Electrochemical Conversion of the Lignin Model Veratryl Alcohol to Veratryl Aldehyde Using Manganese(III)-Schiff Base Homogeneous Catalysts

 ,
, 
Abstract
:Featured Application
Abstract
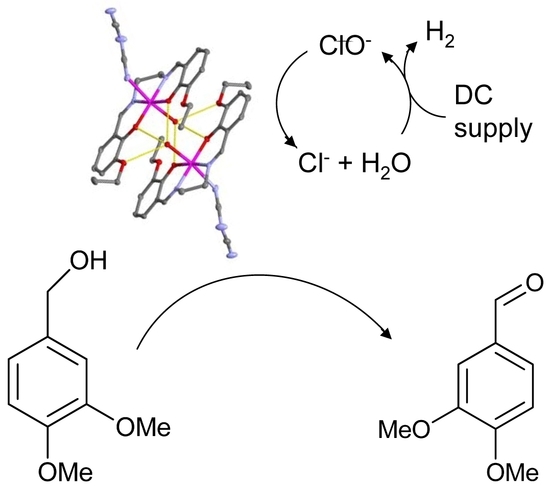
1. Introduction
2. Materials and Methods
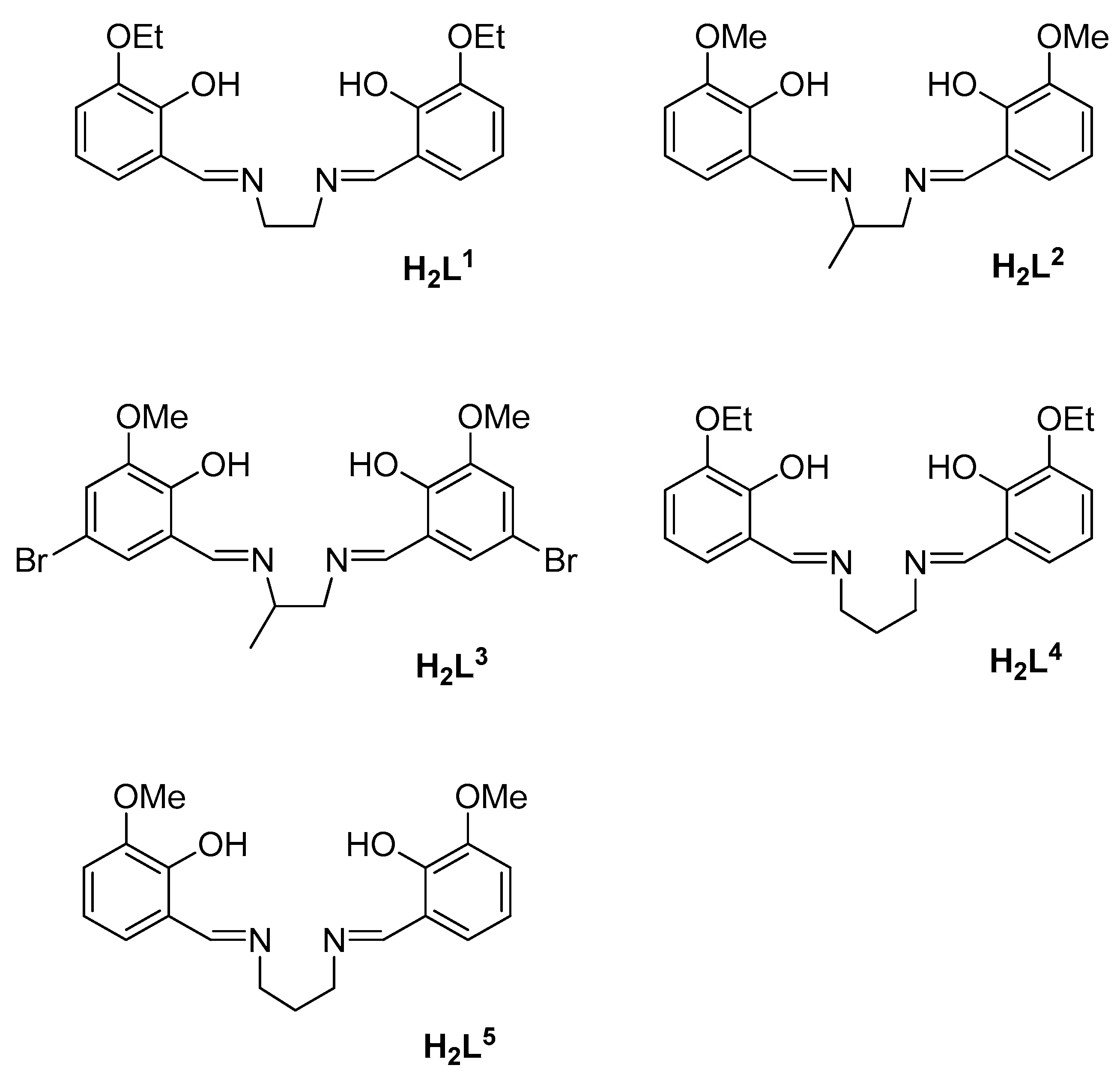
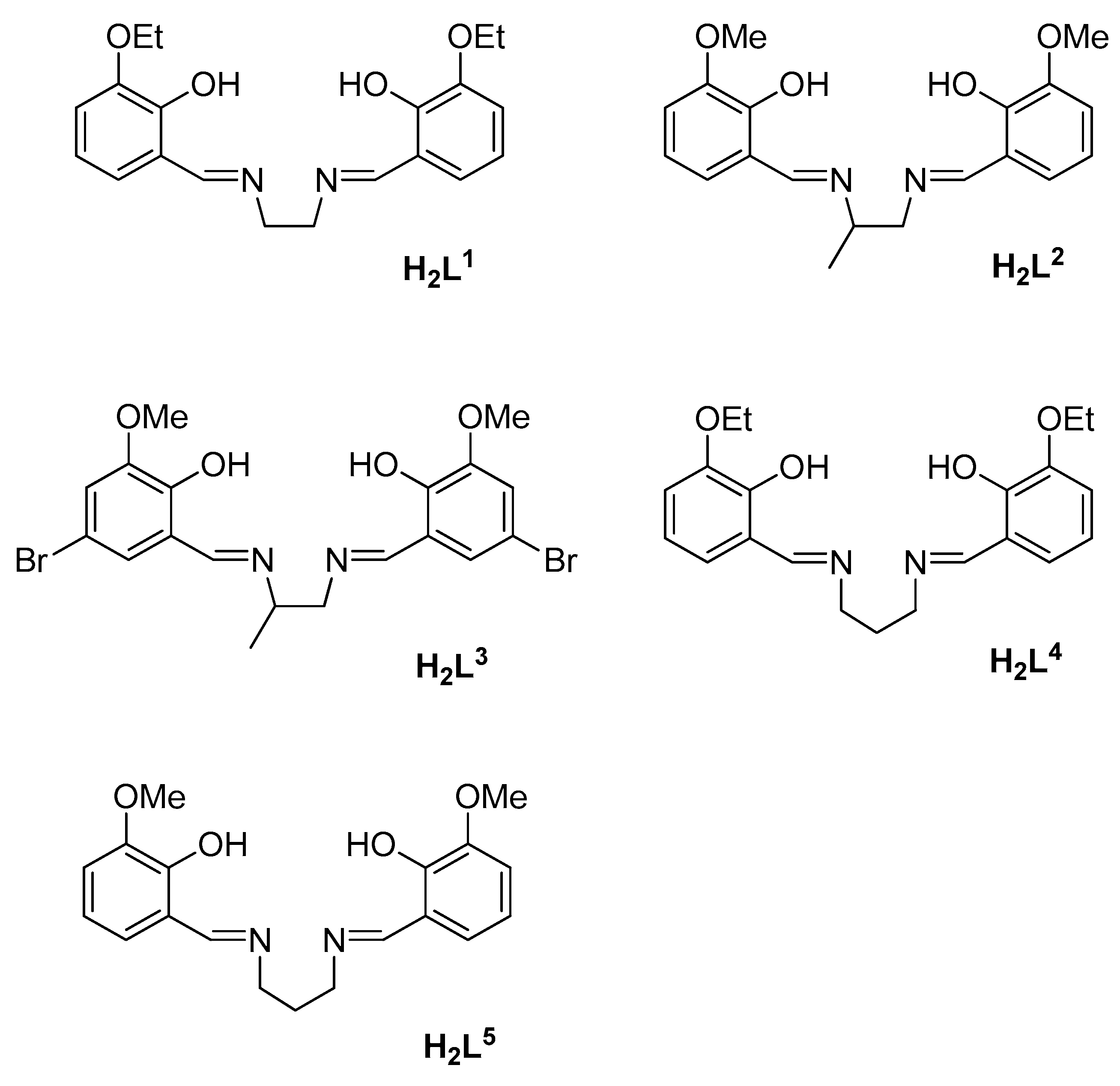
2.1. Synthesis of the Complexes
2.2. Peroxidase Probes
2.3. Electrochemical Oxidations of Veratryl Alcohol (VA)
3. Results
3.1. Synthesis and Characterization of the Complexes
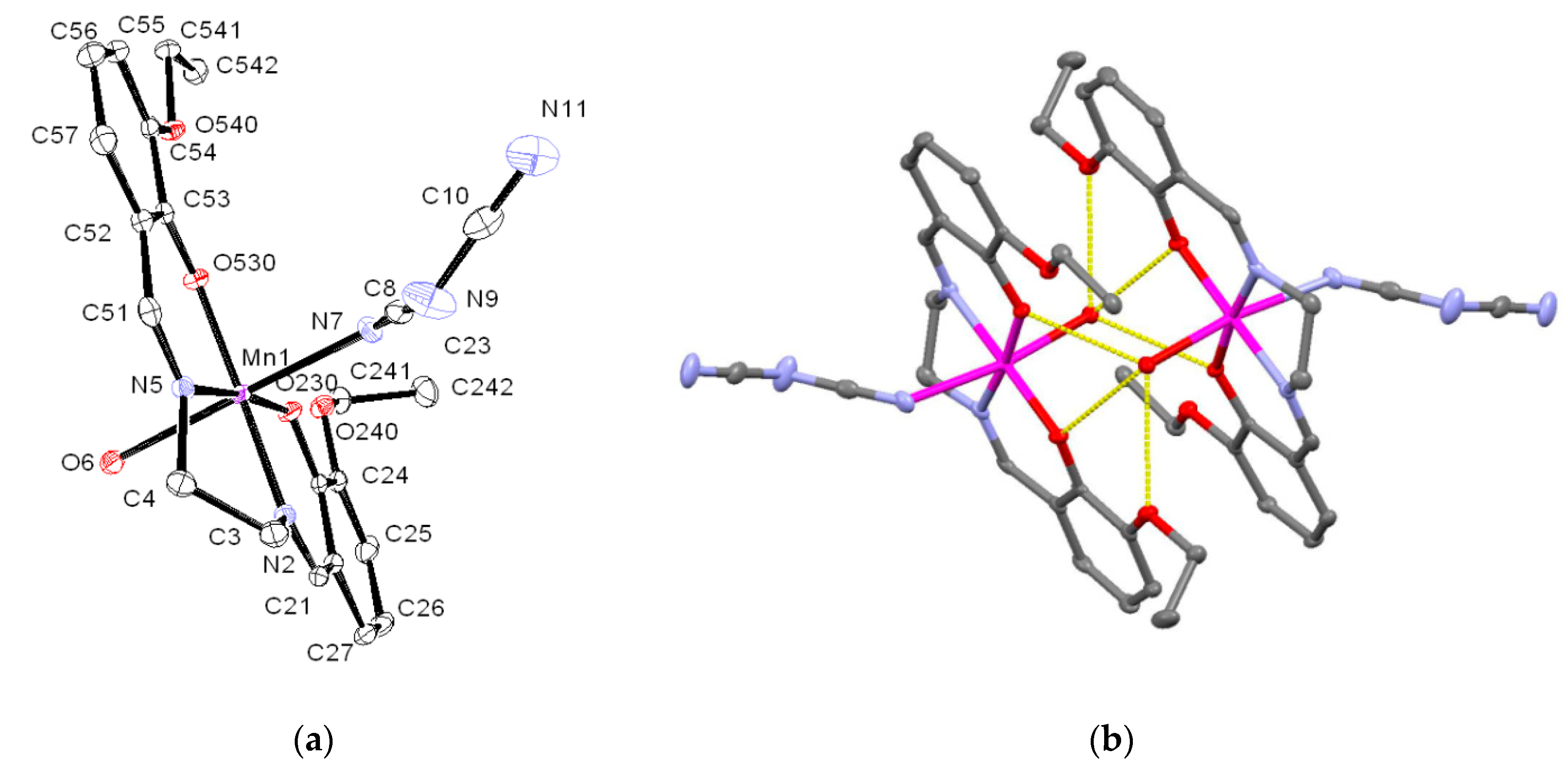
3.2. Crystal Structure of 1
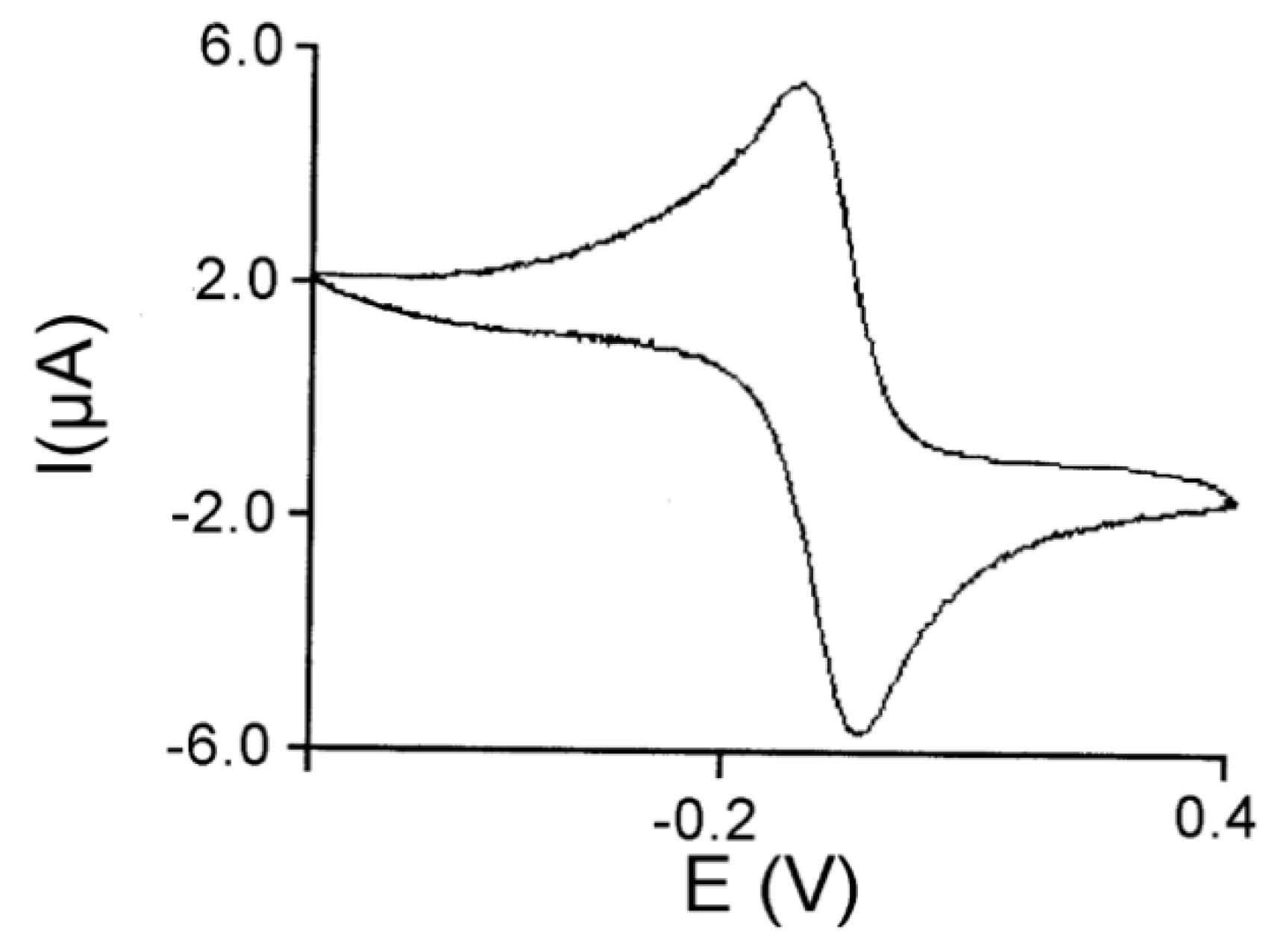
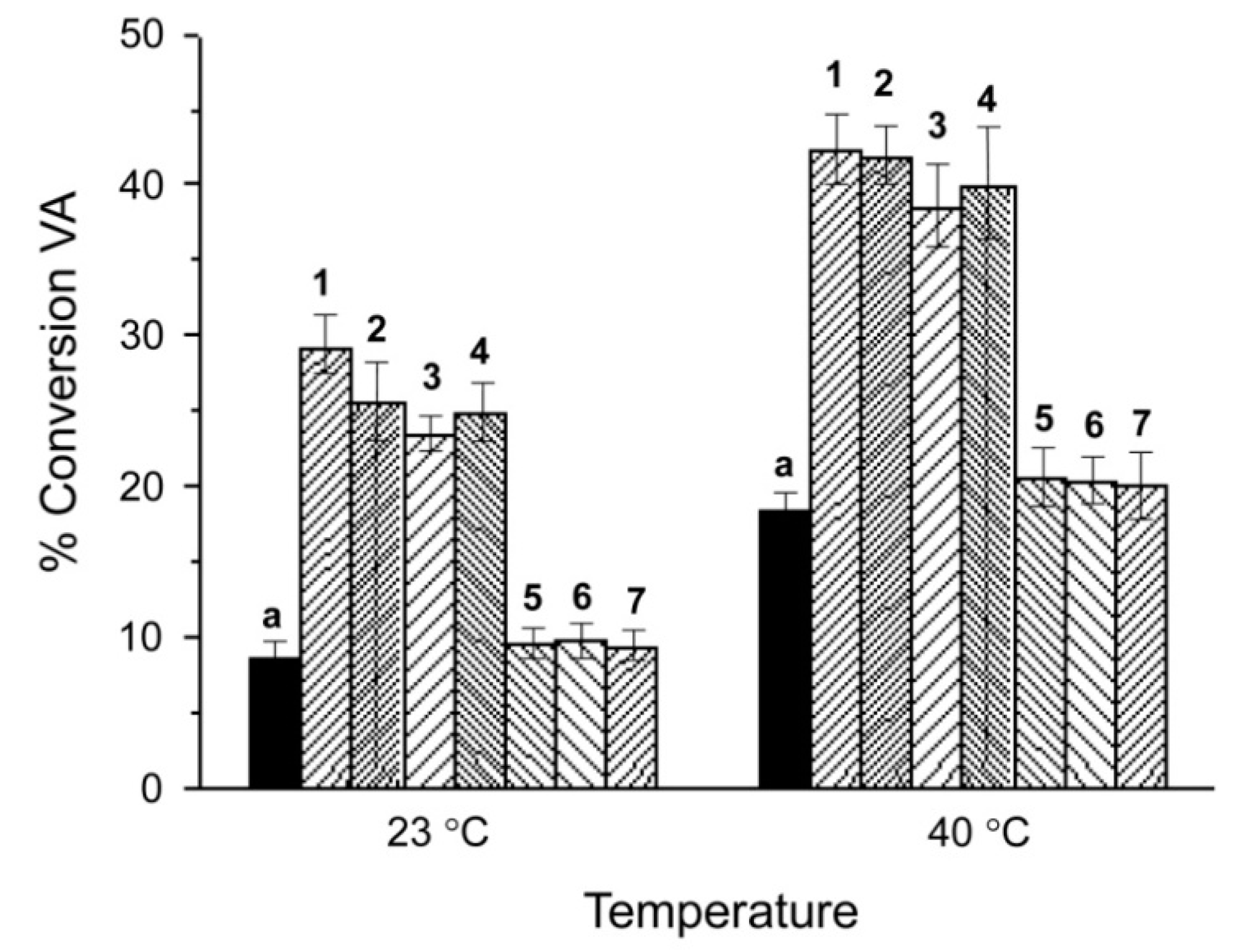
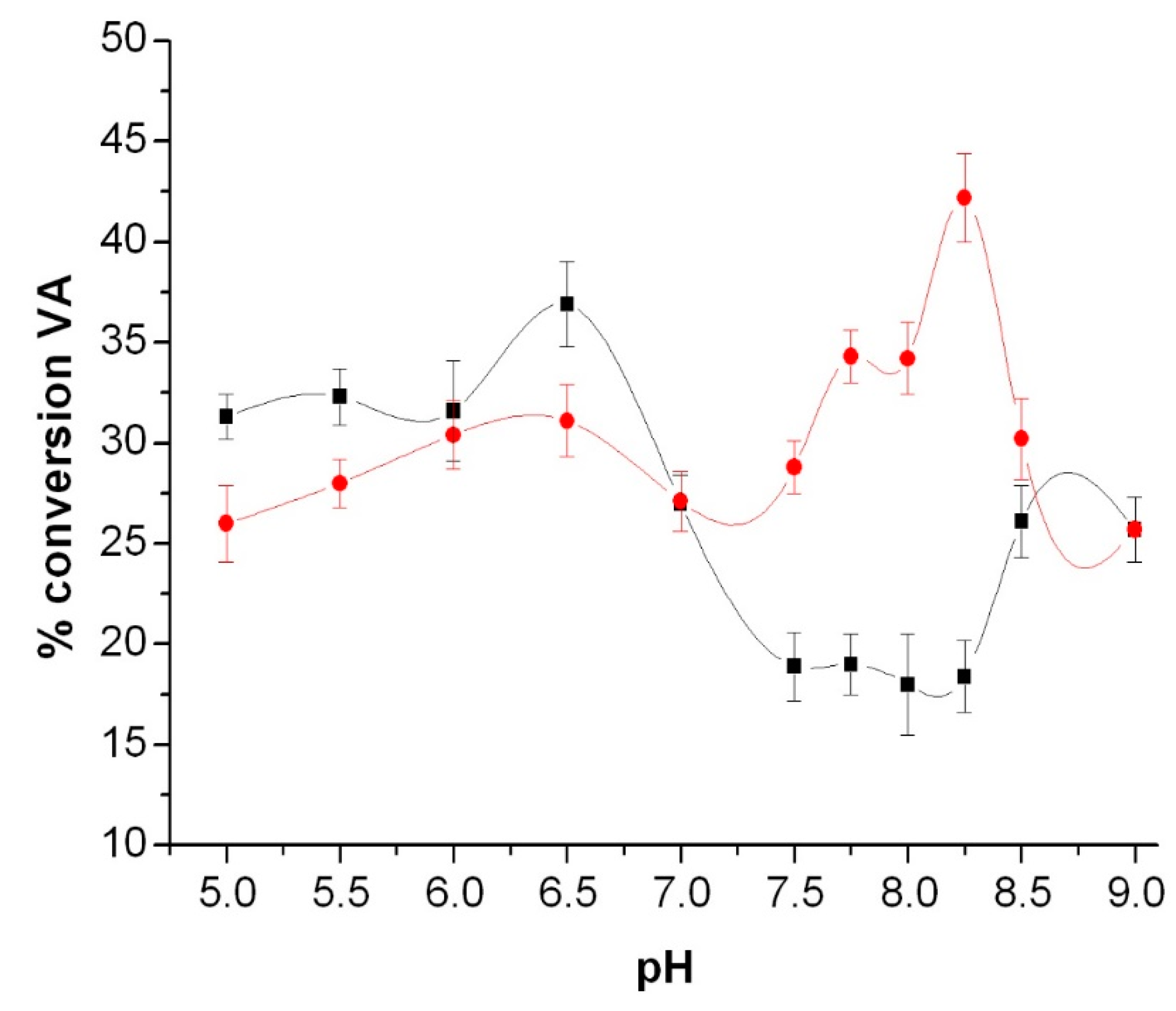
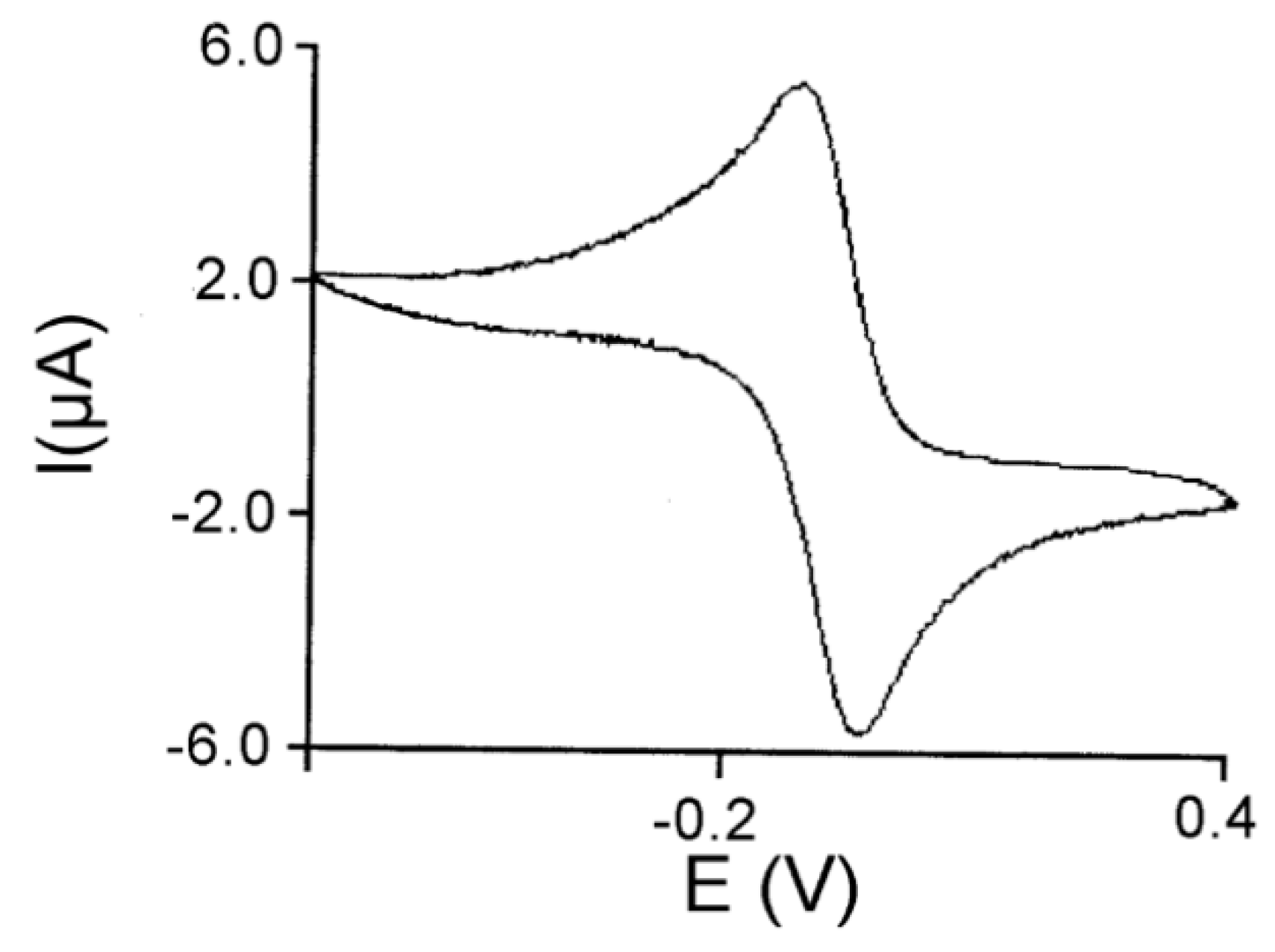
3.3. Peroxidase and Electrolytical Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Behling, R.; Valange, S.; Chatel, G. Heterogeneous catalytic oxidation for lignin valorization into valuable chemicals: What results? What limitations? What trends? Green Chem. 2016, 18, 1839–1854. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Matsuura, B.S.; Monos, T.M.; Magallanes, G.; Stephenson, C.R. Transition-metal catalyzed valorization of lignin: The key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. [Google Scholar] [CrossRef] [PubMed]
- Haritash, A.K.; Kaushik, C.P. Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): A review. J. Hazard. Mat. 2009, 169, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Isroi, I.; Millati, R.; Syamsiah, S.; Niklasson, C.; Cahyanto, M.N.; Ludquist, K.; Taherzadeh, M.J. Biological pretreatment of lignocelluloses with white-rot fungi and its applications: A review. BioResources 2011, 6, 5224–5259. [Google Scholar] [CrossRef]
- Pokhrel, D.; Viraraghavan, T. Treatment of pulp and paper mill wastewater—A review. Sci. Total Environ. 2004, 333, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.; Rieradevall, J.; Torrades, F.; Peral, J.; Domènech, X. Environmental assessment of different advanced oxidation processes applied to a bleaching Kraft mill effluent. Chemosphere 2006, 62, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Capanema, E.A.; Gracz, H.S. Reaction mechanisms in delignification of pine Kraft-AQ pulp with hydrogen peroxide using Mn (IV)-Me4DTNE as catalyst. J. Agric. Food Chem. 2003, 51, 1932–1941. [Google Scholar] [CrossRef]
- Rochefort, D.; Leech, D.; Bourbonnais, R. Electron transfer mediator systems for bleaching of paper pulp. Green Chem. 2004, 6, 14–24. [Google Scholar] [CrossRef]
- Hage, R.; Lienke, A. Applications of transition-metal catalysts to textile and wood-pulp bleaching. Angew. Chem. Int. Ed. 2006, 45, 206–222. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Cai, J.; Wang, Y.; Zhang, J.; Yu, W.; Xu, J. Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ. Sci. 2013, 6, 994–1007. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T. Knocking on wood: Base metal complexes as catalysts for selective oxidation of lignin models and extracts. Acc. Chem. Res. 2015, 48, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; De, S.; Yan, N. Rational control of nano-scale metal-catalysts for biomass conversion. Chem. Commun. 2016, 52, 6210–6224. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Bista, G.; Maharjan, R.; Poudyal, P.; Mainali, M.; Sreerama, L.; Joshi, J. Secretory laccase from Pestalotiopsis Species CDBT-F-G1 fungal strain isolated from high altitude: Optimization of its production and characterization. Appl. Sci. 2019, 9, 340. [Google Scholar] [CrossRef]
- Eckard, A.D.; Muthukumarappan, K.; Gibbons, W. A review of the role of amphiphiles in biomass to ethanol conversion. Appl. Sci. 2013, 3, 396–419. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Fernández, M.Á.; Bermejo, M.R.; Fernández-García, M.I.; González-Riopedre, G.; Rodríguez-Doutón, M.J.; Maneiro, M. Influence of the geometry around the manganese ion on the peroxidase and catalase activities of Mn (III)–Schiff base complexes. J. Inorg. Biochem. 2011, 105, 1538–1547. [Google Scholar] [CrossRef]
- González-Riopedre, G.; Fernández-García, M.I.; Gómez-Fórneas, E.; Maneiro, M. Biomimetic catalysts for oxidation of veratryl alcohol, a lignin model compound. Catalysts 2013, 3, 232–246. [Google Scholar] [CrossRef]
- González-Riopedre, G.; Bermejo, M.R.; Fernández-García, M.I.; González-Noya, A.M.; Pedrido, R.; Rodríguez-Doutón, M.J.; Maneiro, M. Alkali-metal-ion-directed self-assembly of redox-active manganese(III) supramolecular boxes. Inorg. Chem. 2015, 54, 2512–2521. [Google Scholar] [CrossRef]
- Rouco, L.; Fernández-García, M.; Pedrido, R.; Botana, L.; Esteban-Gómez, D.; Platas-Iglesias, C.; Maneiro, M. Modeling the OEC with two new biomimetic models: Preparations, structural characterization, and water photolysis studies of a Ba–Mn box type complex and a Mn4N6 planar-diamond cluster. Catalysts 2018, 8, 382. [Google Scholar] [CrossRef]
- Liu, W.; Groves, J.T. Manganese porphyrins catalyze selective C− H bond halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. [Google Scholar] [CrossRef] [PubMed]
- Palopoli, C.; Duhayon, C.; Tuchagues, J.P.; Signorella, S. Synthesis, characterization, and reactivity studies of a water-soluble bis (alkoxo)(carboxylato)-bridged diMn III complex modeling the active site in catalase. Dalton Trans. 2014, 43, 17145–17155. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2014, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.M.; Storr, T. The chemistry and applications of multimetallic salen complexes. Dalton Trans. 2014, 43, 9380–9391. [Google Scholar] [CrossRef]
- Collinson, S.R.; Thielemans, W. The catalytic oxidation of biomass to new materials focusing on starch, cellulose and lignin. Coord. Chem. Rev. 2010, 254, 1854–1870. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Fernández, M.Á.; Fernández-García, M.I.; González-Noya, A.M.; Maneiro, M.; Bermejo, M.R.; Rodríguez-Doutón, M.J. Supramolecular networks of Mn (III)–Schiff base complexes assembled by nitrate counterions: X-ray crystal structures of 1D chains and 2D networks. Polyhedron 2012, 31, 379–385. [Google Scholar] [CrossRef]
- Bermejo, M.R.; Carballido, R.; Fernández-García, M.I.; González-Noya, A.M.; González-Riopedre, G.; Maneiro, M.; Rodríguez-Silva, L. Synthesis, Characterization, and Catalytic Studies of Mn (III)-Schiff Base-Dicyanamide Complexes: Checking the Rhombicity Effect in Peroxidase Studies. J. Chem. 2017. [Google Scholar] [CrossRef]
- Liberato, A.; Fernández-Trujillo, M.J.; Máñez, A.; Maneiro, M.; Rodríguez-Silva, L.; Basallote, M.G. Pitfalls in the ABTS Peroxidase Activity Test: Interference of Photochemical. Proc. Inorg. Chem. 2018, 57, 14471–14475. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX-97 (Shelxs 97 and Shelxl 97), Programs for Crystal Structure Analyses; University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. MERCURY CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Majumder, A.; Pilet, G.; Garlan, M.T.; Mitra, S. Synthesis and structural characterisation of three dicyanamide complexes with Mn (II), Zn (II) and Cd (II): Supramolecular architectures stabilised by hydrogen bonding. Polyhedron 2006, 25, 2550–2558. [Google Scholar] [CrossRef]
- González-Riopedre, G.; Fernández-García, M.I.; González-Noya, A.M.; Vázquez-Fernández, M.A.; Bermejo, M.R.; Maneiro, M. Manganese-Schiff base complexes as catalysts for water photolysis. Phys. Chem. Chem. Phys. 2011, 13, 18069–18077. [Google Scholar] [CrossRef] [PubMed]
- Bonadies, J.A.; Maroney, M.L.; Pecoraro, V.L. Structurally diverse manganese (III) Schiff base complexes: Solution speciation via paramagnetic 1H NMR spectroscopy and electrochemistry. Inorg. Chem. 1989, 28, 2044–2051. [Google Scholar] [CrossRef]
- Hendrich, M.P.; Debrunner, P.G. Integer-spin electron-paramagnetic resonance of iron proteins. Biophys. J. 1989, 56, 489–506. [Google Scholar] [CrossRef]
- Talsi, E.P.; Bryliakov, K.P. X-band perpendicular-mode EPR spectra of’EPR-silent’manganese (III) porphyrins. Mendel. Commun. 2004, 14, 111–112. [Google Scholar] [CrossRef]
- Tyryshkin, A.M.; Watt, R.K.; Baranov, S.V.; Dasgupta, J.; Hendrich, M.P.; Dismukes, G.C. Spectroscopic evidence for Ca2+ involvement in the assembly of the Mn4Ca cluster in the photosynthetic water-oxidizing complex. Biochemistry 2006, 45, 12876–12889. [Google Scholar] [CrossRef]
- Childs, R.E.; Bardsley, W.G. The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem. J. 1975, 145, 93–103. [Google Scholar] [CrossRef]
- Liu, S.W.; Xu, N.H.; Tan, C.Y.; Fang, W.; Tan, Y.; Jiang, Y.Y. A sensitive colorimetric aptasensor based on trivalent peroxidase-mimic DNAzyme and magnetic nanoparticles. Anal. Chim. Acta 2018, 1018, 86–93. [Google Scholar] [CrossRef]
- Díaz-Rubio, L.; Hernández-Martínez, R.; Estolano-Cobián, A.; Chávez-Velasco, D.; Salazar-Aranda, R.; Waksman de Torres, N.; Rivero, I.A.; García-González, V.; Ramos, M.A.; Córdova-Guerrero, I. Synthesis, biological evaluation and docking studies of chalcone and flavone analogs as antioxidants and acetylcholinesterase inhibitors. Appl. Sci. 2019, 9, 410. [Google Scholar] [CrossRef]
- Rochefort, D.; Bourbonnais, R.; Leech, D.; Paice, M.G. Oxidation of lignin model compounds by organic and transition metal-based electron transfer mediators. Chem. Commun. 2002, 11, 1182–1183. [Google Scholar] [CrossRef]
- Wu, X.; Guo, S.; Zhang, J. Selective oxidation of veratryl alcohol with composites of Au nanoparticles and graphene quantum dots as catalysts. Chem. Commun. 2015, 51, 6318–6321. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Huang, X.; Song, S.; Wang, D.; Xuemei, L.; Qu, Y.; Gao, P. Kinetics of the H2O2-dependent ligninase-catalyzed oxidation of veratryl alcohol in the presence of cationic surfactant studied by spectrophotometric technique. Spectrochim. Acta Part A 2003, 59, 2547–2551. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Garedew, M.; Lam, C.H.; Jackson, J.E.; Miller, D.J.; Saffron, C.M. Mild electrocatalytic hydrogenation and hydrodeoxygenation of bio-oil derived phenolic compounds using ruthenium supported on activated carbon cloth. Green Chem. 2012, 14, 2540–2549. [Google Scholar] [CrossRef]
- Sippola, V.O.; Krause, A.O.I. Bis (o-phenanthroline) copper-catalysed oxidation of lignin model compounds for oxygen bleaching of pulp. Catal. Today 2005, 100, 237–242. [Google Scholar] [CrossRef]
- Van Doorslaer, S.; Caretti, I.; Fallis, I.A.; Murphy, D.M. The power of electron paramagnetic resonance to study asymmetric homogeneous catalysts based on transition-metal complexes. Coord. Chem. Rev. 2009, 253, 2116–2130. [Google Scholar] [CrossRef]
- Hine, F. Electrode Processes and Electrochemical Engineering; Plenum Press: New York, NY, USA, 1985. [Google Scholar]
- Mukimin, A.; Wijaya, K.; Kuncaka, A. Oxidation of remazol brilliant blue r (RB. 19) with in situ electro-generated active chlorine using Ti/PbO2 electrode. Sep. Purif. Technol. 2012, 95, 1–9. [Google Scholar] [CrossRef]
- Chen, Z.; Concepcion, J.J.; Song, N.; Meyer, T.J. Chloride-assisted catalytic water oxidation. Chem. Commun. 2014, 50, 8053–8056. [Google Scholar] [CrossRef]
- Szpyrkowicz, L.; Juzzolino, C.; Kaul, S.N.; Daniele, S.; de Faveri, M.D. Electrochemical oxidation of dyeing baths bearing disperse dyes. Indus. Eng. Chem. Res. 2000, 39, 3241–3248. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TON a | mmol VA Converted to VAH | Amount (mg) of VA Oxidized | Ef b/mol F−1 |
|---|---|---|---|---|
| 1 | 84 | 5.70 × 10−2 | 9.580 | 0.31 |
| 2 | 84 | 5.64 × 10−2 | 9.485 | 0.30 |
| 3 | 68 | 4.57 × 10−2 | 7.686 | 0.25 |
| 4 | 80 | 5.39 × 10−2 | 9.065 | 0.29 |
| 5 | 40 | 2.73 × 10−2 | 4.592 | 0.15 |
| 6 | 40 | 2.73 × 10−2 | 4.591 | 0.15 |
| 7 | 40 | 2.67 × 10−2 | 4.491 | 0.14 |
| Empirical formula | C22 H24 Mn N5 O5 |
| Formula weight | 493.40 |
| Temperature [K] | 100 (2) |
| Wavelength [Å] | 0.71069 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a [Å] | 13.0012(3) |
| b [Å] | 13.1768(4) |
| c [Å] | 13.7748(4) |
| α [°] | 90 |
| β [°] | 110.325(2) |
| γ [°] | 90 |
| Volume [Å3] | 2212.89(11) |
| Z | 4 |
| Density (calculated) [g cm−3] | 1.481 |
| Absorption coefficient [mm−1] | 0.641 |
| F(000) | 1024 |
| Theta range for data collection [°] | 1.67 to 27.87 |
| Reflections collected | 34,661 |
| Independent reflections | 5273 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0299; wR2 = 0.0689 |
| R indices (all data) | R1 = 0.0410; wR2 = 0.0722 |
| Compound | VA to VAH a | Peroxidase Activity b | Mn-Oaxial c | A||d | Eox (V) | Ered (V) |
|---|---|---|---|---|---|---|
| 1 | 42.2 ± 4 | 65 ± 5 | 2.2495(11) | 43 | −0.085 | −0.165 |
| 2 | 41.8 ± 3 | 35 ± 1 | 2.257(2) | 42 | −0.010 | −0.098 |
| 3 | 38.5 ± 5 | 54 ± 5 | un. | 43 | −0.034 | −0.114 |
| 4 | 39.9 ± 6 | 50 ± 3 | 2.270(6) | 42 | −0.154 | −0.237 |
| 5 | 20.2 ± 3 | 2.2 ± 0.4 | un. | 49 | −0.116 | −0.256 |
| 6 | 20.1 ± 2 | 2.4 ± 0.3 | un. | 49 | un. | un. |
| 7 | 19.8 ± 3 | 1.9 ± 0.4 | un. | un. | −0.034 | −0.144 |
| Mn1-O230 | 1.8837(10) | Mn1-O530 | 1.8895(10) |
| Mn1-N2 | 1.9834(12) | Mn1-N5 | 1.9828(13) |
| Mn1-O6 | 2.2495(11) | Mn1-N7 | 2.3037(13) |
| C4-N5 | 1.473(19) | N2-C3 | 1.474(19) |
| N5-C51 | 1.290(19) | N2-C21 | 1.287(2) |
| N7-C8 | 1.162(2) | C8-N9 | 1.301(2) |
| N9-C10 | 1.305(2) | C10-N11 | 1.154(2) |
| O230-Mn1-O530 | 94.19(4) | N2-Mn1-N5 | 82.26(5) |
| O230-Mn1-O6 | 93.45(5) | N5-Mn1-N7 | 85.62(5) |
| O230-Mn1-N2 | 92.11(5) | N7-Mn1-O6 | 168.90(5) |
| O230-Mn1-N5 | 173.87(5) | Mn1-N2-C3 | 113.35(9) |
| O230-Mn1-N7 | 96.56(5) | Mn1-N2-C21 | 125.68(11) |
| O530-Mn1-O6 | 92.79(5) | Mn1-N5-C4 | 113.15(9) |
| O530-Mn1-N5 | 91.48(5) | Mn1-N5-C51 | 126.00(10) |
| O530-Mn1-N7 | 91.25(5) | N2-Mn1-N7 | 87.15(5) |
| O530-Mn1-N2 | 173.64(5) | N2-Mn1-N5 | 82.26(5) |
| N2-Mn1-O6 | 87.70(5) | C21-N2-C3 | 120.97(13) |
| N5-Mn1-O6 | 83.95(5) | C51-N5-C4 | 120.84(13) |
| O230-Mn1-O530 | 94.19(4) |
| D-H…A | d(D-H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| O6–H6A..O530 [a] | 0.79(4) | 2.18(3) | 2.8509(17) | 143(2) |
| O6–H6A..O540 [a] | 0.79(4) | 2.37(2) | 3.0630(16) | 148(3)’ |
| O6–H6B..O230 [a] | 0.75(2) | 2.30(2) | 2.9300(16) | 142(2) |
| O6–H6B..O240 [a] | 0.75(2) | 2.35(2) | 3.0169(16) | 149(2)´ |
| C4–H4A..N11 [b] | 0.97 | 2.58 | 3.247(2) | 126 |
| C51–H51..N11 [c] | 0.934(19) | 2.337(19) | 3.229(2) | 159,5(15) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rouco, L.; Fernández-García, M.I.; González-Noya, A.M.; González-Riopedre, G.; Tyryshkin, A.M.; Maneiro, M. Electrochemical Conversion of the Lignin Model Veratryl Alcohol to Veratryl Aldehyde Using Manganese(III)-Schiff Base Homogeneous Catalysts. Appl. Sci. 2019, 9, 3430. https://doi.org/10.3390/app9163430
Rouco L, Fernández-García MI, González-Noya AM, González-Riopedre G, Tyryshkin AM, Maneiro M. Electrochemical Conversion of the Lignin Model Veratryl Alcohol to Veratryl Aldehyde Using Manganese(III)-Schiff Base Homogeneous Catalysts. Applied Sciences. 2019; 9(16):3430. https://doi.org/10.3390/app9163430
Chicago/Turabian StyleRouco, Lara, M. Isabel Fernández-García, Ana M. González-Noya, Gustavo González-Riopedre, Alexei M. Tyryshkin, and Marcelino Maneiro. 2019. "Electrochemical Conversion of the Lignin Model Veratryl Alcohol to Veratryl Aldehyde Using Manganese(III)-Schiff Base Homogeneous Catalysts" Applied Sciences 9, no. 16: 3430. https://doi.org/10.3390/app9163430
APA StyleRouco, L., Fernández-García, M. I., González-Noya, A. M., González-Riopedre, G., Tyryshkin, A. M., & Maneiro, M. (2019). Electrochemical Conversion of the Lignin Model Veratryl Alcohol to Veratryl Aldehyde Using Manganese(III)-Schiff Base Homogeneous Catalysts. Applied Sciences, 9(16), 3430. https://doi.org/10.3390/app9163430