
TREM2 Regulates High Glucose-Induced Microglial Inflammation via the NLRP3 Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot Analysis
2.3. qRT-PCR
2.4. Generation of CRISPR/Cas9-Mediated Knockout (KO) Cell Line
2.5. Generation of TREM2-Overexpressing (OE) Cell Line
2.6. ELISA
2.7. GST Pull-Down Assay
2.8. Coimmunoprecipitation
2.9. Statistical Analysis
3. Results
3.1. High Glucose Enhances the Expression of TREM2 and the IL-1β Proinflammatory Cytokine
3.2. High Glucose Promotes the Interaction of TREM2 with NLRP3
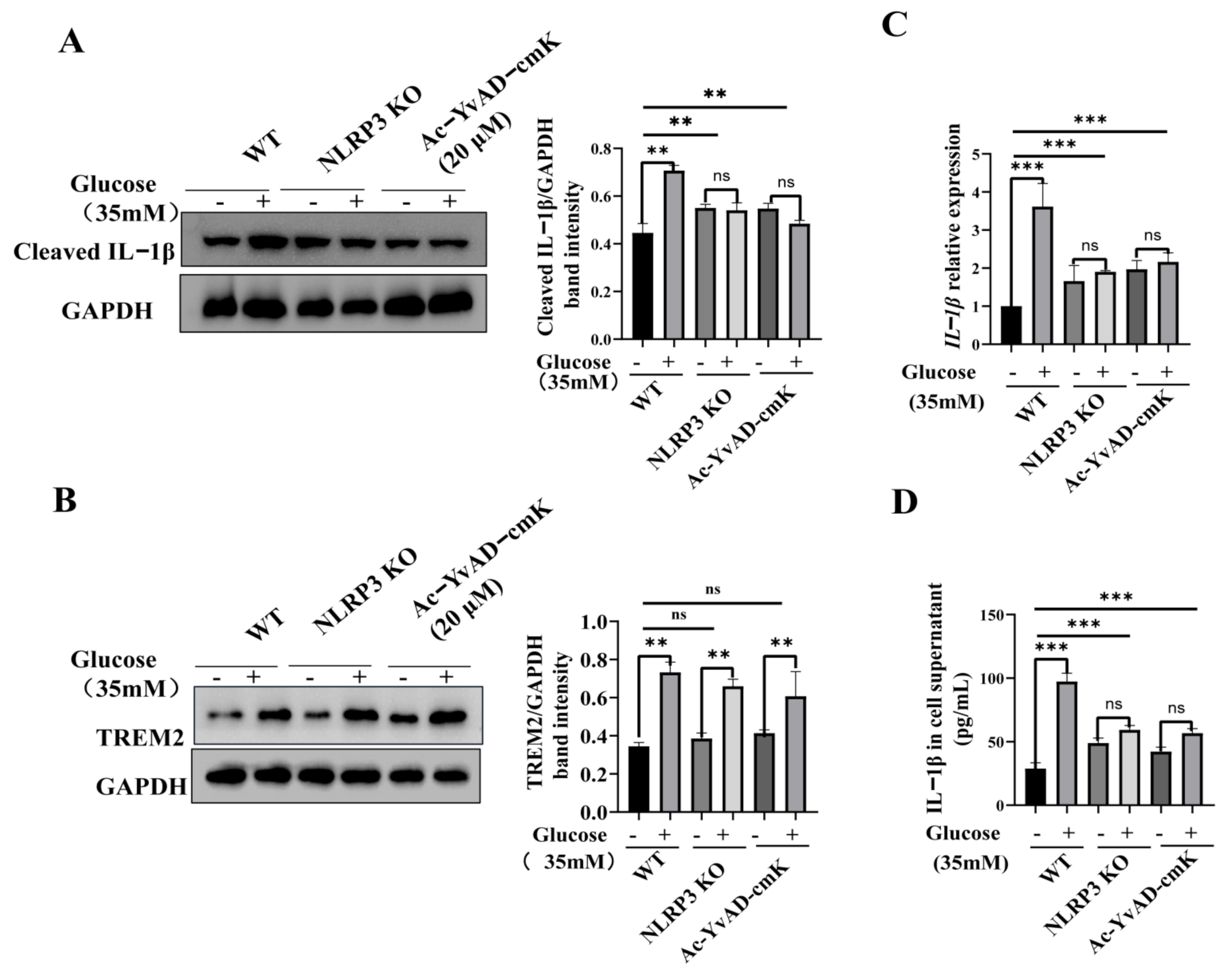
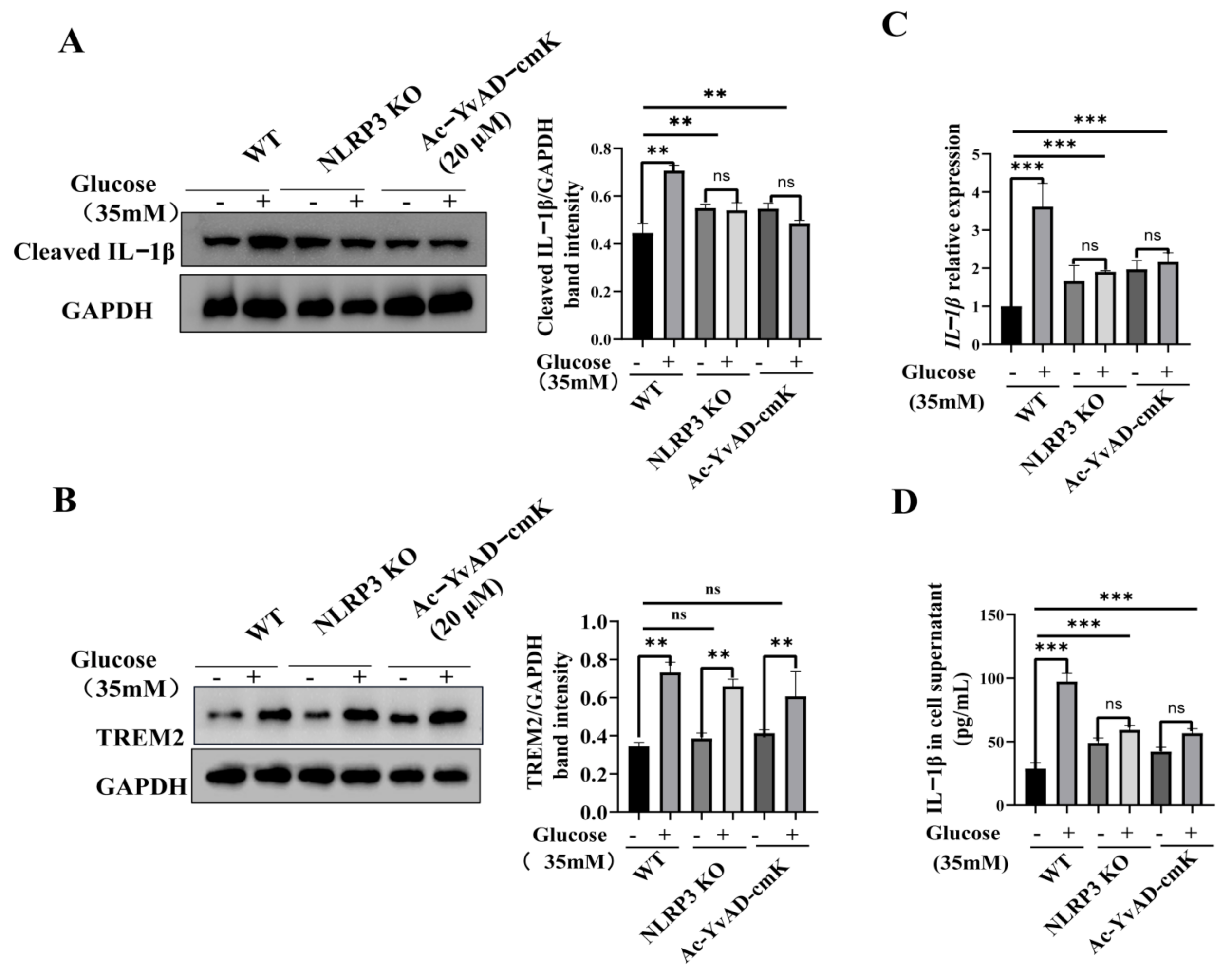
3.3. TREM2-Regulated Microglial Inflammation Is Mediated by the NLRP3 Inflammasome Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TREM2 | triggering receptor expressed on myeloid cells-2; |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor protein 3; |
| qPCR | real-time quantitative polymerase chain reaction; |
| ELISA | enzyme-linked immunosorbent assay; |
| Co-IP | co-Immunoprecipitation; |
| IL-1β | interlukin-1β; |
| KO | knockout; |
| OE | overexpression; |
| ROS | reactive oxygen species; |
| DAP12 | DNAX activation protein 12; |
| NHD | Nasu-Hakola disease; |
| TLR2 | TLR4, toll like receptor2,4; |
| LPS | lipopolysaccharide; |
| PLOSL | Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy; |
| NEB | New England Biolabs; |
References
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-W.; Zhang, J.; Li, X.; Wang, Y.; Fu, Y.-H.; Gao, X.-Y. A new research hot spot: The role of NLRP3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci. 2020, 240, 117138. [Google Scholar] [CrossRef] [PubMed]
- Diedisheim, M.; Carcarino, E.; Vandiedonck, C.; Roussel, R.; Gautier, J.-F.; Venteclef, N. Regulation of inflammation in diabetes: From genetics to epigenomics evidence. Mol. Metab. 2020, 41, 101041. [Google Scholar] [CrossRef]
- Bing, C. Is interleukin-1beta a culprit in macrophage-adipocyte crosstalk in obesity? Adipocyte 2015, 4, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, S.; Nie, K.; Li, Y.; Gao, Y.; Gan, R. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2018, 499, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xu, W.; Cheng, H.; Yuan, H.; Tan, X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav. Immun. 2019, 81, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Li, F.; Chen, X.; Jia, J.; Sun, S.; Zhou, D.; Ma, L.; Jiang, T.; Bai, F.; Xiong, L.; et al. Triggering Receptor Expressed on Myeloid Cells 2, a Novel Regulator of Immunocyte Phenotypes, Confers Neuroprotection by Relieving Neuroinflammation. Anesthesiology 2017, 127, 98–110. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Luo, Y.; Du, Y.; Zhang, X.; Fu, J. Curcumin inhibits LPS-induced neuroinflammation by promoting microglial M2 polarization via TREM2/TLR4/NF-kappaB pathways in BV2 cells. Mol. Immunol. 2019, 116, 29–37. [Google Scholar] [CrossRef]
- He, G.-L.; Luo, Z.; Shen, T.-T.; Wang, Z.-Z.; Li, P.; Luo, X.; Yang, J.; Tan, Y.-L.; Wang, Y.; Gao, P.; et al. TREM2 Regulates Heat Acclimation-Induced Microglial M2 Polarization Involving the PI3K-Akt Pathway Following EMF Exposure. Front. Cell. Neurosci. 2020, 13, 591. [Google Scholar] [CrossRef] [Green Version]
- Minami, Y.; Sonoda, N.; Hayashida, E.; Makimura, H.; Ide, M.; Ikeda, N. p66Shc Signaling Mediates Diabetes-Related Cogni-tive Decline. Sci. Rep. 2018, 8, 3213. [Google Scholar] [CrossRef]
- Klein, J.P.; Hains, B.C.; Craner, M.J.; Black, J.A.; Waxman, S.G. Apoptosis of vasopressinergic hypothalamic neurons in chronic dia-betes mellitus. Neurobiol. Dis. 2004, 15, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Musen, G.; Lyoo, I.K.; Sparks, C.R.; Weinger, K.; Hwang, J.; Ryan, C.M.; Jimerson, D.C.; Hennen, J.; Renshaw, P.F.; Jacobson, A.M. Effects of Type 1 Diabetes on Gray Matter Density as Measured by Voxel-Based Morphometry. Diabetes 2006, 55, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Yang, J.-M.; Wang, J.-Y.; Tao, P.-L.; Yang, S.N. Synergistic apoptosis induced by bacterial endotoxin lipopolysaccharide and high glucose in rat microglia. Neurosci. Lett. 2001, 304, 177–180. [Google Scholar] [CrossRef]
- Quan, Y.; Jiang, C.T.; Xue, B.; Zhu, S.G.; Wang, X. High glucose stimulates TNFalpha and MCP-1 expression in rat microglia via ROS and NF-kappaB pathways. Acta Pharmacol. Sin. 2011, 32, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S. Minocycline Reduces Proinflammatory Cytokine Expression, Microglial Activation, and Caspase-3 Activation in a Rodent Model of Diabetic Retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B. The Microglial Innate Immune Receptor TREM2 Is Re-quired for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H. Lipid-Associated Macrophages Control Metabolic Ho-meostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L. TREM2 Maintains Microglial Metabolic Fit-ness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Gritsenko, A.; Green, J.P.; Brough, D.; Lopez-Castejon, G. Mechanisms of NLRP3 priming in inflammaging and age related dis-eases. Cytokine Growth Factor Rev. 2020, 55, 15–25. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Xu, H.; Wang, C.; An, Y.; Luan, W.; Wang, X.; Li, S.; Ma, F.; Ni, L.; et al. Cordycepin Modulates Body Weight by Reducing Prolactin Via an Adenosine A1 Receptor. Curr. Pharm. Des. 2018, 24, 3240–3249. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Karstoft, K.; Pedersen, B.K. Exercise and type 2 diabetes: Focus on metabolism and inflammation. Immunol. Cell Biol. 2015, 94, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mathew, V.; Farkouh, M.E. Targeting Inflammation in the Prevention and Treatment of Type 2 Diabetes: Insights From CANTOS. J. Am. Coll. Cardiol. 2018, 71, 2402–2404. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Du, J.; Wang, X. High glucose stimulates GRO secretion from rat microglia via ROS, PKC, and NF-κB pathways. J. Neurosci. Res. 2007, 85, 3150–3159. [Google Scholar] [CrossRef]
- Wang, L.Q.; Zhou, H.J. LncRNA MALAT1 promotes high glucose-induced inflammatory response of microglial cells via pro-voking MyD88/IRAK1/TRAF6 signaling. Sci. Rep. 2018, 8, 8346. [Google Scholar] [CrossRef] [PubMed]
- Baptista, F.I.; Aveleira, C.A.; Castilho, A.F.; Ambrosio, A.F. Elevated Glucose and Interleukin-1beta Differentially Affect Retinal Microglial Cell Proliferation. Mediat. Inflamm. 2017, 2017, 4316316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef]
- Wang, M.; Gao, X.; Zhao, K.; Chen, H.; Xu, M.; Wang, K. Effect of TREM2 on Release of Inflammatory Factor from LPS-stimulated Microglia and Its Possible Mechanism. Ann. Clin. Lab. Sci. 2019, 49, 249–256. [Google Scholar]
- Correale, C.; Genua, M.; Vetrano, S.; Mazzini, E.; Martinoli, C.; Spinelli, A.; Arena, V.; Peyrin-Biroulet, L.; Caprioli, F.; Passini, N.; et al. Bacterial Sensor Triggering Receptor Expressed on Myeloid Cells-2 Regulates the Mucosal Inflammatory Response. Gastroenterology 2013, 144, 346–356.e3. [Google Scholar] [CrossRef]
- Sharif, O.; Gawish, R.; Warszawska, J.M.; Martins, R.; Lakovits, K.; Hladik, A. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumo-nia. PLoS Pathog. 2014, 10, e1004167. [Google Scholar] [CrossRef]
- Li, C.; Zhao, B.; Lin, C.; Gong, Z.; An, X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-kappaB signaling. Cell Biol. Int. 2019, 43, 360–372. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, X.; Dong, W.; Wang, X.; He, S.; Zhang, H. TREM2 suppresses the proinflammatory response to facilitate PRRSV infection via PI3K/NF-kappaB signaling. PLoS Pathog. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Long, H.; Zhong, G.; Wang, C.; Zhang, J.; Zhang, Y.; Luo, J. TREM2 Attenuates Abeta1-42-Mediated Neuroinflammation in BV-2 Cells by Downregulating TLR Signaling. Neurochem. Res. 2019, 44, 1830–1839. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, W.; Zhang, M.; Tian, X.; Li, Y.; Lu, Y. Imbalance of Microglial TLR4/TREM2 in LPS-Treated APP/PS1 Transgenic Mice: A Potential Link between Alzheimer’s Disease and Systemic Inflammation. Neurochem. Res. 2019, 44, 1138–1151. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Y.; Huang, Y.; Liu, W.; Cai, G.; Huang, S. Resveratrol mediates mechanical allodynia through modulating in-flammatory response via the TREM2-autophagy axis in SNI rat model. J. Neuroinflammation. 2020, 17, 311. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Y.D.; Chen, Q.; Gao, Q.; Zhu, X.C.; Zhou, J.S. TREM2 modifies microglial phenotype and provides neuropro-tection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomol. 2019, 9, 850. [Google Scholar] [CrossRef] [Green Version]
- Qu, W.; Wang, Y.; Wu, Y.; Liu, Y.; Chen, K.; Liu, X. Triggering Receptors Expressed on Myeloid Cells 2 Promotes Corneal Re-sistance Against Pseudomonas aeruginosa by Inhibiting Caspase-1-Dependent Pyroptosis. Front. Immunol. 2018, 9, 1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S. NLRP3 inflammasome inhibition with MCC950 improves diabe-tes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef]
- Chen, W.; Guo, C.; Huang, S.; Jia, Z.; Wang, J.; Zhong, J.; Ge, H.; Yuan, J.; Chen, T.; Liu, X.; et al. MitoQ attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the NLRP3 inflammasome after ICH. Pharmacol. Res. 2020, 161, 105122. [Google Scholar] [CrossRef]
- Ma, D.C.; Zhang, N.N.; Zhang, Y.N.; Chen, H.S. Salvianolic Acids for Injection alleviates cerebral ischemia/reperfusion injury by switching M1/M2 phenotypes and inhibiting NLRP3 inflammasome/pyroptosis axis in microglia in vivo and in vitro. J. Eth-nopharmacol. 2021, 270, 113776. [Google Scholar] [CrossRef]
- Su, X.-Q.; Wang, X.-Y.; Gong, F.-T.; Feng, M.; Bai, J.-J.; Zhang, R.-R.; Dang, X.-Q. Oral treatment with glycyrrhizin inhibits NLRP3 inflammasome activation and promotes microglial M2 polarization after traumatic spinal cord injury. Brain Res. Bull. 2020, 158, 1–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Long, W.; Gao, M.; Jiao, F.; Chen, Z.; Liu, M.; Yu, L. TREM2 Regulates High Glucose-Induced Microglial Inflammation via the NLRP3 Signaling Pathway. Brain Sci. 2021, 11, 896. https://doi.org/10.3390/brainsci11070896
Li Y, Long W, Gao M, Jiao F, Chen Z, Liu M, Yu L. TREM2 Regulates High Glucose-Induced Microglial Inflammation via the NLRP3 Signaling Pathway. Brain Sciences. 2021; 11(7):896. https://doi.org/10.3390/brainsci11070896
Chicago/Turabian StyleLi, Yuan, Weihong Long, Menghan Gao, Fangtai Jiao, Zecai Chen, Mingyuan Liu, and Lu Yu. 2021. "TREM2 Regulates High Glucose-Induced Microglial Inflammation via the NLRP3 Signaling Pathway" Brain Sciences 11, no. 7: 896. https://doi.org/10.3390/brainsci11070896