
Protein Kinase C (PKC) in Neurological Health: Implications for Alzheimer’s Disease and Chronic Alcohol Consumption

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PKC and Brain
3. PKC and AD
4. PKC and Neuroinflammation
5. Dual Role of PKC in Neuroprotection and Neurodegeneration
5.1. Restoring Ion Channel Function and Reduced Neuronal Excitability
5.2. Maintaining Calcium Homeostasis and Promoting Neuronal Survival
5.3. PKC Isoforms That Have Been Implicated in AD Pathology
5.4. PKC in Regulating Mitochondrial Function and Dynamics in AD
5.5. Involvement of PKC Signalling Pathways in Synaptic Dysfunction
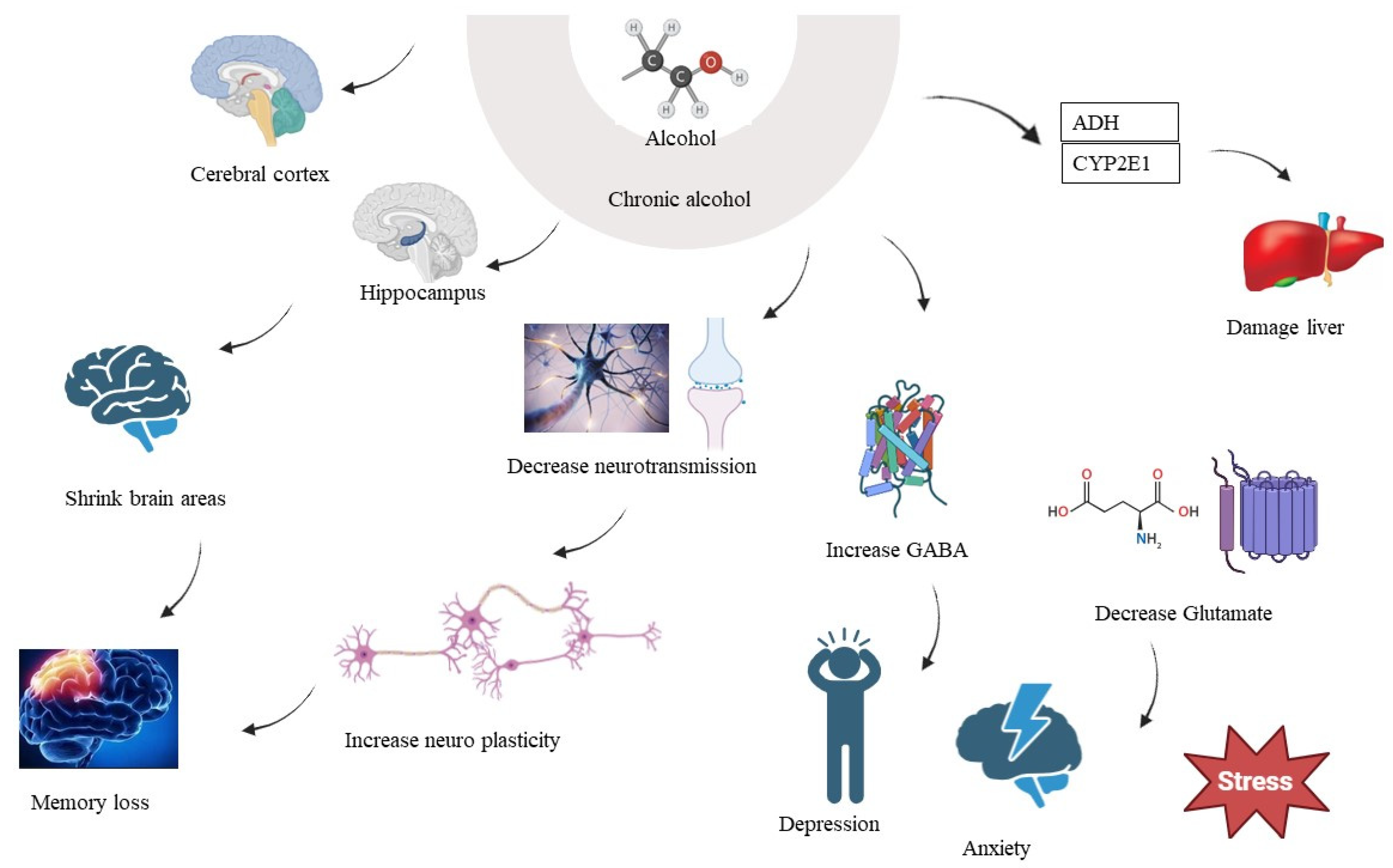
6. Chronic Alcohol Consumption and Brain
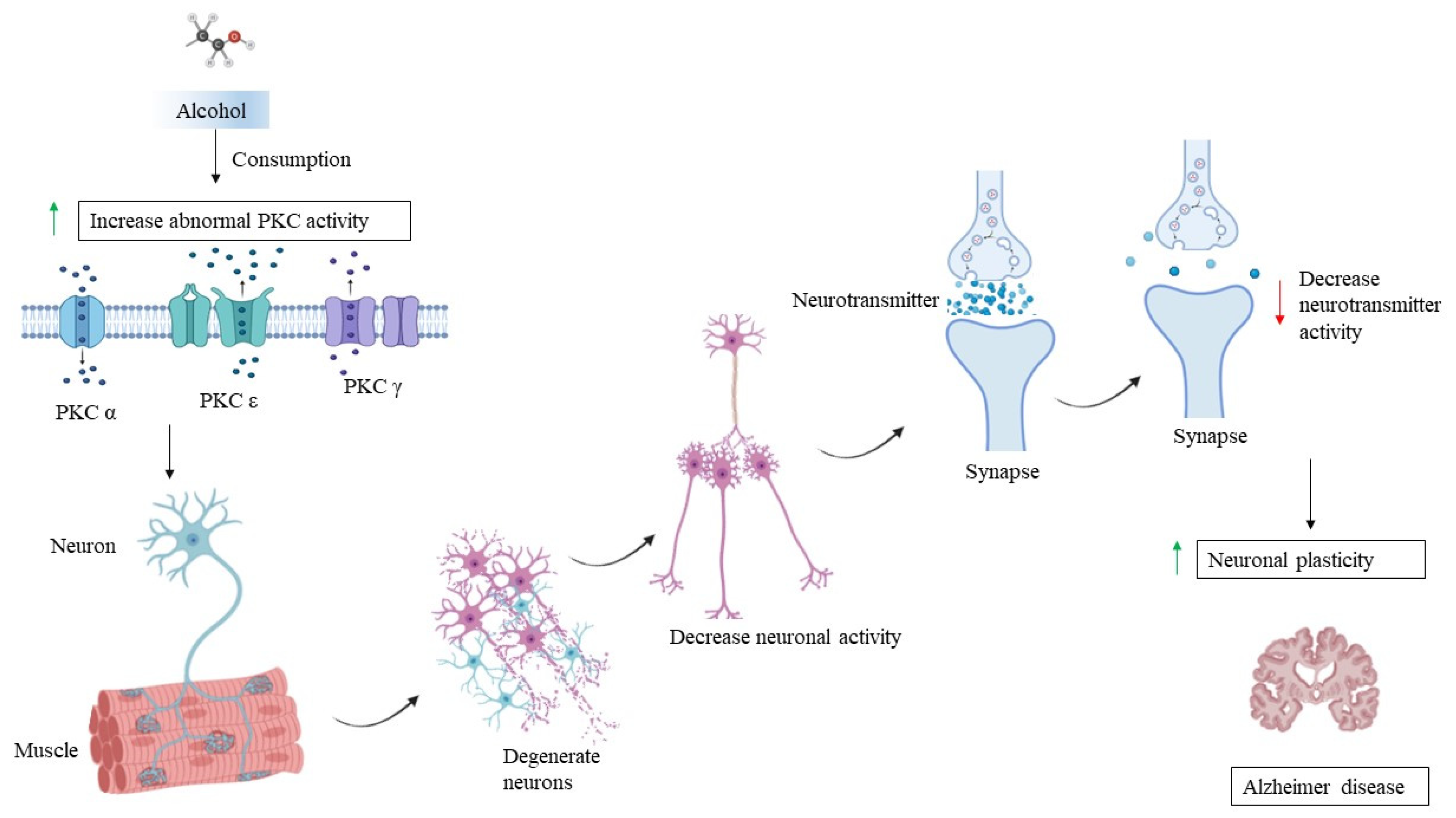
7. PKC and Chronic Alcohol Consumption
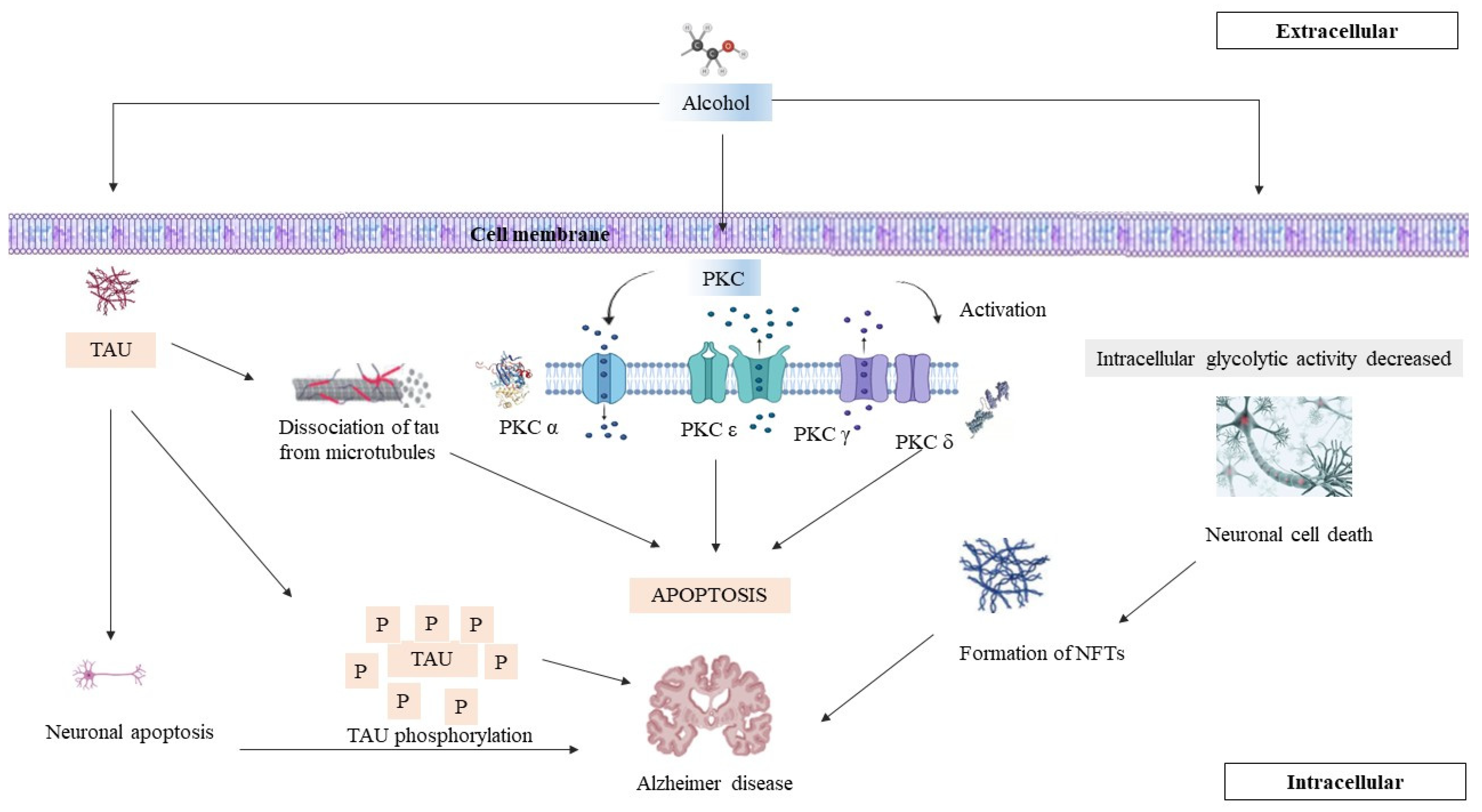
8. PKC, AD, and Chronic Alcohol Consumption
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolpaw, J.R.; Birbaumer, N.; Heetderks, W.J.; McFarland, D.J.; Peckham, P.H.; Schalk, G.; Donchin, E.; Quatrano, L.A.; Robinson, C.J.; Vaughan, T.M.; et al. Brain-computer interface technology: A review of the first international meeting. IEEE Trans. Rehabil. Eng. 2000, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, Y.; Koide, H.; Ogita, K.; Nishizuka, Y. The protein kinase C family for the regulation of cellular functions. Annu. Rev. Biomed. 1992, 1, 1–6. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Buoso, E.; Galbiati, V.; Racchi, M. Role of Protein Kinase C in immune cell activation and its implication chemical-induced immunotoxicity. In Protein Kinase-Mediated Decisions between Life and Death; Springer: Cham, Switzerland, 2021; pp. 151–163. [Google Scholar]
- Tyagi, K.; Roy, A. Evaluating the current status of protein kinase C (PKC)-protein kinase D (PKD) signaling axis as a novel therapeutic target in ovarian cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188496. [Google Scholar]
- Hashimoto, K.; Mikawa, K.; Kuroda, T.; Ase, K.; Kishimoto, A. Calpains and regulation of protein kinase C. In Intracellular Calcium-Dependent Proteolysis; CRC Press: Boca Raton, FL, USA, 2019; pp. 181–190. [Google Scholar]
- Farah, C.A.; Sossin, W.S. The role of C2 domains in PKC signaling. In Calcium Signaling; Springer: Dordrecht, The Netherlands, 2012; pp. 663–683. [Google Scholar]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef] [PubMed]
- Gahmberg, C.G.; Grönholm, M.; Madhavan, S.; Jahan, F.; Mikkola, E.; Viazmina, L.; Koivunen, E. Regulation of cell adhesion: A collaborative effort of integrins, their ligands, cytoplasmic actors, and phosphorylation. Q. Rev. Biophys. 2019, 5, e10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.A.; Hoppe, A.D. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2004, 76, 1093–1103. [Google Scholar] [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Agrawal, Y.; Santra, M.K.; Mishra, V.; Pathak, C. Molecular insights of NADPH oxidases and its pathological consequences. Cell Biochem. Funct. 2021, 39, 218–234. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Javed, T.; Shattat, G.F. Cardiovascular pharmacology. In Advanced Drug Formulation Design to Optimize Therapeutic Outcomes; CRC Press: Boca Raton, FL, USA, 2007; Volume 9, p. 359. [Google Scholar]
- Egan, B.T.; Szeiffova, B.B.; Viczenczova, C.; Diez, E.R.; Barancik, M.; Tribulova, N. Protection of cardiac cell-to-cell coupling attenuate myocardial remodeling and proarrhythmia induced by hypertension. Physiol. Res. 2016, 65, S29–S42. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Sutton, C.R.; Rao, S. Protein kinase C in the immune system: From signaling to chromatin regulation. Immunology 2015, 146, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Kacar, E.; Ercan, Z.; Serhatlioglu, I.; Sumer, A.; Kelestimur, H.; Kutlu, S. The effects of apelin on myometrium contractions in pregnant rats. Cell. Mol. Biol. 2018, 64, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, A.; Xiong, W.; Lin, H.; Xiao, W.; Huang, J.; Zhang, S.; Liu, Z. Catechins enhance skeletal muscle performance. Crit. Rev. Food Sci. Nutr. 2020, 60, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeong, S.K.; Ahn, S.K. An update of the defensive barrier function of skin. Yonsei Med. J. 2006, 47, 293–306. [Google Scholar] [CrossRef] [PubMed]
- de Castro, G.S.; Calder, P.C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 2018, 37, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, F. Synaptic plasticity and the neurobiology of learning and memory. Acta Biomed. 2007, 78 (Suppl. S1), 58–66. [Google Scholar] [PubMed]
- Mattson, M.P.; Liu, D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 2003, 304, 539–549. [Google Scholar] [CrossRef]
- Jellinger, K.A. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162. [Google Scholar] [CrossRef]
- Pascale, A.; Amadio, M.; Govoni, S.; Battaini, F. The aging brain, a key target for the future: The protein kinase C involvement. Pharmacol. Res. 2007, 55, 560–569. [Google Scholar] [CrossRef]
- Atwal, J.K. Signalling Mechanisms Underlying Trk Function in Neonatal Sympathetic Neurons. Ph.D. Thesis, McGill University, Montreal, QC, Canada, 2001. [Google Scholar]
- Sossin, W.S. An autonomous kinase generated during long-term facilitation in Aplysia is related to the Ca(2+)-independent protein kinase C Apl II. Learn. Mem. 1997, 3, 389–401. [Google Scholar] [CrossRef]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.H.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The critical modulators regulating gut–brain axis. J. Cell. Physiol. 2017, 232, 2359–2372. [Google Scholar] [CrossRef]
- Banasiak, K.J.; Xia, Y.; Haddad, G.G. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. 2000, 62, 215–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, X. The Roles of Par3, Par6, and aPKC Polarity Proteins in Normal Neurodevelopment and in Neurodegenerative and Neuropsychiatric Disorders. J. Neurosci. Res. 2022, 42, 4774–4793. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Gosens, R.; Zaagsma, J.; Bromhaar, M.G.; Nelemans, A.; Meurs, H. Acetylcholine: A novel regulator of airway smooth muscle remodelling? Eur. J. Pharmacol. 2004, 500, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hessel, E.V.; Staal, Y.C.; Piersma, A.H. Design and validation of an ontology-driven animal-free testing strategy for developmental neurotoxicity testing. Toxicol. Appl. Pharmacol. 2018, 354, 136–152. [Google Scholar] [CrossRef]
- Maioli, M.; Ventura, C. Protein kinase C control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2001, 11, 243–267. [Google Scholar]
- Battaini, F.; Pascale, A. Protein kinase C signal transduction regulation in physiological and pathological aging. Ann. N. Y. Acad. Sci. 2005, 1057, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Nisa, F.Y.; Rahman, M.A.; Hossen, M.A.; Khan, M.F.; Khan, M.A.; Majid, M.; Sultana, F.; Haque, M.A. Role of neurotoxicants in the pathogenesis of AD: A mechanistic insight. Ann. Med. 2021, 53, 1479–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin modulates excitatory synaptic transmission and synaptic plasticity in the mouse hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Bin Ibrahim, M.Z.; Benoy, A.; Sajikumar, S. Long-term plasticity in the hippocampus: Maintaining within and ‘tagging’ between synapses. FEBS J. 2022, 289, 2176–2201. [Google Scholar] [CrossRef] [PubMed]
- Köles, L.; Kató, E.; Hanuska, A.; Zádori, Z.S.; Al-Khrasani, M.; Zelles, T.; Rubini, P.; Illes, P. Modulation of excitatory neurotransmission by neuronal/glial signalling molecules: Interplay between purinergic and glutamatergic systems. Purinergic Signal. 2016, 12, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Hayashi, T. Post-translational palmitoylation of ionotropic glutamate receptors in excitatory synaptic functions. Br. J. Pharmacol. 2021, 178, 784–797. [Google Scholar] [CrossRef]
- Frick, A.; Magee, J.; Johnston, D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 2004, 7, 126–135. [Google Scholar] [CrossRef]
- Keeler, A.B.; Molumby, M.J.; Weiner, J.A. Protocadherins branch out: Multiple roles in dendrite development. Cell Adhes. Migr. 2015, 9, 214–226. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef]
- Yamauchi, T. Neuronal Ca2+/calmodulin-dependent protein kinase II—Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory. Biol. Pharm. Bull. 2005, 28, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in AD: A review on emergent natural polyphenolic therapeutics. Complement. Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.; Atwood, C.S.; Laws, S.M.; Gandy, S.E.; Martins, R.N. The role of beta amyloid in AD: Still a cause of everything or the only one who got caught? Pharmacol. Res. 2004, 50, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Alkon, D.L.; Sun, M.K.; Nelson, T.J. PKC signaling deficits: A mechanistic hypothesis for the origins of AD. Trends Pharmacol. Sci. 2007, 28, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and tau in the pathogenesis of AD. Int. J. Biol. Sci. 2021, 17, 2181. [Google Scholar] [CrossRef]
- Wu, H.Y.; Kuo, P.C.; Wang, Y.T.; Lin, H.T.; Roe, A.D.; Wang, B.Y.; Han, C.L.; Hyman, B.T.; Chen, Y.J.; Tai, H.C. β-Amyloid induces pathology-related patterns of tau hyperphosphorylation at synaptic terminals. J. Neuropathol. Exp. Neurol. 2018, 77, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated tau in AD and other tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Talman, V.; Pascale, A.; Jäntti, M.; Amadio, M.; Tuominen, R.K. Protein kinase C activation as a potential therapeutic strategy in AD: Is there a role for embryonic lethal abnormal vision-like proteins? Basic Clin. Pharmacol. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Baldini, C.; Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Redox control of protein kinase C: Cell-and disease-specific aspects. Antioxid. Redox Signal. 2010, 13, 1051–1085. [Google Scholar] [CrossRef]
- Iyer, D.N.; Faruq, O.; Zhang, L.; Rastgoo, N.; Liu, A.; Chang, H. Pathophysiological roles of myristoylated alanine-rich C-kinase substrate (MARCKS) in hematological malignancies. Biomark. Res. 2021, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Mi, K.; Johnson, G.V. The role of tau phosphorylation in the pathogenesis of AD. Curr. Alzheimer Res. 2006, 3, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Meco, M.T.; Moscat, J. The atypical PKCs in inflammation: NF-κB and beyond. Immunol. Rev. 2012, 246, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Parker, P.J. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochem. J. 2003, 376, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.J.; Shea, T.B. Phosphorylation events mediated by protein kinase Cα and ε participate in regulation of tau steady-state levels and generation of certain “Alzheimer-like” phospho-epitopes. Int. J. Dev. Neurosci. 1997, 15, 295–307. [Google Scholar] [CrossRef]
- Hampel, H.; Blennow, K.; Shaw, L.M.; Hoessler, Y.C.; Zetterberg, H.; Trojanowski, J.Q. Total and phosphorylated tau protein as biological markers of AD. Exp. Gerontol. 2010, 45, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human AD brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Zuo, L.; Zhang, M.; Cheng, P.; Guo, Z.; Yang, J.; Li, C.; Wu, J. High glucose induces tau hyperphosphorylation in hippocampal neurons via inhibition of ALKBH5-mediated Dgkh m6A demethylation: A potential mechanism for diabetic cognitive dysfunction. Cell Death Discov. 2023, 14, 385. [Google Scholar] [CrossRef]
- Sharma, L.; Sharma, A.; Kumar, D.; Asthana, M.K.; Lalhlenmawia, H.; Kumar, A.; Bhattacharyya, S.; Kumar, D. Promising protein biomarkers in the early diagnosis of Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 1727–1744. [Google Scholar] [CrossRef]
- Mylroie, H.; Dumont, O.; Bauer, A.; Thornton, C.C.; Mackey, J.; Calay, D.; Hamdulay, S.S.; Choo, J.R.; Boyle, J.J.; Samarel, A.M.; et al. PKCε-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc. Res. 2015, 106, 509–519. [Google Scholar] [CrossRef]
- Matta, C.; Mobasheri, A. Regulation of chondrogenesis by protein kinase C: Emerging new roles in calcium signalling. Cell Signal. 2014, 26, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Sharma, L. Potential Enzymatic Targets in Alzheimer’s: A Comprehensive Review. Curr. Drug Targets 2019, 20, 316–339. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Simpkins, J.W.; Alkon, D.L.; Smith, K.E.; Chen, Y.W.; Tan, Z.; Huber, J.D.; Rosen, C.L. Common mechanisms of AD and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimers Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, A.; Sharma, L. A Comprehensive Review of Alzheimer’s Association with Related Proteins: Pathological Role and Therapeutic Significance. Curr. Neuropharmacol. 2020, 18, 674–695. [Google Scholar] [CrossRef]
- Tam, J.H.; Seah, C.; Pasternak, S.H. The Amyloid Precursor Protein is rapidly transported from the Golgi apparatus to the lysosome and where it is processed into beta-amyloid. Mol. Brain 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Autotaxin–lysophosphatidic acid signaling in AD. Int. J. Mol. Sci. 2018, 9, 1827. [Google Scholar] [CrossRef] [PubMed]
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent models of neuroinflammation for AD. J. Neuroinflamm. 2015, 12, 74. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Figueiredo-Pereira, M.E. Relationship between tau pathology and neuroinflammation in AD. Mount Sinai J. Med. 2010, 77, 50–58. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in AD: Interplay mechanisms and clinical translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial activation in metal neurotoxicity: Impact in neurodegenerative diseases. BioMed Res. Int. 2023, 2023, 7389508. [Google Scholar] [CrossRef]
- Goel, A.; Singh, S. Emerging approaches for the treatment of Alzheimer disease: Targeting NF-κB pathway. Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Y.; Li, C.; Zheng, Q.; Tian, J.; Li, Z.; Huang, T.Y.; Zhang, W.; Xu, H. Inhibition of PKCδ reduces amyloid-β levels and reverses Alzheimer disease phenotypes. J. Exp. Med. 2018, 215, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Nelson, T.J.; Verma, V.A.; Wender, P.A.; Alkon, D.L. A cellular model of AD therapeutic efficacy: PKC activation reverses Aβ-induced biomarker abnormality on cultured fibroblasts. Neurobiol. Dis. 2009, 34, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, A.; Rotem-Dai, N.; Strominger, I.; Anto, N.P.; Isakov, N.; Monsonego, A.; Livneh, E. Protein kinase C eta is activated in reactive astrocytes of an AD mouse model: Evidence for its immunoregulatory function in primary astrocytes. Glia 2021, 69, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Kaleli, H.N.; Ozer, E.; Kaya, V.O.; Kutlu, O. Protein kinase C isozymes and autophagy during neurodegenerative disease progression. Cells 2020, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Lordén, G.; Newton, A.C. Conventional protein kinase C in the brain: Repurposing cancer drugs for neurodegenerative treatment? Neuronal Signal. 2021, 5, NS20210036. [Google Scholar] [CrossRef] [PubMed]
- Lordén, G.; Wozniak, J.M.; Doré, K.; Dozier, L.E.; Cates-Gatto, C.; Patrick, G.N.; Gonzalez, D.J.; Roberts, A.J.; Tanzi, R.E.; Newton, A.C. Enhanced activity of Alzheimer disease-associated variant of protein kinase Cα drives cognitive decline in a mouse model. Nat. Commun. 2022, 13, 7200. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.L.; Godoy, J.; Alvarez, A.; Bronfman, M.; Inestrosa, N.C. Protein kinase C inhibits amyloid β-peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002, 16, 1982–1984. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carvajal, A.; González-Muñiz, R.; Fernández-Ballester, G.; Ferrer-Montiel, A. Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Expert Opin. Investig. Drugs 2020, 29, 1209–1222. [Google Scholar] [CrossRef]
- Payal, N.; Sharma, L.; Sharma, A.; Hobanii, Y.H.; Hakami, M.A.; Ali, N.; Rashid, S.; Sachdeva, M.; Gulati, M.; Yadav, S.; et al. Understanding the Therapeutic Approaches for Neuroprotection. Curr. Pharm. Des. 2023, 29, 3368–3384. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Kar, S.; Aliter, K.F. Potential anti-COVID-19 therapeutics that block the early stage of the viral life cycle: Structures, mechanisms, and clinical trials. Int. J. Mol. Sci. 2020, 21, 5224. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Bu, F.Q.; Wang, F.; Liu, L.; Zhang, S.; Wang, G.; Hu, X.Y. Recent advances on the molecular mechanisms of exercise-induced improvements of cognitive dysfunction. Transl. Neurodegener. 2023, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010, 91, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Rozemuller, J.M.; Van Haastert, E.S.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase-1 and-2 in the different stages of AD pathology. Curr. Pharm. Des 2008, 14, 1419–1427. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K. Inflammatory processes in AD. J. Neuroimmunol. 2007, 184, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Basheer, N.; Smolek, T.; Hassan, I.; Liu, F.; Iqbal, K.; Zilka, N.; Novak, P. Does modulation of tau hyperphosphorylation represent a reasonable therapeutic strategy for Alzheimer’s disease? From preclinical studies to the clinical trials. Mol. Psychiatry 2023, 28, 2197–2214. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Blood-brain barrier integrity and glial support: Mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr. Pharm. Des. 2012, 18, 3624–3644. [Google Scholar] [CrossRef]
- Gharibani, P.; Abramson, E.; Shanmukha, S.; Smith, M.D.; Godfrey, W.H.; Lee, J.J.; Hu, J.; Baydyuk, M.; Dorion, M.F.; Deng, X.; et al. PKC modulator bryostatin-1 therapeutically targets CNS innate immunity to attenuate neuroinflammation and promote remyelination. bioRxiv 2023, 555084. [Google Scholar] [CrossRef]
- Abrial, E.; Lucas, G.; Scarna, H.; Haddjeri, N.; Lambás-Señas, L. A role for the PKC signaling system in the pathophysiology and treatment of mood disorders: Involvement of a functional imbalance? Mol. Neurobiol. 2011, 44, 407–419. [Google Scholar] [CrossRef]
- Dagda, R.K.; Das Banerjee, T. Role of protein kinase A in regulating mitochondrial function and neuronal development: Implications to neurodegenerative diseases. Rev. Neurosci. 2015, 26, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Vetreno, R.P.; Ramos, R.L.; Anzalone, S.; Savage, L.M. Brain and behavioral pathology in an animal model of Wernicke’s encephalopathy and Wernicke–Korsakoff Syndrome. Brain Res. 2012, 1436, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shi, G.; Yang, K.C.; Gu, L.; Kanthasamy, A.G.; Anantharam, V.; Dudley, S.C., Jr. Role of protein kinase C in metabolic regulation of the cardiac Na+ channel. Heart Rhythm. 2017, 14, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Jubaidi, F.F.; Zainalabidin, S.; Taib, I.S.; Abdul Hamid, Z.; Mohamad Anuar, N.N.; Jalil, J.; Mohd Nor, N.A.; Budin, S.B. The Role of PKC-MAPK Signalling Pathways in the Development of Hyperglycemia-Induced Cardiovascular Complications. Int. J. Mol. Sci. 2022, 23, 8582. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Wu, E.Q.; Guérin, A.; Andrew, P.Y.; Tsaneva, M.; Gupta, S.R.; Bao, Y.; Mulani, P.M. Risks of developing psychiatric disorders in pediatric patients with psoriasis. J. Am. Acad. Dermatol. 2012, 67, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.H.; Slater, M.A.; Patterson, T.L.; Grant, I.; Garfin, S.R. Prevalence, onset, and risk of psychiatric disorders in men with chronic low back pain: A controlled study. Pain 1991, 45, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Huang, C.L.; Tsai, C.J.; Chou, P.H.; Lin, C.C.; Chang, C.K. Alcohol-related dementia: A systemic review of epidemiological studies. Psychosomatics 2017, 58, 331–342. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef]
- Crabb, D.W.; Im, G.Y.; Szabo, G.; Mellinger, J.L.; Lucey, M.R. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2020, 71, 306–333. [Google Scholar] [CrossRef]
- Zima, T.; Kalousová, M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 110S–115S. [Google Scholar] [CrossRef]
- Lu, W.; Tang, S.; Li, A.; Huang, Q.; Dou, M.; Zhang, Y.; Hu, X.; Chang, R.C.; Wong, G.T.; Huang, C. The role of PKC/PKR in aging, AD, and perioperative neurocognitive disorders. Front. Aging Neurosci. 2022, 14, 973068. [Google Scholar] [CrossRef]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial dysfunction in AD and the rationale for bioenergetics based therapies. Aging Dis. 2016, 7, 201–213. [Google Scholar] [CrossRef]
- Wills, L.; Kenny, P.J. Addiction-related neuroadaptations following chronic nicotine exposure. J. Neurochem. 2021, 157, 1652–1673. [Google Scholar] [CrossRef]
- Jakob-Roetne, R.; Jacobsen, H. AD: From pathology to therapeutic approaches. Angew. Chem. Int. Ed. 2009, 48, 3030–3059. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Kuhad, A.; Chopra, K. Suppression of neuro-inflammatory signaling cascade by tocotrienol can prevent chronic alcohol-induced cognitive dysfunction in rats. Behav. Brain Res. 2009, 203, 296–303. [Google Scholar] [CrossRef]
- Peng, B.; Yang, Q.; Joshi, R.B.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of alcohol drinking in AD, Parkinson’s disease, and amyotrophic lateral sclerosis. Int. J. Mol. Sci 2020, 21, 2316. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Lana, D.; Pepeu, G. The integrated role of ACh, ERK and mTOR in the mechanisms of hippocampal inhibitory avoidance memory. Neurobiol. Learn. Mem. 2020, 171, 107215. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Song, Z.; Li, Y.; Yao, Y.; Fang, M.; Bai, C.; An, P.; Chen, H.; Chen, Z.; Tang, B.; et al. Activation of PKCδ reduces tau phosphorylation in cellular and mouse models of tauopathy. Aging Cell 2021, 20, e13323. [Google Scholar] [CrossRef]
- Li, J.; Deng, J.; Sheng, W.; Zuo, Z. Protein Kinase C Activator Attenuates Neuroinflammation in LPS-Stimulated BV-2 Microglial Cells by Inhibiting MAPK Pathways. Neurotox. Res. 2021, 41, 238–249. [Google Scholar]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Protein kinase C activation improves blood-brain barrier integrity in AD via regulation of tight junction proteins. Aging Cell 2023, 22, e13522. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and AD. Trends. Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in AD. J. Alzheimers Dis. 2018, 62, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Peterson, C. Calcium and the aging nervous system. Neurochem. Int. 2015, 88, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of AD. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodríguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; LaFerla, F.M. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of AD. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an AD mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P. Abnormal mitochondrial dynamics and synaptic degeneration as early events in AD: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Eckert, A.; Schulz, K.L.; Rhein, V.; Götz, J. Convergence of amyloid-β and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 2010, 41, 107–111. [Google Scholar] [CrossRef]
- Ma, T.; Chen, Y.; Vingtdeux, V.; Zhao, H.; Viollet, B.; Marambaud, P.; Klann, E. Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. J. Neurosci. Res. 2014, 34, 12230–12238. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and β-amyloid protein in AD. Neuromol. Med. 2012, 13, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in AD and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Nixon, K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Pfefferbaum, A. Neurocircuitry in alcoholism: A substrate of disruption and repair. Psychopharmacology 2005, 180, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kril, J.J.; Harper, C.G. Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol. Rev. 2012, 22, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Pfefferbaum, A.; Pitel, A.-L. Hippocampal volume recovery with abstinence from alcohol. Psychiatry Res. Neuroimaging 2013, 214, 202–209. [Google Scholar]
- Oscar-Berman, M.; Marinkovic, K. Alcoholism and the brain: An overview. Alcohol Res. Health 2003, 27, 125–133. [Google Scholar] [PubMed]
- Ritz, L.; Segobin, S.; Lannuzel, C.; Boudehent, C.; Vabret, F.; Eustache, F.; Pitel, A.-L. Direct voxel-based comparisons between grey matter shrinkage and glucose hypometabolism in chronic alcoholism. Neuropsychol. Rev. 2016, 36, 1625–1641. [Google Scholar] [CrossRef]
- Harper, C.; Matsumoto, I. Ethanol and brain damage. Curr. Opin. Pharmacol. 2005, 5, 73–78. [Google Scholar] [CrossRef]
- Harper, C.G.; Kril, J.J.; Holloway, R.L. Brain shrinkage in chronic alcoholics: A pathological study. Br. Med. J. (Clin. Res. Ed.) 1985, 290, 501–504. [Google Scholar] [CrossRef]
- Kril, J.J.; Halliday, G.M.; Svoboda, M.D.; Cartwright, H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience 1997, 79, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Oscar-Berman, M.; Marinkovic, K. Alcohol: Effects on neurobehavioral functions and the brain. Neuropsychol. Rev. 2007, 17, 239–257. [Google Scholar] [CrossRef]
- Sullivan, E.V.; Rosenbloom, M.J.; Pfefferbaum, A. Pattern of motor and cognitive deficits in detoxified alcoholic men. Alcohol. Clin. Exp. Res. 2000, 24, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Zahr, N.M.; Mayer, D.; Vinco, S.; Orduna, J.; Luong, R.; Sullivan, E.V.; Pfefferbaum, A. In vivo evidence for alcohol-induced neurochemical changes in rat brain without protracted withdrawal, pronounced thiamine deficiency, or severe liver damage. Neuropsychopharmacology 2009, 34, 1427–1442. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Nixon, K.; Crews, F.T. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J. Neurochem. 2002, 83, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Sullivan, E.V.; Mathalon, D.H.; Shear, P.K.; Rosenbloom, M.J.; Lim, K.O. Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol. Clin. Exp. Res. 1995, 19, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.K.; Ahmad, M.H.; Sahu, M.R.; Subba, R.; Mondal, A.C. Detrimental effects of alcohol-induced inflammation on brain health: From neurogenesis to neurodegeneration. Cell. Mol. Neurobiol. 2023, 43, 1885–1904. [Google Scholar] [CrossRef] [PubMed]
- Lanquetin, A.; Leclercq, S.; de Timary, P.; Segobin, S.; Naveau, M.; Coulbault, L.; Maccioni, P.; Lorrai, I.; Colombo, G.; Vivien, D.; et al. Role of inflammation in alcohol-related brain abnormalities: A translational study. Brain Commun. 2021, 3, fcab154. [Google Scholar] [CrossRef]
- Ramos, A.; Joshi, R.S.; Szabo, G. Innate immune activation: Parallels in alcohol use disorder and AD. Front. Mol. Neurosci. 2022, 15, 910298. [Google Scholar] [CrossRef]
- Hansen, N. New insights into the role of the locus coeruleus-noradrenergic system in memory and perception dysfunction. J. Alzheimers Dis. 2017, 15, 653–658. [Google Scholar]
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol use disorder, neurodegeneration, Alzheimer’s and Parkinson’s disease: Interplay between oxidative stress, neuroimmune response and excitotoxicity. Front. Cell. Neurosci. 2020, 14, 282. [Google Scholar] [CrossRef]
- Crews, F.T.; Nixon, K.; Kim, D.; Joseph, J.; Shukitt-Hale, B.; Qin, L.; Zou, J. BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol. Clin. Exp. Res. 2006, 30, 1938–1949. [Google Scholar] [CrossRef]
- Sullivan, E.V.; Pfefferbaum, A. Neuroimaging of the Wernicke–Korsakoff syndrome. Alcohol Alcohol. 2011, 46, 158–167. [Google Scholar] [CrossRef]
- Piano, M.R. Alcoholic cardiomyopathy: Incidence, clinical characteristics, and pathophysiology. Chest 2017, 151, 181–190. [Google Scholar]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2011, 18, 2905–2916. [Google Scholar] [CrossRef]
- Taouis, M.; Chen, J.; Daviaud, C.; Dupont, J. Cloning and characterization of the rat leptin receptor Ob-Ra in pancreatic islets. Biochem. Biophys. Res. Commun. 1999, 256, 637–641. [Google Scholar]
- Mukamal, K.J.; Chiuve, S.E.; Rimm, E.B. Alcohol consumption and risk for coronary heart disease in men with healthy lifestyles. Arch. Intern. Med. 2006, 166, 2145–2150. [Google Scholar] [CrossRef]
- Crews, F.T.; Zou, J.; Qin, L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 2005, 19, 427–440. [Google Scholar] [CrossRef]
- Pascual, M.; Blanco, A.M.; Cauli, O.; Minarro, J.; Guerri, C. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur. J. Neurosci. 2007, 25, 541–550. [Google Scholar] [CrossRef]
- Qayyum, A.; Zai, C.C.; Hirata, Y.; Tiwari, A.K.; Cheema, S.; Nowrouzi, B.; Beitchman, J.H.; Kennedy, J.L. The Role of the Catechol-o-Methyltransferase (COMT) GeneVal158Met in Aggressive Behavior, a Review of Genetic Studies. Curr. Neuropharmacol. 2015, 13, 802–814. [Google Scholar] [CrossRef]
- Baik, J.H. Dopamine signaling in food addiction: Role of dopamine D2 receptors. BMB Rep. 2013, 46, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Miller, A. The role of benzodiazepines in the treatment of insomnia: Guidelines for the use of hypnotic drugs. J. Pract. Nurs. 2013, 63, 35–38. [Google Scholar]
- Zahr, N.M.; Kaufman, K.L.; Harper, C.G. Clinical and pathological features of alcohol-related brain damage. Nat. Rev. Neurol. 2011, 7, 284–294. [Google Scholar] [CrossRef]
- Zou, J.Y.; Crews, F.T. TNFα potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NFκB inhibition. Brain Res. 2006, 1073–1074, 293–302. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Mathew, S.J.; D’Souza, D.C.; Garakani, A.; Gunduz-Bruce, H.; Charney, D.S. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs 2010, 24, 669–693. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Crews, F.T.; Qin, L.; Sheedy, D.; Vetreno, R.P.; Zou, J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatry 2013, 73, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.P.; Huang, F.L.; Jäger, T. PKC isoforms in synaptic plasticity and memory. Cell Sci. Rev. 2004, 1, 1–18. [Google Scholar]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef]
- Kessels, H.W.; Malinow, R. Synaptic AMPA receptor plasticity and behavior. Neuron 2009, 61, 340–350. [Google Scholar] [CrossRef]
- Crews, F.T.; Vetreno, R.P. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 2016, 233, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Horn, K.H.; Zhou, F.C. Alcohol teratogenesis: Mechanisms of damage and strategies for intervention. Exp. Biol. Med. 2005, 230, 394–406. [Google Scholar] [CrossRef]
- Nixon, K.; Kim, D.H.; Potts, E.N.; He, J.; Crews, F.T. Distinct cell proliferation events during abstinence after alcohol dependence: Microglia proliferation precedes neurogenesis. Neurobiol. Dis. 2008, 31, 218–229. [Google Scholar] [CrossRef]
- Kril, J.J.; Halliday, G.M. Brain shrinkage in alcoholics: A decade on and what have we learned? Prog. Neurobiol. 1999, 58, 381–387. [Google Scholar] [CrossRef]
- Lowe, P.P.; Morel, C.; Ambade, A.; Iracheta-Vellve, A.; Kwiatkowski, E.; Satishchandran, A.; Furi, I.; Cho, Y.; Gyongyosi, B.; Catalano, D.; et al. Chronic alcohol-induced neuroinflammation involves CCR2/5-dependent peripheral macrophage infiltration and microglia alterations. J. Neuroinflamm. 2020, 17, 296. [Google Scholar] [CrossRef]
- Marshall, S.A.; McClain, J.A.; Wooden, J.I.; Nixon, K. Microglia dystrophy following binge-like alcohol exposure in adolescent and adult male rats. Front. Neuroanat. 2020, 14, 52. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Nandy, S.K.; Jyoti, A.; Saxena, J.; Sharma, A.; Siddiqui, A.J.; Sharma, L. Protein Kinase C (PKC) in Neurological Health: Implications for Alzheimer’s Disease and Chronic Alcohol Consumption. Brain Sci. 2024, 14, 554. https://doi.org/10.3390/brainsci14060554
Singh N, Nandy SK, Jyoti A, Saxena J, Sharma A, Siddiqui AJ, Sharma L. Protein Kinase C (PKC) in Neurological Health: Implications for Alzheimer’s Disease and Chronic Alcohol Consumption. Brain Sciences. 2024; 14(6):554. https://doi.org/10.3390/brainsci14060554
Chicago/Turabian StyleSingh, Nishtha, Shouvik Kumar Nandy, Anupam Jyoti, Juhi Saxena, Aditi Sharma, Arif Jamal Siddiqui, and Lalit Sharma. 2024. "Protein Kinase C (PKC) in Neurological Health: Implications for Alzheimer’s Disease and Chronic Alcohol Consumption" Brain Sciences 14, no. 6: 554. https://doi.org/10.3390/brainsci14060554