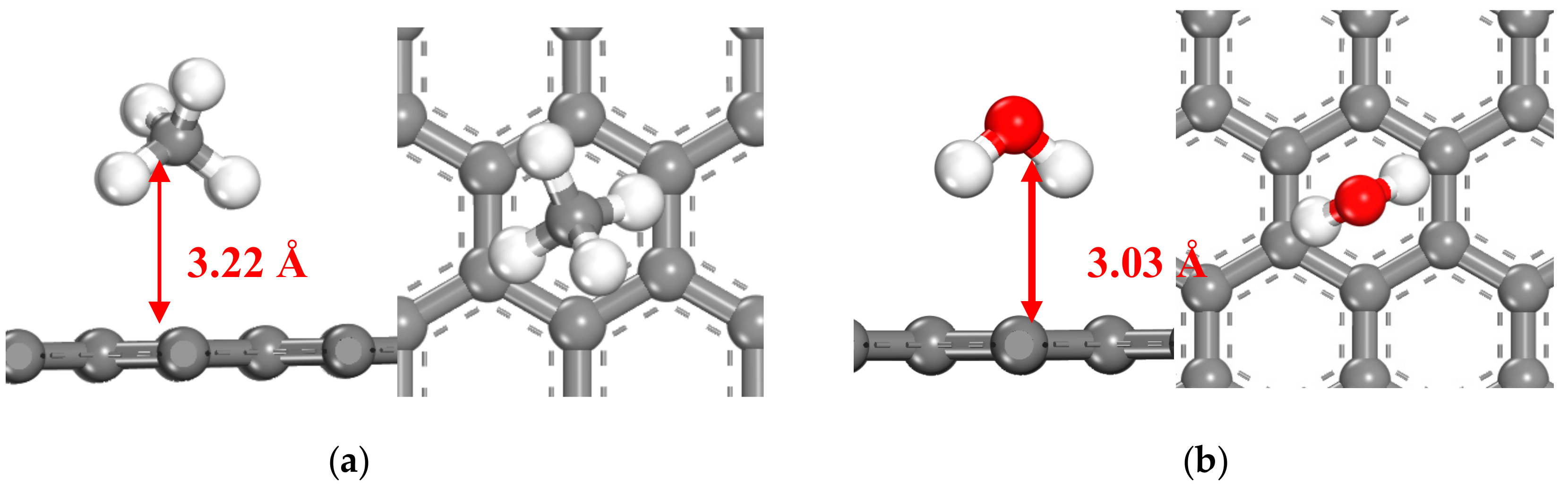
3.1.1. CH4 and H2O Adsorption on Graphene
3.1.2. CH4 and H2O Adsorption on Coal Molecules
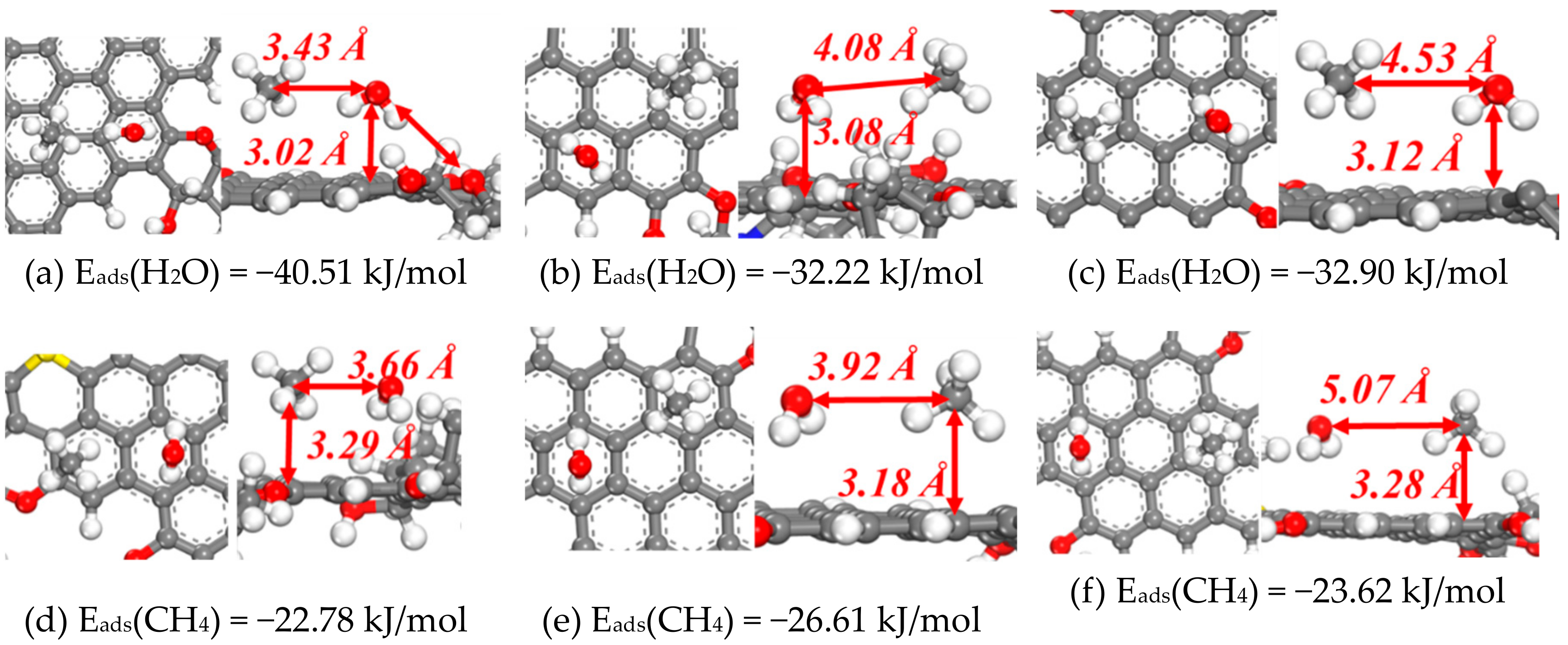
- The most stable adsorption site for CH4 is above aromatic planes, especially if a CH4 molecule interacts with more than one aromatic planes, such as the case in row 1 of Table 2. Polycyclic aromatic planes, which resemble the graphene plane, have slightly stronger bind affinity with CH4, than monocyclic aromatic planes.
- The most stable adsorption site for H2O is N atom sites with H2O can form strong hydrogen bonds with N. However, considering that the percentage of the N element in coal is rather low, the amount of H2O molecule that can be attracted in coal by N atoms by hydrogen bonding is limited.
- Water can form hydrogen bonds with O atoms or be adsorbed on aromatic planes, both of which have similar adsorption energies (around −33 kJ/mol). The adsorption of H2O on aromatic planes is slightly stronger than CH4 adsorption, which has Eads values around 20 kJ/mol. However, this difference is not large enough to allow the coal surface to have a strong binding preference with H2O.
- The presence of substituents on the aromatic rings, such as hydroxyl, carbonyl, -O-, -N=, and -S -groups, often have little influence (<5 kJ/mol) on the adsorption energies of CH4 and H2O.
- The least favorable adsorption sites for both CH4 and H2O are alkane groups and chains. It suggests that the molecular surfaces of anthracite coals, which have high carbon concentrations, should have stronger binding affinity with CH4 and H2O than bituminous coals, subbituminous coal, and lignite. This observation is consistent with previous findings that methane binding is stronger on larger aromatic planes [5]. However, this observation does not necessarily suggest that anthracite coals have stronger methane holding capacity, since other morphological factors, including pore sizes and specific surface areas, can also influence the methane sorption behaviors of coals.
3.1.3. Co-Adsorption of CH4 and H2O on Coal Molecules
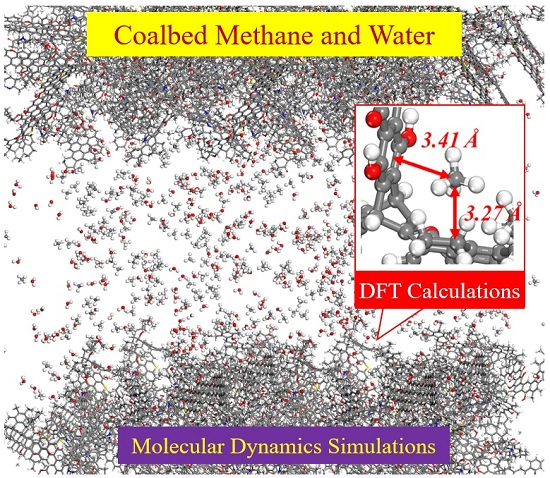
3.2. MD Simulation of CH4 Desorption and Diffusion in Coalbed
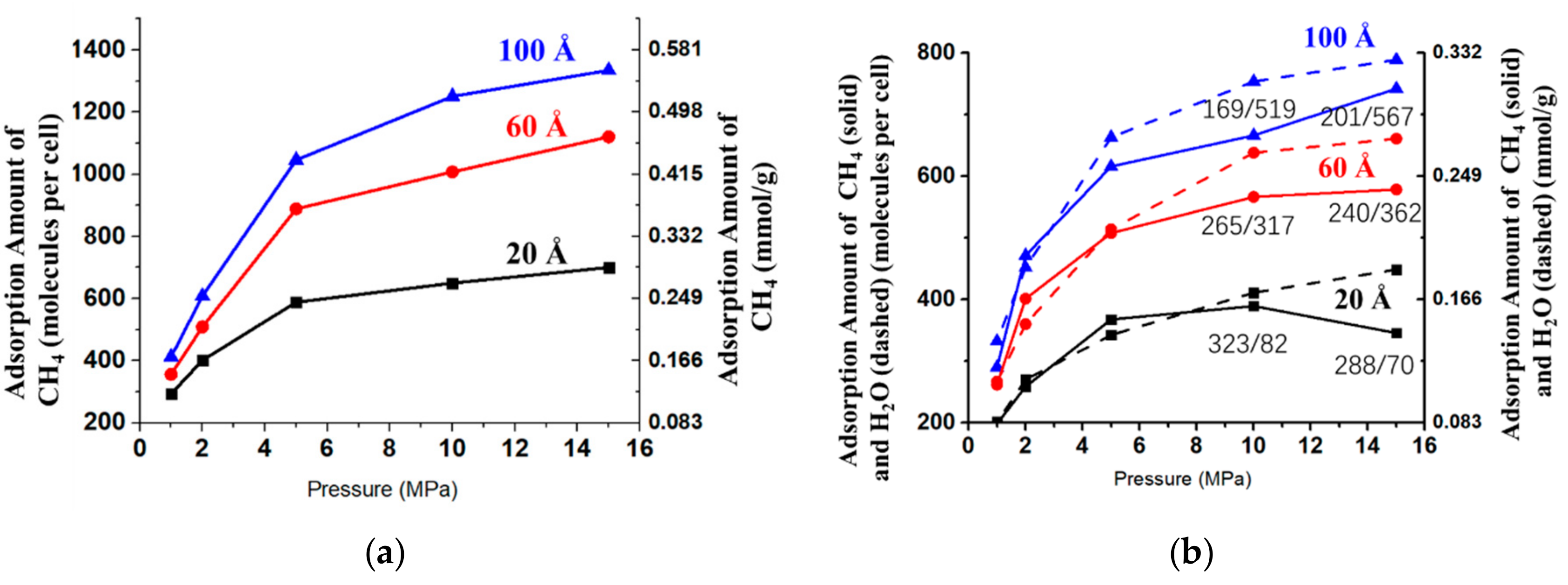
3.2.1. Adsorption Isotherms
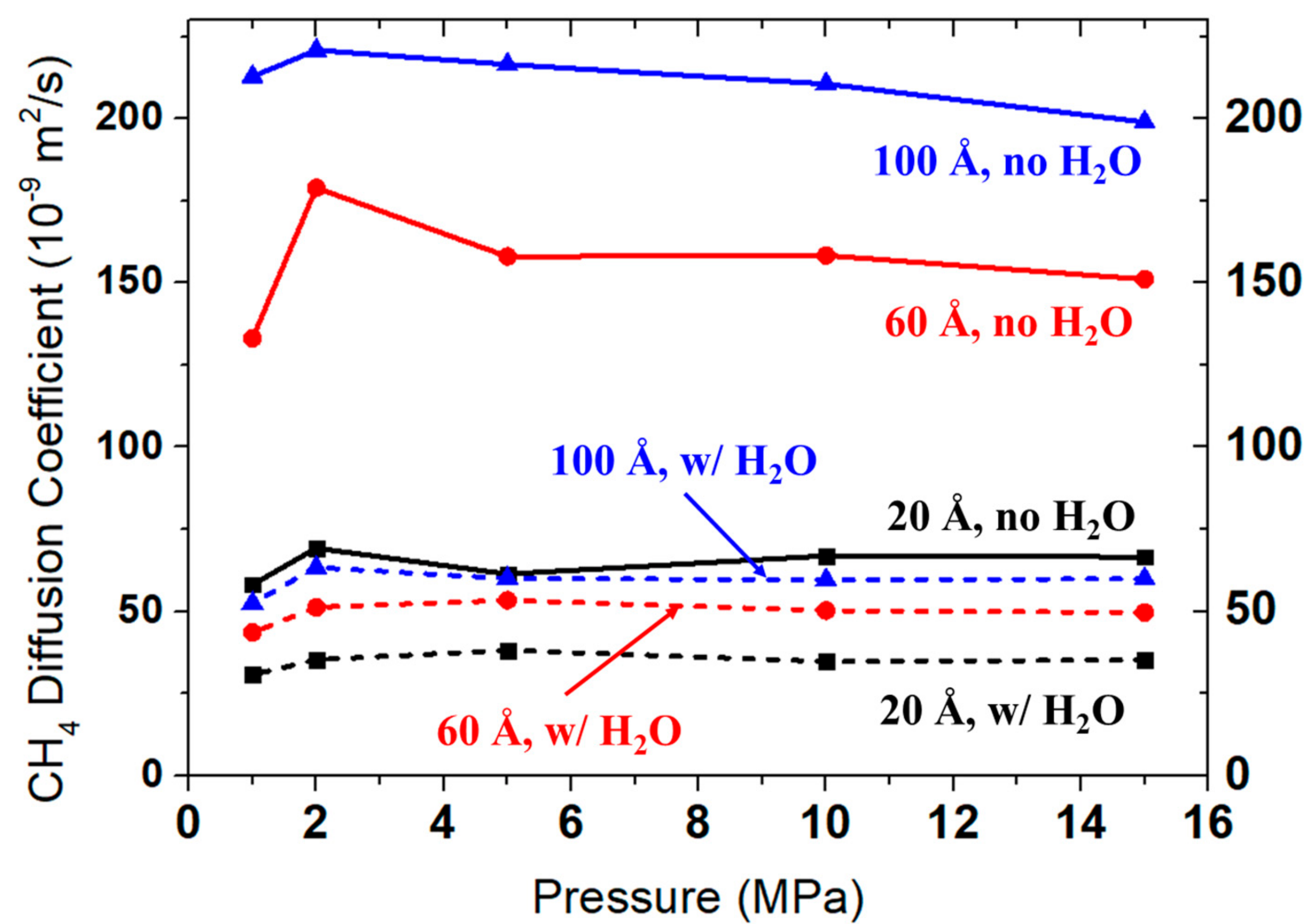
3.2.2. Diffusivity of CH4
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore, T.A. Coalbed Methane: A Review. Int. J. Coal Geol. 2012, 101, 36–81. [Google Scholar] [CrossRef]
- Mastalerz, M. Chapter 7—Coal Bed Methane: Reserves, Production and Future Outlook. In Future Energy, 2nd ed.; Letcher, T.M., Ed.; Elsevier: Boston, MA, USA, 2014; pp. 145–158. [Google Scholar]
- Qin, Y.; Moore, T.A.; Shen, J.; Yang, Z.; Shen, Y.; Wang, G. Resources and Geology of Coalbed Methane in China: A Review. Int. Geol. Rev. 2018, 60, 777–812. [Google Scholar] [CrossRef]
- Su, X.; Wang, Q.; Lin, H.; Song, J.; Guo, H. A Combined Stimulation Technology for Coalbed Methane Wells: Part 1. Theory and Technology. Fuel 2018, 233, 592–603. [Google Scholar] [CrossRef]
- Qiu, N.-X.; Xue, Y.; Guo, Y.; Sun, W.-J.; Chu, W. Adsorption of Methane on Carbon Models of Coal Surface Studied by the Density Functional Theory Including Dispersion Correction (DFT-D3). Comput. Theor. Chem. 2012, 992, 37–47. [Google Scholar] [CrossRef]
- Yu, S.; Bo, J.; Fengjuan, L. Competitive Adsorption of CO2/N2/CH4 onto Coal Vitrinite Macromolecular: Effect of Electrostatic Interactions and Oxygen Functionalities. Fuel 2019, 235, 23–38. [Google Scholar] [CrossRef]
- Yutong, F.; Yu, S. CO2-Adsorption Promoted Ch4-Desorption onto Low-Rank Coal Vitrinite by Density Functional Theory Including Dispersion Correction (DFT-D3). Fuel 2018, 219, 259–269. [Google Scholar] [CrossRef]
- Xu, H.; Chu, W.; Huang, X.; Sun, W.; Jiang, C.; Liu, Z. CO2 Adsorption-Assisted CH4 Desorption on Carbon Models of Coal Surface: A DFT Study. Appl. Surf. Sci. 2016, 375, 196–206. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Xue, Y.; Tian, Z.-Y.; Mo, J.-J.; Qiu, N.-X.; Chu, W.; Xie, H.-P. Adsorption of CH4 on Nitrogen- and Boron-Containing Carbon Models of Coal Predicted by Density-Functional Theory. Appl. Surf. Sci. 2013, 285, 190–197. [Google Scholar] [CrossRef]
- Pini, R.; Ottiger, S.; Storti, G.; Mazzotti, M. Prediction of Competitive Adsorption on Coal by a Lattice DFT model. Adsorption 2010, 16, 37–46. [Google Scholar] [CrossRef]
- Yu, S.; Yan-ming, Z.; Wu, L. Macromolecule Simulation and CH4 Adsorption Mechanism of Coal Vitrinite. Appl. Surf. Sci. 2017, 396, 291–302. [Google Scholar] [CrossRef]
- Hu, H.; Du, L.; Xing, Y.; Li, X. Detailed Study on Self- and Multicomponent Diffusion of CO2-CH4 Gas Mixture in Coal by Molecular Simulation. Fuel 2017, 187, 220–228. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, H.; Zhang, Z.; Chen, H.; Liu, X. Molecular Simulation of CO2/CH4/H2O Competitive Adsorption and Diffusion in Brown Coal. RSC Adv. 2019, 9, 3004–3011. [Google Scholar] [CrossRef]
- Dang, Y.; Zhao, L.; Lu, X.; Xu, J.; Sang, P.; Guo, S.; Zhu, H.; Guo, W. Molecular Simulation of CO2/CH4 Adsorption in Brown Coal: Effect of Oxygen-, Nitrogen-, and Sulfur-Containing Functional Groups. Appl. Surf. Sci. 2017, 423, 33–42. [Google Scholar] [CrossRef]
- Xiang, J.; Zeng, F.; Liang, H.; Li, B.; Song, X. Molecular Simulation of the CH4/CO2/H2O Adsorption onto the Molecular Structure of Coal. Sci. China Earth Sci. 2014, 57, 1749–1759. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Clennell, M.B.; Dewhurst, D.N.; Pervukhina, M. Molecular Simulation of CO2–CH4 Competitive Adsorption and Induced Coal Swelling. Fuel 2015, 160, 309–317. [Google Scholar] [CrossRef]
- Huang, W.; Lei, M.; Qiu, Z.; Leong, Y.-K.; Zhong, H.; Zhang, S. Damage Mechanism and Protection Measures of a Coalbed Methane Reservoir in the Zhengzhuang Block. J. Nat. Gas Sci. Eng. 2015, 26, 683–694. [Google Scholar] [CrossRef]
- Su, X.; Wang, Q.; Song, J.; Chen, P.; Yao, S.; Hong, J.; Zhou, F. Experimental Study of Water Blocking Damage on Coal. J. Pet. Sci. Eng. 2017, 156, 654–661. [Google Scholar] [CrossRef]
- Ni, G.; Cheng, W.; Lin, B.; Zhai, C. Experimental Study on Removing Water Blocking Effect (WBE) from Two Aspects of the Pore Negative Pressure and Surfactants. J. Nat. Gas Sci. Eng. 2016, 31, 596–602. [Google Scholar] [CrossRef]
- Ni, G.; Li, Z.; Xie, H. The Mechanism and Relief Method of the Coal Seam Water Blocking Effect (WBE) Based on the Surfactants. Powder Technol. 2018, 323, 60–68. [Google Scholar] [CrossRef]
- Delley, B. Ground-State Enthalpies: Evaluation of Electronic Structure Approaches with Emphasis on the Density Functional Method. J. Phys. Chem. A 2006, 110, 13632–13639. [Google Scholar] [CrossRef]
- Delley, B. Time Dependent Density Functional Theory with Dmol3. J. Phys. Condens. Matter 2010, 22, 384208. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Becke, A.D. A Multicenter Numerical Integration Scheme for Polyatomic Molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, P.; Mathews, A.L.C. The Molecular Representations of Coal—A Review. Fuel 2012, 96, 1–14. [Google Scholar]
- Given, P. Advances in organic geochemistry the Chemical Study of Coal Macerals. In The Chemical Study of Coal Macerals, Proceedings of the International Meeting in Milan, Milan, Italy, 8–11 September 2019; Hobson, G.D., Speers, G.C., Eds.; Macmillan: New York, NY, USA, 1962; pp. 39–48. [Google Scholar]
- Given, P. The Distribution of Hydrogen in Coals. Fuel 1960, 39, 147–153. [Google Scholar]
- Fuchs, W.; Sandhoff, A.G. Theory of Coal Pyrolysis. Ind. Eng. Chem. 1942, 34, 567–571. [Google Scholar] [CrossRef]
- Sun, H. Compass: An Ab Initio Force-Field Optimized for Condensed-Phase Applications overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Hoover, W. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.; Wang, L.; Li, X.; Wang, C.; Li, L. Simulation of the Interaction of Methane, Carbon Dioxide and Coal. Int. J. Min. Sci. Technol. 2013, 23, 919–923. [Google Scholar] [CrossRef]
- Vidali, G.; Ihm, G.; Kim, H.-Y.; Cole, M.W. Potentials of Physical Adsorption. Surf. Sci. Rep. 1991, 12, 135–181. [Google Scholar] [CrossRef]
- Li, K.; Li, H.; Yan, N.; Wang, T.; Zhao, Z. Adsorption and Dissociation of CH4 on Graphene: A Density Functional Theory Study. Appl. Surf. Sci. 2018, 459, 693–699. [Google Scholar] [CrossRef]
- Alfarge, D.K.; Wei, M.; Bai, B. Numerical simulation study of factors affecting relative permeability modification for water-shutoff treatments. Fuel 2017, 207, 226–239. [Google Scholar] [CrossRef]
- Jenness, G.R.; Karalti, O.; Jordan, K.D. Benchmark Calculations of Water–Acene Interaction Energies: Extrapolation to the Water–Graphene Limit and Assessment of Dispersion–Corrected Dft Methods. Phys. Chem. Chem. Phys. 2010, 12, 6375–6381. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | CH4 | H2O | ||
|---|---|---|---|---|
| Eads (kJ/mol) | Distance (Å) | Eads (kJ/mol) | Distance (Å) | |
| PBE+DFT-D | −24.90 | 3.22 | −31.63 | 3.03 |
| PBE (no dispersion) | −8.24 | 3.58 | −7.12 | 3.19 |
| BLYP+DFT-D | −29.24 | 3.19 | −28.26 | 3.03 |
| BLYP (no dispersion) | −0.63 | 4.60 | −12.54 | 3.30 |
| RPBE | −4.62 | 3.96 | −6.68 | 3.64 |
| m11-L | −29.47 | 2.87 | −29.48 | 3.10 |
| revtpss | −4.59 | 3.22 | −11.37 | 3.17 |
| COMPASS forcefield | −5.38 | 3.47 | −20.19 | 3.36 |
| Literature (DFT) | −31.8 [36] | 3.36 [36] | −17.4 [37] | 3.25 [37] |
| Experimental | −13.5 [38] | 3.03 [38] | −19.0 [39] | - |
| No. | Adsorption Site | Representative Adsorption Structures and Energies | ||
|---|---|---|---|---|
| 1 | Sandwiched by aromatic planes |  −37.92 kJ/mol, G2 |  −27.37 kJ/mol, G2 | |
| 2 | Above polycyclic aromatic planes such as  or or  |  −24.06 kJ/mol, FS |  −22.05 kJ/mol, FS |  −21.44 kJ/mol, G2 |
| 3 | Above an aromatic ring  with or without substituents |  −19.77 kJ/mol, G1 |  −18.97 kJ/mol, G1 |  −17.46 kJ/mol, G2 |
| 4 | Beside O, N, and S atoms, for example  , ,  , ,  , and , and  |  −16.28 kJ/mol, G1 |  −14.36 kJ/mol, FS |  −13.26 kJ/mol, G1 |
| 5 | Beside alkane groups or chains, such as -CH3, -C2H5, -C2H4,-,… |  −7.96 kJ/mol, G2 |  −7.25 kJ/mol, G1 |  −5.90 kJ/mol, G1 |
| No. | Adsorption Site | Representative Adsorption Structures and Energies | ||
|---|---|---|---|---|
| 1 | Hydrogen bonding with  or or  as proton donor as proton donor |  −54.33 kJ/mol, G2 |  −48.48 kJ/mol, G1 |  −46.06 kJ/mol, FS |
| 2 | Sandwiched by aromatic planes |  −41.30 kJ/mol, G2 |  −33.87 kJ/mol, G2 | |
| 3 | Hydrogen bonding with  , ,  or or  as proton donor or acceptor as proton donor or acceptor |  −33.54 kJ/mol, FS |  −33.49 kJ/mol, G2 |  −31.39 kJ/mol, G1 |
| 4 | Above monocyclic or polycyclic aromatic planes |  −33.49 kJ/mol, G1 |  −32.40 kJ/mol, FS |  −29.47 kJ/mol, G2 |
| 5 | Beside alkane groups or chains such as ‒CH3, ‒C2H5, ‒C2H4‒,… |  −9.38 kJ/mol, G2 |  −9.19 kJ/mol, G1 |  −8.04 kJ/mol, G1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Lin, L.; Li, M.; Zhang, X.; Yang, C.; Wang, Y.; Fan, B.; Chen, C.; Luo, W. A Multi-Scale Modeling of CH4 and H2O Adsorption on Coal Molecules and the Water Blocking Effect in Coalbed Methane Extraction. Appl. Sci. 2019, 9, 3421. https://doi.org/10.3390/app9163421
Yang Y, Lin L, Li M, Zhang X, Yang C, Wang Y, Fan B, Chen C, Luo W. A Multi-Scale Modeling of CH4 and H2O Adsorption on Coal Molecules and the Water Blocking Effect in Coalbed Methane Extraction. Applied Sciences. 2019; 9(16):3421. https://doi.org/10.3390/app9163421
Chicago/Turabian StyleYang, Yanhui, Ling Lin, Mengxi Li, Xueying Zhang, Chunli Yang, Yuting Wang, Bin Fan, Congmei Chen, and Wenjia Luo. 2019. "A Multi-Scale Modeling of CH4 and H2O Adsorption on Coal Molecules and the Water Blocking Effect in Coalbed Methane Extraction" Applied Sciences 9, no. 16: 3421. https://doi.org/10.3390/app9163421
APA StyleYang, Y., Lin, L., Li, M., Zhang, X., Yang, C., Wang, Y., Fan, B., Chen, C., & Luo, W. (2019). A Multi-Scale Modeling of CH4 and H2O Adsorption on Coal Molecules and the Water Blocking Effect in Coalbed Methane Extraction. Applied Sciences, 9(16), 3421. https://doi.org/10.3390/app9163421