Integrity of Cerebellar Fastigial Nucleus Intrinsic Neurons Is Critical for the Global Ischemic Preconditioning


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Four-Vessel Occlusion
2.3. Regional Cortical CBF
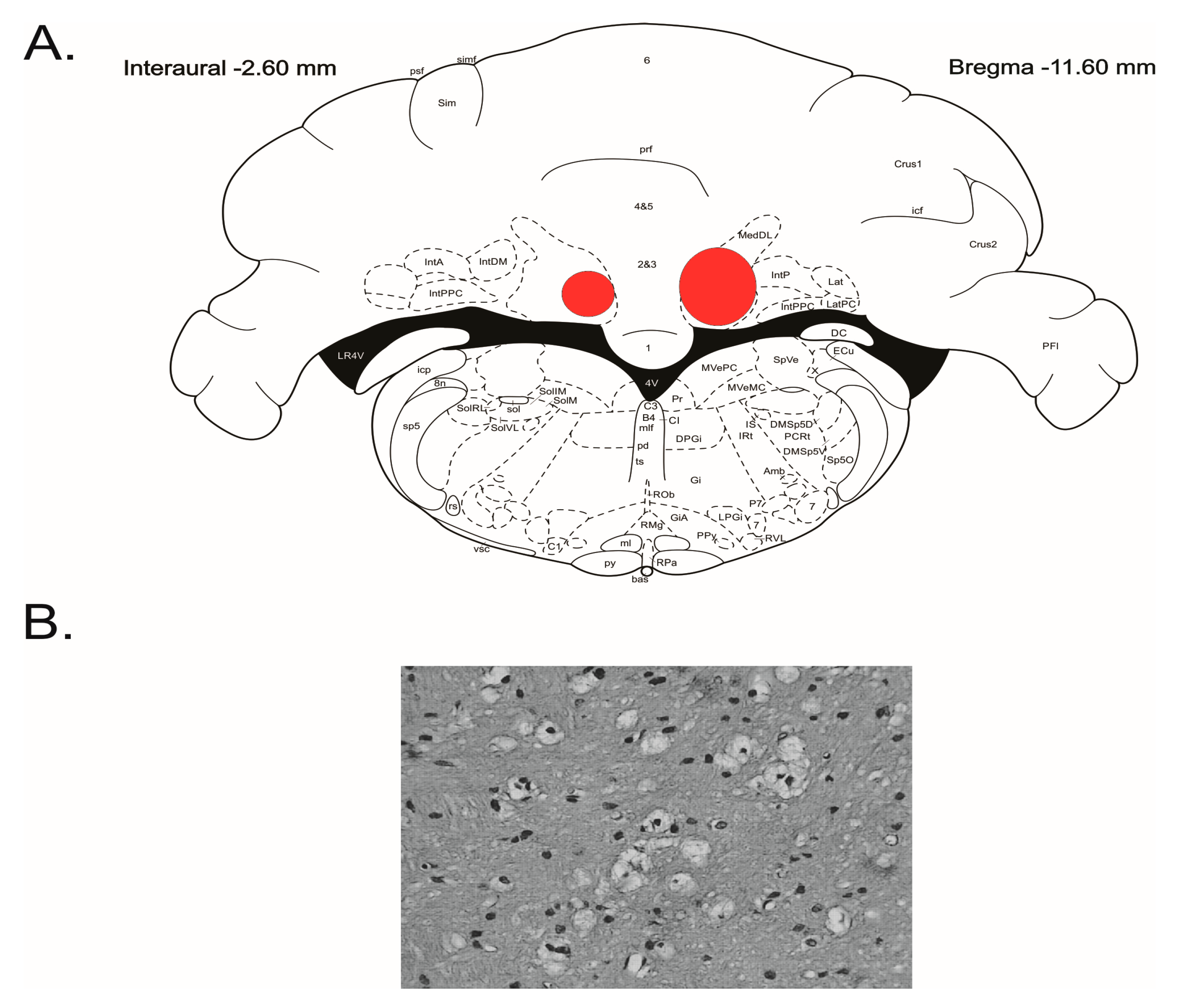
2.4. Fastigial Neurons Lesion
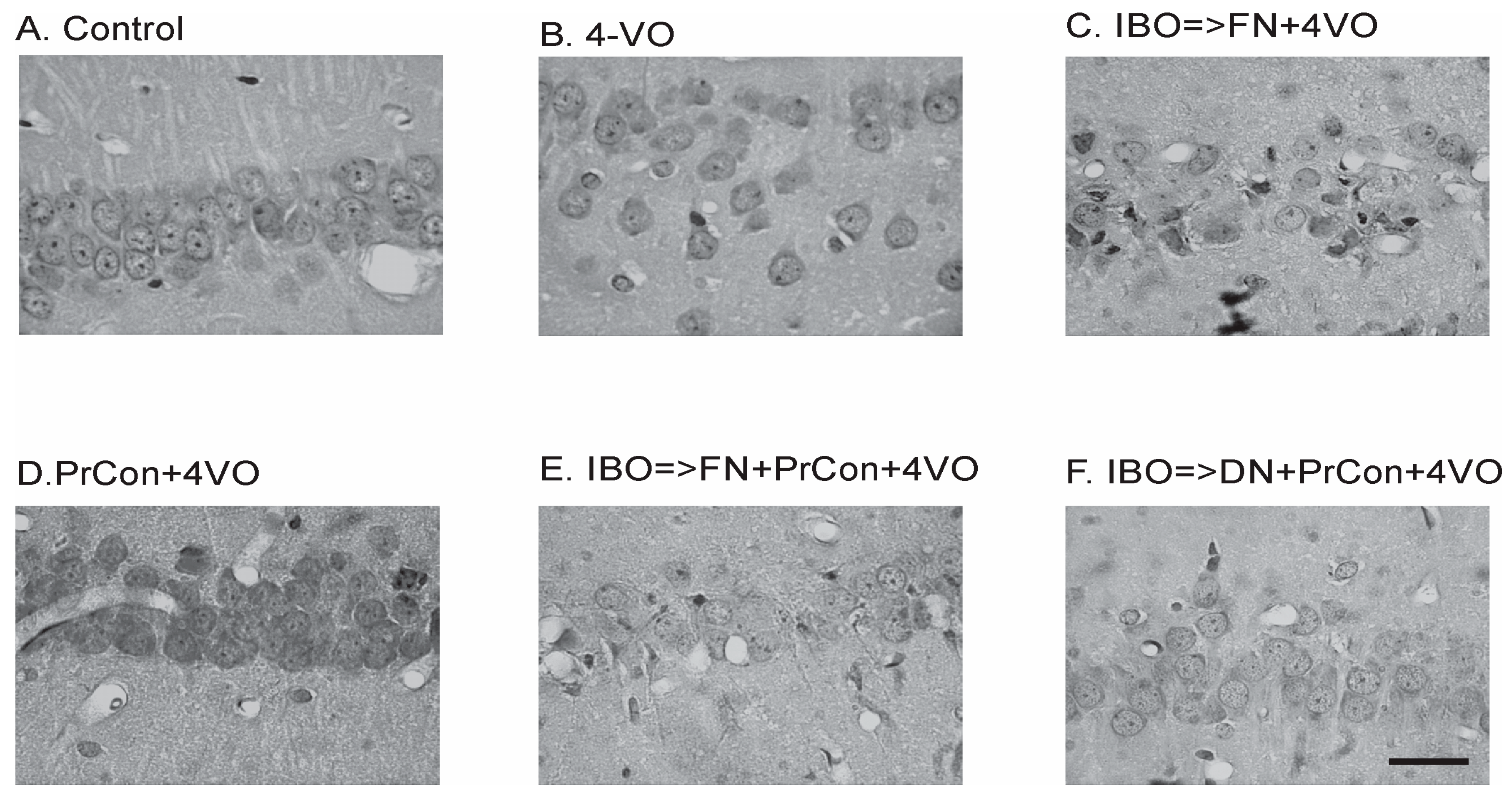
2.5. Histological Processing
2.6. Experiments and Experimental Groups
2.7. Statistical Procedures
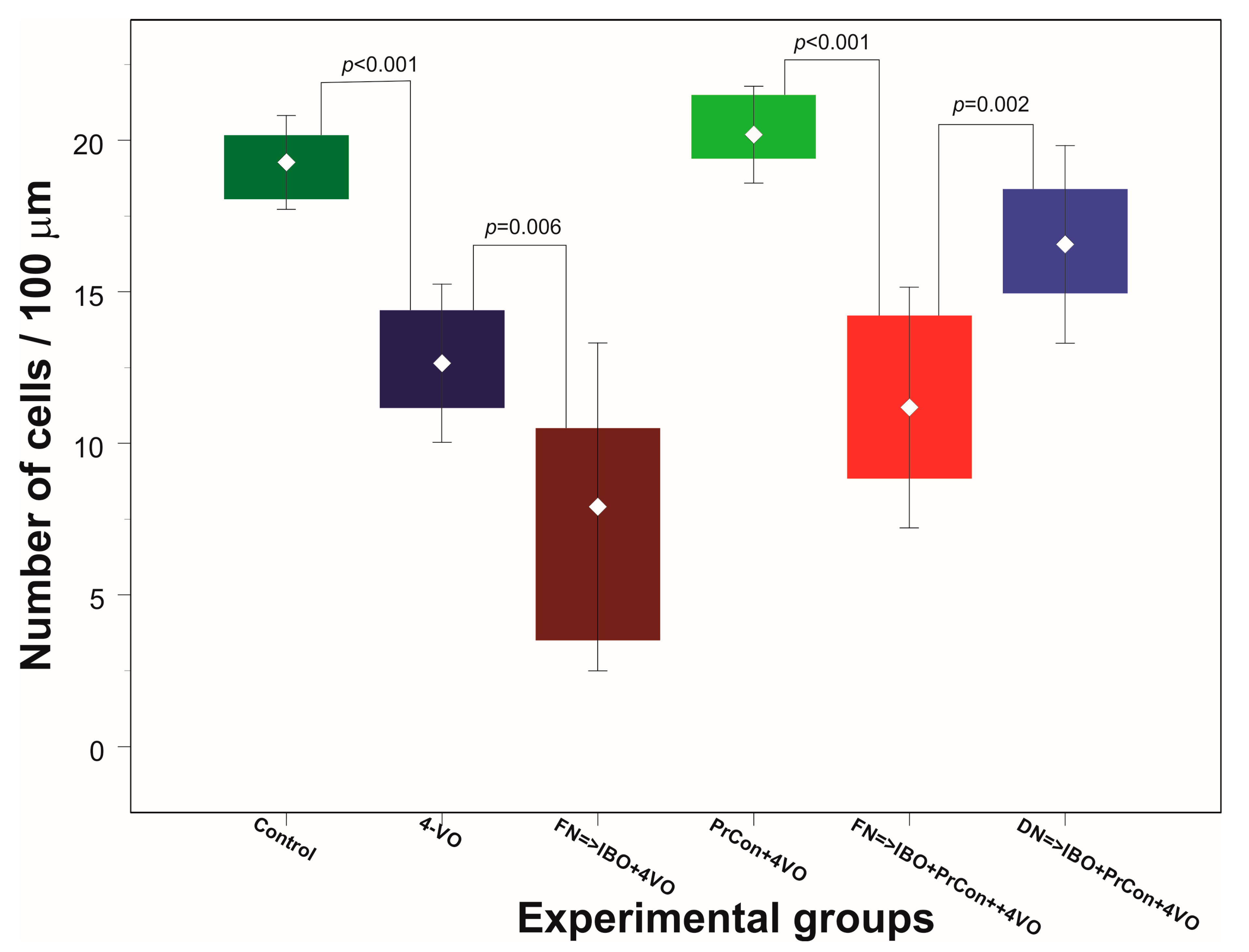
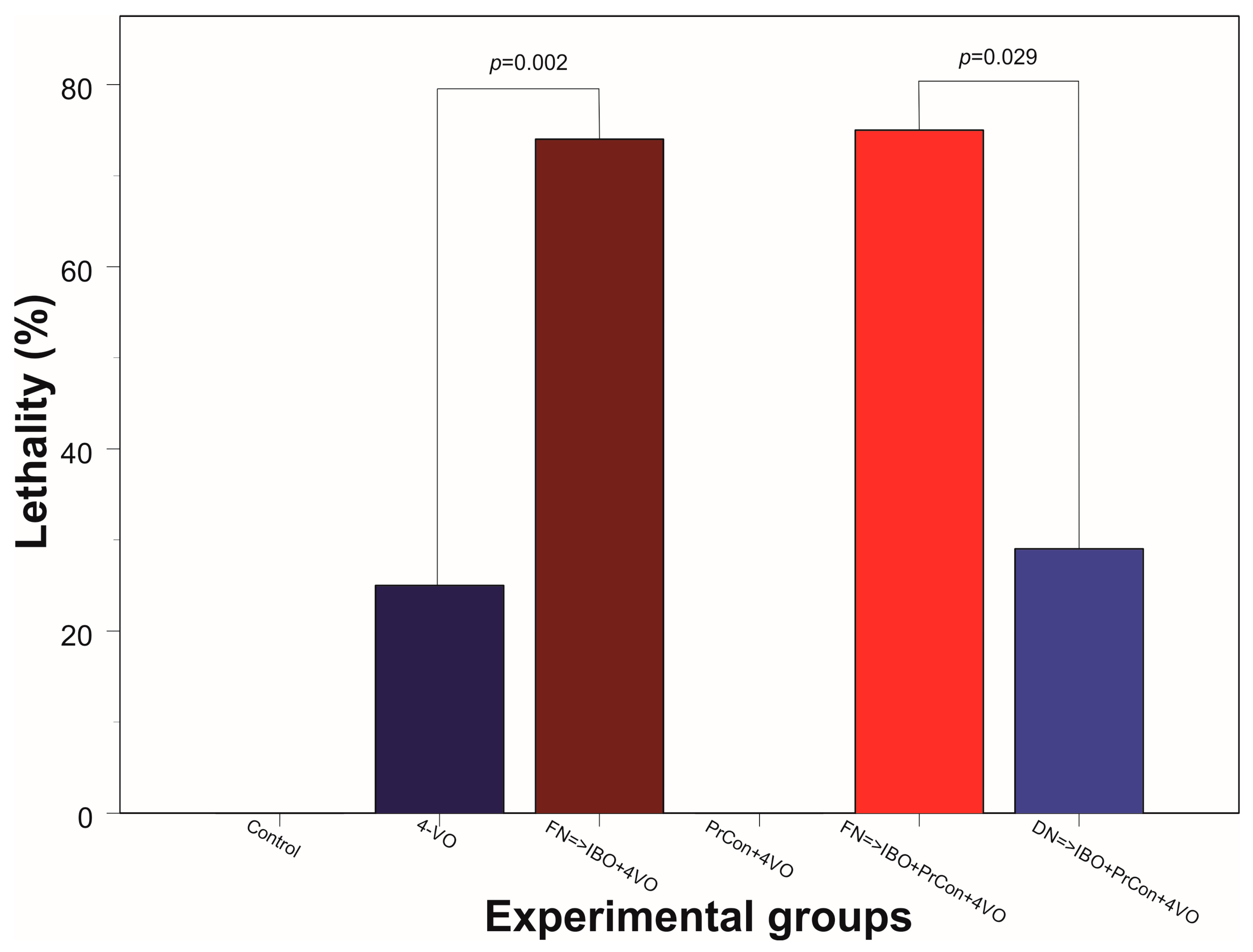
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, J.; Dong, W.W.; Zhang, W.H.; Zheng, J.; Wang, X. Electrical stimulation of cerebellar fastigial nucleus: Mechanism of neuroprotection and prospects for clinical application against cerebral ischemia. CNS Neurosci. Ther. 2014, 20, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.; Fonoff, E.T.; Bor-Seng-Shu, E.; Teixeira, M.J.; Chadi, G. Neurogenic neuroprotection: Future perspectives. Transl. Neurosci. 2012, 3, 399–412. [Google Scholar] [CrossRef]
- Golanov, E.V.; Zhou, P. Neurogenic neuroprotection. Cell. Mol. Neurobiol. 2003, 23, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Golanov, E.V.; Reis, D.J. Reductions in focal ischemic infarctions elicited from cerebellar fastigial nucleus do not result from elevations in cerebral blood flow. J. Cereb. Blood Flow Metab. 1993, 13, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Reis, D.J.; Berger, S.B.; Underwood, M.D.; Khayata, M. Electrical stimulation of cerebellar fastigial nucleus reduces ischemic infarction elicited by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 1991, 11, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.B.; Ballon, D.; Graham, M.; Underwood, M.D.; Khayata, M.; Leggiero, R.D.; Koutcher, J.A.; Reis, D.J. Magnetic resonance imaging demonstrates that electric stimulation of cerebellar fastigial nucleus reduces cerebral infarction in rats. Stroke 1990, 21, III172–III176. [Google Scholar] [PubMed]
- Underwood, M.D.; Berger, S.B.; Khayata, M.; Reis, D.J. Fastigial nucleus stimulation reduces the volume of cerebral infarction produced by occlusion of the middle cerebral artery in rat. J. Cereb. Blood Flow Metab. 1989, 9 (Suppl. 1), S32. [Google Scholar]
- Glickstein, S.B.; Golanov, E.V.; Reis, D.J. Intrinsic neurons of fastigial nucleus mediate neurogenic neuroprotection against excitotoxic and ischemic neuronal injury in rat. J. Neurosci. 1999, 19, 4142–4154. [Google Scholar] [PubMed]
- Golanov, E.V.; Liu, F.; Reis, D.J. Stimulation of cerebellum protects hippocampal neurons from global ischemia. Neuro Rep. 1998, 9, 819–824. [Google Scholar] [CrossRef]
- Reis, D.J.; Feinstein, D.; Galea, E.; Golanov, E.V. Central neurogenic neuroprotection: Protection of brain from focal ischemia by cerebellar stimulation. Fundam. Clin. Pharmacol. 1997, 11, 39s–43s. [Google Scholar] [CrossRef]
- Golanov, E.V.; Yamamoto, S.; Reis, D.J. Electrical stimulation of cerebellar fastigial nucleus fails to rematch blood flow and metabolism in focal ischemic infarctions. Neurosci. Lett. 1996, 210, 181–184. [Google Scholar] [CrossRef]
- Glickstein, S.B.; Ilch, C.P.; Reis, D.J.; Golanov, E.V. Stimulation of the subthalamic vasodilator area and fastigial nucleus independently protects the brain against focal ischemia. Brain Res. 2001, 912, 47–59. [Google Scholar] [CrossRef]
- Glickstein, S.B.; Ilch, C.P.; Golanov, E.V. Electrical stimulation of the dorsal periaqueductal gray decreases volume of the brain infarction independently of accompanying hypertension and cerebrovasodilation. Brain Res. 2003, 994, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Kuwabara, K.; Tagaya, M.; Ohtsuki, T.; Hata, R.; Ueda, H.; Handa, N.; Kimura, K.; Kamada, T. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res. 1991, 561, 203–211. [Google Scholar] [CrossRef]
- Perez-Pinzon, M.A.; Xu, G.P.; Dietrich, W.D.; Rosenthal, M.; Sick, T.J. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Heurteaux, C.; Lauritzen, I.; Widmann, C.; Lazdunski, M. Essential role of adenosine, adenosine a1 receptors, and atp-sensitive k+ channels in cerebral ischemic preconditioning. Proc. Natl. Acad. Sci. USA 1995, 92, 4666–4670. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Hakim, A.M. Transient forebrain ischemia protects against subsequent focal cerebral ischemia without changing cerebral perfusion. Stroke 1995, 26, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Simon, R.P.; Hallenbeck, J.M. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003, 26, 248–254. [Google Scholar] [CrossRef]
- Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 2006, 7, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef]
- Meller, R.; Simon, R.P. A critical review of mechanisms regulating remote preconditioning-induced brain protection. J. Appl. Physiol. 2015, 119, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Schwarcz, R.; Hokfelt, T.; Fuxe, K.; Jonsson, G.; Goldstein, M.; Terenius, L. Ibotenic acid-induced neuronal degeneration: A morphological and neurochemical study. Exp. Brain Res. 1979, 37, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Zwerus, R.; Absalom, A. Update on anesthetic neuroprotection. Curr. Opin. Anaesthesiol. 2015, 28, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.B.; Ren, P.C.; Chao, Y.; Wang, Q.Y.; Kuai, J.K.; Lv, M.M.; Chen, L.; Gao, C.J.; Sun, X.D. Protective role of isoflurane pretreatment in rats with focal cerebral ischemia and the underlying molecular mechanism. Mol. Med. Rep. 2015, 12, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W. Mechanism and evolution of hypoxia-tolerance in humans. J. Exp. Biol. 1998, 201, 1243–1254. [Google Scholar] [PubMed]
- Gooden, B.A. The evolution of asphyxial defense. Integr. Physiol. Behav. Sci. 1993, 28, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Elsner, R.; Gooden, B. Diving and asphyxia. A comparative study of animals and man. Monogr. Physiol. Soc. 1983, 40, 1–168. [Google Scholar] [PubMed]
- Hochachka, P.W.; Lutz, P.L. Mechanism, origin, and evolution of anoxia tolerance in animals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2001, 130, 435–459. [Google Scholar] [CrossRef]
- Blix, A.S.; Folkow, B. Cardiovascular adjustments to diving in mammals and birds. In Sect 2, the Cardiovascular System; Renkin, E.M., Michel, C.C., Eds.; American Physiological Society: Bethesda, MD, USA, 1983; Volume 4, Part 2, pp. 917–945. [Google Scholar]
- Butler, P.J.; Jones, D.R. Physiology of diving of birds and mammals. Physiol. Rev. 1997, 77, 837–899. [Google Scholar] [PubMed]
- Elsner, R.; Shurley, J.T.; Hammond, D.D.; Brooks, R.E. Cerebral tolerance to hypoxemia in asphyxiated weddell seals. Respir. Physiol. 1970, 9, 287–297. [Google Scholar] [CrossRef]
- Castellini, J.M. Life under water: Physiological adaptations to diving and living at sea. In Comprehensive Physiology; American Physiological society: Bethesda, MD, USA, 2012; Volume 2, pp. 1889–1919. [Google Scholar]
- Gooden, B.A. Mechanism of the human diving response. Integr. Physiol. Behav. Sci. 1994, 29, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.E.; Sheel, A.W. The human diving response, its function, and its control. Scand. J. Med. Sci. Sports 2005, 15, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Panneton, W.M. The mammalian diving response: An enigmatic reflex to preserve life? Physiology 2013, 28, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Powell, F.L.; Kim, B.C.; Johnson, S.R.; Fu, Z. Oxygen sensing in the brain—Invited article. Adv. Exp. Med. Biol. 2009, 648, 369–376. [Google Scholar] [PubMed]
- Guyenet, P.G.; Stornetta, R.L.; Bayliss, D.A. Central respiratory chemoreception. J. Comp. Neurol. 2010, 518, 3883–3906. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.A.; Sunderram, J. Oxygen-sensing neurons in the central nervous system. J. Appl. Physiol. 2004, 96, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Reis, D.J.; Golanov, E.V.; Ruggiero, D.A.; Sun, M.-K. Sympatho-excitatory neurons of the rostral ventrolateral medulla are oxygen sensors and essential elements in the tonic and reflex control of the systemic and cerebral circulations. J. Hypertens. 1994, 12, S159–S180. [Google Scholar]
- Larson, J.; Drew, K.L.; Folkow, L.P.; Milton, S.L.; Park, T.J. No oxygen? No problem! Intrinsic brain tolerance to hypoxia in vertebrates. J. Exp. Biol. 2014, 217, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hafeez, A.; Noorulla, F.; Geng, X.; Shao, G.; Ren, C.; Lu, G.; Zhao, H.; Ding, Y.; Ji, X. Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 2017, 157, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bonilla, L.; Benakis, C.; Moore, J.; Iadecola, C.; Anrather, J. Immune mechanisms in cerebral ischemic tolerance. Front. Neurosci. 2014, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Wang, J.J.; Zhu, J.N. Cerebellar fastigial nucleus: From anatomic construction to physiological functions. Cerebellum Ataxias 2016, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Uusisaari, M.Y.; Knopfel, T. Diversity of neuronal elements and circuitry in the cerebellar nuclei. Cerebellum 2012, 11, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Husson, Z.; Rousseau, C.V.; Broll, I.; Zeilhofer, H.U.; Dieudonne, S. Differential gabaergic and glycinergic inputs of inhibitory interneurons and purkinje cells to principal cells of the cerebellar nuclei. J. Neurosci. 2014, 34, 9418–9431. [Google Scholar] [CrossRef] [PubMed]
- Ito, M. Cerebellar circuitry as a neuronal machine. Prog. Neurobiol. 2006, 78, 272–303. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, D.A.; Anwar, M.; Golanov, E.V.; Reis, D.J. The pedunculopontine tegmental nucleus issues collaterals to the fastigial nucleus and rostral ventrolateral reticular nucleus in the rat. Brain Res. 1997, 760, 272–276. [Google Scholar] [CrossRef]
- Gerrits, N.M.; Voogd, J. The projection of the nucleus reticularis tegmenti pontis and adjacent regions of the pontine nuclei to the central cerebellar nuclei in the cat. J. Comp. Neurol. 1987, 258, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Mihailoff, G.A. Cerebellar nuclear projections from the basilar pontine nuclei and nucleus reticularis tegmenti pontis as demonstrated with pha-l tracing in the rat. J. Comp. Neurol. 1993, 330, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.S.; Sugihara, I.; Shinoda, Y. Projection patterns of single mossy fibers originating from the lateral reticular nucleus in the rat cerebellar cortex and nuclei. J. Comp. Neurol. 1999, 411, 97–118. [Google Scholar] [CrossRef]
- Brodal, A.; Taber, E.; Walberg, F. The raphe nuclei of the brain stem in the cat. Ii. Efferent connections. J. Comp. Neurol. 1960, 114, 239–259. [Google Scholar] [CrossRef]
- Matsushita, M.; Xiong, G. Uncrossed and crossed projections from the upper cervical spinal cord to the cerebellar nuclei in the rat, studied by anterograde axonal tracing. J. Comp. Neurol. 2001, 432, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Dietrichs, E.; Haines, D.E.; Roste, G.K.; Roste, L.S. Hypothalamocerebellar and cerebellohypothalamic projections—Circuits for regulating nonsomatic cerebellar activity? Histol. Histopathol. 1994, 9, 603–614. [Google Scholar] [PubMed]
- Homma, Y.; Nonaka, S.; Matsuyama, K.; Mori, S. Fastigiofugal projection to the brainstem nuclei in the cat: An anterograde pha-l tracing study. Neurosci. Res. 1995, 23, 89–102. [Google Scholar] [CrossRef]
- Harper, J.W.; Heath, R.G. Anatomic connections of the fastigial nucleus to the rostral forebrain in the cat. Exp. Neurol. 1973, 39, 285–292. [Google Scholar] [CrossRef]
- Katoh, Y.Y.; Arai, R.; Benedek, G. Bifurcating projections from the cerebellar fastigial neurons to the thalamic suprageniculate nucleus and to the superior colliculus. Brain Res. 2000, 864, 308–311. [Google Scholar] [CrossRef]
- Steriade, M. Two channels in the cerebellothalamocortical system. J. Comp. Neurol. 1995, 354, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Watt, C.B.; Mihailoff, G.A. The cerebellopontine system in the rat. I. Autoradiographic studies. J. Comp. Neurol. 1983, 215, 312–330. [Google Scholar] [CrossRef] [PubMed]
- Çavdar, S.; Tangül, Ş.A.N.; Aker, R.; Şehirli, Ü.; Onat, F. Cerebellar connections to the dorsomedial and posterior nuclei of the hypothalamus in the rat. J. Anat. 2001, 198, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.G.; Harper, J.W. Ascending projections of the cerebellar fastigial nucleus to the hippocampus, amygdala, and other temporal lobe sites: Evoked potential and histological studies in monkeys and cats. Exp. Neurol. 1974, 45, 268–287. [Google Scholar] [CrossRef]
- Katafuchi, T.; Koizumi, K. Fastigial inputs to paraventricular neurosecretory neurones studied by extra- and intracellular recordings in rats. J. Physiol. 1990, 421, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, T.J.H.; Sillitoe, R.V.; Voogd, J. Cerebellum and cerebellar connections. In The Rat Nervous System, 4th ed.; Paxinos, G., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 133–205. [Google Scholar]
- Hata, N.; Miura, M. The inhibitory effect of the cerebellar fastigial stimulation on adh secretion. J. Physiol. 1974, 242, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, A.; Sved, A.F.; Reis, D.J. Fastigial stimulation releases vasopressin in amounts that elevate arterial pressure. Am. J. Physiol. 1983, 244, H687–H694. [Google Scholar] [PubMed]
- Xu, F.; Frazier, D.T. Role of the cerebellar deep nuclei in respiratory modulation. Cerebellum 2002, 1, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Nisimaru, N. Cardiovascular modules in the cerebellum. Jpn. J. Physiol. 2004, 54, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.J.; Pascoe, J.P.; Paton, J.F.R.; Spyer, K.M. Cardiovascular and respiratory responses evoked from the posterior cerebellar cortex and fastigial nucleus in the cat. J. Physiol. 1987, 393, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D. The cerebrocerebellar system: Anatomic substrates of the cerebellar contribution to cognition and emotion. Int. Rev. Psychiatry 2001, 13, 247–260. [Google Scholar] [CrossRef]
- Lutherer, L.O.; Lutherer, B.C.; Dormer, K.J.; Janssen, H.F.; Barnes, C.D. Bilateral lesions of the fastigial nucleus prevent the recovery of blood pressure following hypotension induced by hemorrhage or administration of endotoxin. Brain Res. 1983, 269, 251–257. [Google Scholar] [CrossRef]
- Dietrichs, E.; Røste, G.K.; Røste, L.S.; Qvist, H.L.; Haines, D.E. The hypothalamocerebellar projection in the cat: Branching and nuclear termination. Arch. Ital. Biol. 1994, 132, 25–38. [Google Scholar] [PubMed]
- Horn, E.M.; Waldrop, T.G. Oxygen-sensing neurons in the caudal hypothalamus and their role in cardiorespiratory control. Respir. Physiol. 1997, 110, 219–228. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, Z.; Frazier, D.T. Microinjection of acetazolamide into the fastigial nucleus augments respiratory output in the rat. J. Appl. Physiol. 2001, 91, 2342–2350. [Google Scholar] [PubMed]
- Sillitoe, R.V.; Fu, Y.; Watson, C. Cerebellum. In The Mouse Nervous System; Watson, C., Paxinos, G., Puelles, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 360–397. [Google Scholar]
- Achari, N.K.; Downman, C.B. Autonomic effector responses to stimulation of nucleus fastigius. J. Physiol. 1970, 210, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Reis, D.J. Cerebellum: A pressor response elicited from the fastigial nucleus and its efferent pathway in brainstem. Brain Res. 1969, 13, 595–599. [Google Scholar] [CrossRef]
- Giuditta, M.; Ruggiero, D.A.; Del Bo, A. Anatomical basis for the fastigial pressor response. Blood Press. 2003, 12, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Chida, K.; Iadecola, C.; Underwood, M.D.; Reis, D.J. A novel vasodepressor response elicited from the rat cerebellar fastigial nucleus: The fastigial depressor response. Brain Res. 1986, 370, 378–382. [Google Scholar] [CrossRef]
- Chida, K.; Iadecola, C.; Reis, D.J. Global reduction in cerebral blood flow and metabolism elicited from intrinsic neurons of fastigial nucleus. Brain Res. 1989, 500, 177–192. [Google Scholar] [CrossRef]
- Iadecola, C.; Kraig, R.P. Focal elevations in neocortical interstitial k+ produced by stimulation of the fastigial nucleus in rat. Brain Res. 1991, 563, 273–277. [Google Scholar] [CrossRef]
- Heurteaux, C.; Bertaina, V.; Widmann, C.; Lazdunski, M. K+ channel openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid beta-protein precursor genes and neuronal death in rat hippocampus. Proc. Natl. Acad. Sci. USA 1993, 90, 9431–9435. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, T.; Sapienza, S.; Urbano, A. Eeg and behavioural sleep-like effects induced by the fastigial nucleus in unrestrained, unanaesthetized cats. Arch. Ital. Biol. 1968, 106, 61–72. [Google Scholar] [PubMed]
- Golanov, E.V.; Reis, D.J. Vasodilation evoked from medulla and cerebellum is coupled to bursts of cortical eeg activity in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1995, 268, R454–R467. [Google Scholar]
- Hablitz, J.J.; Rea, G. Cerebellar nuclear stimulation in generalized penicillin epilepsy. Brain Res. Bull. 1976, 1, 599–601. [Google Scholar] [CrossRef]
- Golanov, E.V.; Christensen, J.D.; Reis, D.J. Role of potassium channels in the central neurogenic neuroprotection elicited by cerebellar stimulation in rat. Brain Res. 1999, 842, 496–500. [Google Scholar] [CrossRef]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the ca1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [PubMed]
- Kirino, T. Delayed neuronal death. Neuropathology 2000, 20, S95–S97. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Koizumi, S.; Thura, M.; Ihara, H.; Golanov, E. Electrical stimulation of cerebellar fastigial nucleus up-regulates uncoupling protein 4 and stabilizes mitochondrial membrane potential in the cortex. Neurosci. Res. 2011, 71, e406. [Google Scholar] [CrossRef]
- Yamamoto, S.; Golanov, E.V. Stabilizing effect of cerebellar fastigial nucleus stimulation on neuronal mitochondrial membrane potential. FASEB J. 2004, 18, 3756. [Google Scholar]
- Zhou, P.; Qian, L.; Glickstein, S.B.; Golanov, E.V.; Pickel, V.M.; Reis, D.J. Electrical stimulation of cerebellar fastigial nucleus protects rat brain, in vitro, from staurosporine-induced apoptosis. J. Neurochem. 2001, 79, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Racay, P.; Chomova, M.; Tatarkova, Z.; Kaplan, P.; Hatok, J.; Dobrota, D. Ischemia-induced mitochondrial apoptosis is significantly attenuated by ischemic preconditioning. Cell Mol. Neurobiol. 2009, 29, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yokota, H.; Jover, T.; Cappuccio, I.; Calderone, A.; Simionescu, M.; Bennett, M.V.; Zukin, R.S. Ischemic preconditioning: Neuronal survival in the face of caspase-3 activation. J. Neurosci. 2004, 24, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.R.; Saul, I.; Busto, R.; Ginsberg, M.D.; Sick, T.J.; Perez-Pinzon, M.A. Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. J. Cereb. Blood Flow Metab. 2001, 21, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Reis, D.J.; Kobylarz, K.; Yamamoto, S.; Golanov, E.V. Brief electrical stimulation of cerebellar fastigial nucleus conditions long-lasting salvage from focal cerebral ischemia: Conditioned central neurogenic neuroprotection. Brain Res. 1998, 780, 161–165. [Google Scholar] [CrossRef]
- Feng, L.B.; Pang, X.M.; Zhang, L.; Li, J.P.; Huang, L.G.; Su, S.Y.; Zhou, X.; Li, S.H.; Xiang, H.Y.; Chen, C.Y.; et al. Microrna involvement in mechanism of endogenous protection induced by fastigial nucleus stimulation based on deep sequencing and bioinformatics. BMC Med. Genom. 2015, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.M.; Liu, J.L.; Li, J.P.; Huang, L.G.; Zhang, L.; Xiang, H.Y.; Feng, L.B.; Chen, C.Y.; Li, S.H.; Su, S.Y. Fastigial nucleus stimulation regulates neuroprotection via induction of a novel microrna, rno-mir-676–1, in middle cerebral artery occlusion rats. J. Neurochem. 2015, 133, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, Y.; Jiang, Y.; Li, L.; Li, C.; Li, J. Electrical stimulation of cerebellar fastigial nucleus protects against cerebral ischemic injury by ppargamma upregulation. Neurol. Res. 2017, 39, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Dong, W.; Xie, P.; Cheng, P.; Bai, S.; Ren, Y.; Wang, G.; Chen, X.; Cui, C.; Zhuang, Y.; et al. The effect of pre-condition cerebella fastigial nucleus electrical stimulation within and beyond the time window of thrombolytic on ischemic stroke in the rats. PLoS ONE 2015, 10, e0128447. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome proliferator-activated receptor gamma up-regulates the bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J. Biol. Chem. 2007, 282, 37006–37015. [Google Scholar] [CrossRef] [PubMed]
- Stenzel-Poore, M.P.; Stevens, S.L.; Xiong, Z.; Lessov, N.S.; Harrington, C.A.; Mori, M.; Meller, R.; Rosenzweig, H.L.; Tobar, E.; Shaw, T.E.; et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003, 362, 1028–1037. [Google Scholar] [CrossRef]
- Galea, E.; Feinstein, D.L.; Golanov, E.V.; Glickstein, S.B.; Reis, D.J. Reduction of inflammatory reactivity of brain microvessels by stimulation of the cerebellar fastigial nucleus: Role of mad-3 (nfkb inhibitor). J. Cereb. Blood Flow Metab. 1997, 17, 567. [Google Scholar]
- Galea, E.; Glickstein, S.B.; Feinstein, D.L.; Golanov, E.V.; Reis, D.J. Stimulation of cerebellar fastigial nucleus inhibits interleukin-1beta-induced cerebrovascular inflammation. Am. J. Physiol. 1998, 275, H2053–H2063. [Google Scholar] [PubMed]
- Galea, E.; Golanov, E.V.; Feinstein, D.L.; Kobylarz, K.A.; Glickstein, S.B.; Reis, D.J. Cerebellar stimulation reduces inducible nitric oxide synthase expression and protects brain from ischemia. Am. J. Physiol. Heart Circ. Physiol. 1998, 43, H2035–H2045. [Google Scholar]
- Cao, B.B.; Huang, Y.; Lu, J.H.; Xu, F.F.; Qiu, Y.H.; Peng, Y.P. Cerebellar fastigial nuclear gabaergic projections to the hypothalamus modulate immune function. Brain Behav. Immun. 2013, 27, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.B.; Huang, Y.; Jiang, Y.Y.; Qiu, Y.H.; Peng, Y.P. Cerebellar fastigial nuclear glutamatergic neurons regulate immune function via hypothalamic and sympathetic pathways. J. Neuroimmune Pharmacol. 2015, 10, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, M.; Sui, R.B. Cerebellar fastigial nucleus electrical stimulation alleviates depressive-like behaviors in post-stroke depression rat model and potential mechanisms. Cell Physiol. Biochem. 2017, 41, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Huguet, G.; Kadar, E.; Temel, Y.; Lim, L.W. Electrical stimulation normalizes c-fos expression in the deep cerebellar nuclei of depressive-like rats: Implication of antidepressant activity. Cerebellum 2017, 16, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Y.; Wu, C.J.; Zhu, X.F.; Dong, S.X.; Wei, C.J. Survival and migration of transplanted neural stem cells: Can it elevate the efficiency of transplantation by cerebellar fastigial nucleus stimulation? J. Clin. Rehabil. Tissue Eng. Res. 2010, 14, 985–991. [Google Scholar]
- Jin, Y.L.; Xia, Y.P.; Zhu, X.F. Neuronal differentiation of neural stem cells in co-transplanted rats with middle cerebral artery occlusion under electric simulation of cerebellar fastigial nucleus. J. Clin. Rehabil. Tissue Eng. Res. 2007, 11, 4752–4755. [Google Scholar]
- Wei, Y.; Zhang, R. Preventive effect of fastigial nucleus on oxidative damage in rats undergoing acute myocardial infarction. J. Health Sci. 2008, 54, 330–334. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | paCO2 (mmHg) | paO2 (mmHg) | pH | Glucose (mg/dL) | CBF Drop (% from Baseline) |
|---|---|---|---|---|---|
| 1. Naïve, sham 4-VO | 41.0 ± 6.4 | 111.8 ± 15.1 | 7.4 ± 0.1 | 117.1 ± 16.2 | 0 |
| 2. 20 min 4-VO | 39.8 ± 9.0 | 122.6 ± 15.4 | 7.4 ± 0.1 | 116.1 ± 19.3 | 72.1 ± 1.5 |
| 3. IBO + 20 min 4-VO | 38.7 ± 10.6 | 112.3 ± 11.0 | 7.4 ± 0.1 | 127.2 ± 16.5 | 72.3 ± 1.7 |
| 4. Precondition + 4-VO | 47.6 ± 8.0 | 102.6 ± 16.0 | 7.3 ± 0.1 | 127.4 ± 22.5 | 72.5 ± 1.5 |
| 5. IBO ≥ FN+PrCon + 4-VO | 44.6 ± 12.6 | 113.8 ± 17.6 | 7.3 ± 0.1 | 117.7 ± 15.1 | 72.5 ± 1.2 |
| 6. IBO ≥ DN+PrCon + 4-VO | 44.3 ± 10.7 | 110.0 ± 28.4 | 7.3 ± 0.1 | 104.6 ± 12.5 | 73.6 ± 1.2 |
| ANOVA F(5.59) | 1.004, p = 0.424 | 0.969, p = 0.444 | 1.917, p = 0.105 | 1.831, p = 0.121 | 1.067, p = 0.388 excluding sham |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golanov, E.V.; Regnier-Golanov, A.S.; Britz, G.W. Integrity of Cerebellar Fastigial Nucleus Intrinsic Neurons Is Critical for the Global Ischemic Preconditioning. Brain Sci. 2017, 7, 121. https://doi.org/10.3390/brainsci7100121
Golanov EV, Regnier-Golanov AS, Britz GW. Integrity of Cerebellar Fastigial Nucleus Intrinsic Neurons Is Critical for the Global Ischemic Preconditioning. Brain Sciences. 2017; 7(10):121. https://doi.org/10.3390/brainsci7100121
Chicago/Turabian StyleGolanov, Eugene V., Angelique S. Regnier-Golanov, and Gavin W. Britz. 2017. "Integrity of Cerebellar Fastigial Nucleus Intrinsic Neurons Is Critical for the Global Ischemic Preconditioning" Brain Sciences 7, no. 10: 121. https://doi.org/10.3390/brainsci7100121
APA StyleGolanov, E. V., Regnier-Golanov, A. S., & Britz, G. W. (2017). Integrity of Cerebellar Fastigial Nucleus Intrinsic Neurons Is Critical for the Global Ischemic Preconditioning. Brain Sciences, 7(10), 121. https://doi.org/10.3390/brainsci7100121