
Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.2.1. Study Design
2.2.2. Chronic Intermittent Hypoxia
2.3. Kidney Parameters
2.3.1. Renal Histology
2.3.2. Kidney Weight and Urinary Albumin-to-Creatinine Ratio
2.4. Western-Blot Analysis
2.5. Quantification of Cysteine-Related Thiolomic Profile
2.6. Cell Culture
2.7. Quantitative Real-Time PCR
2.8. Data Analysis
2.8.1. Univariate Analysis
2.8.2. Multivariate Analysis
3. Results
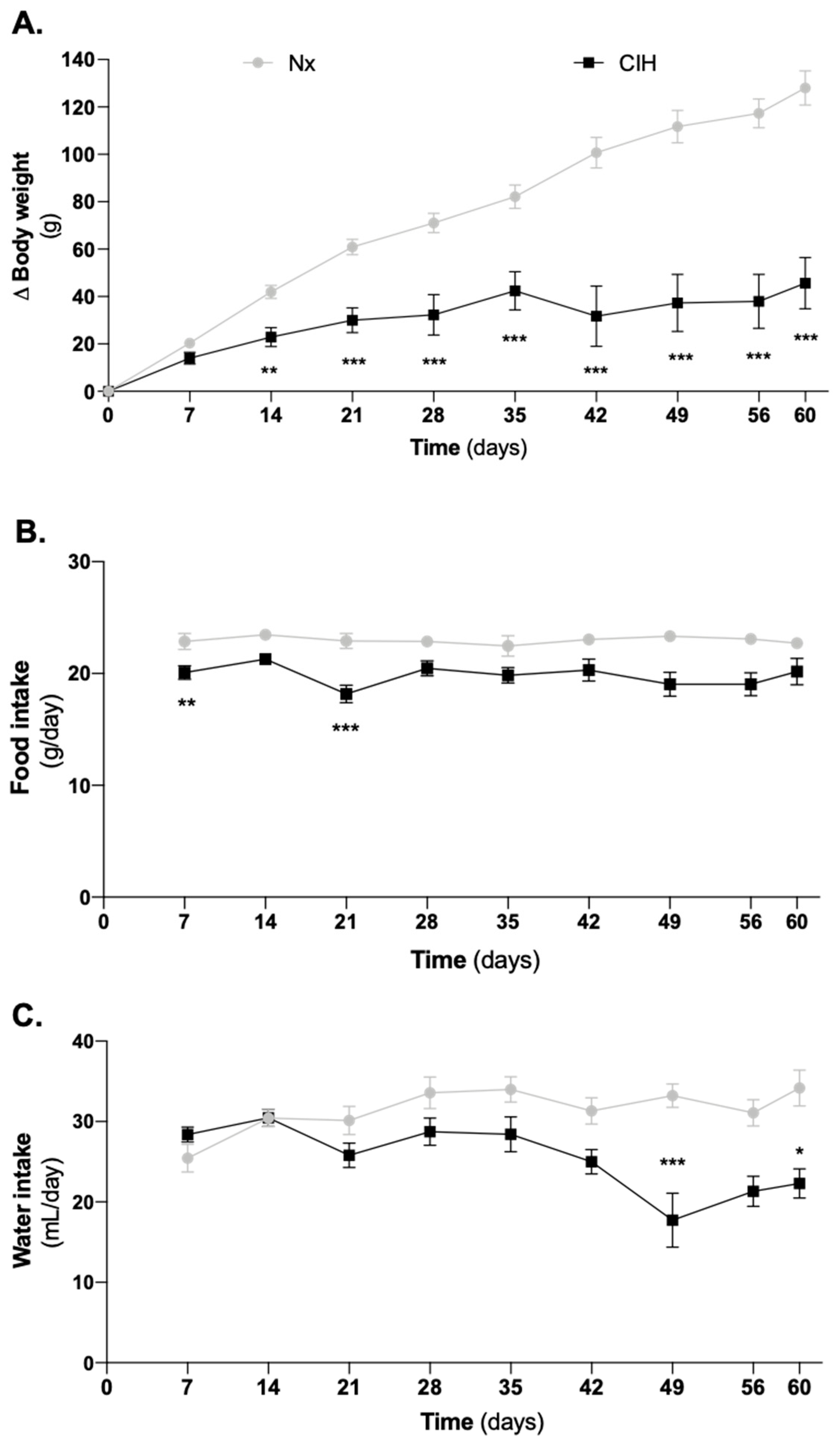
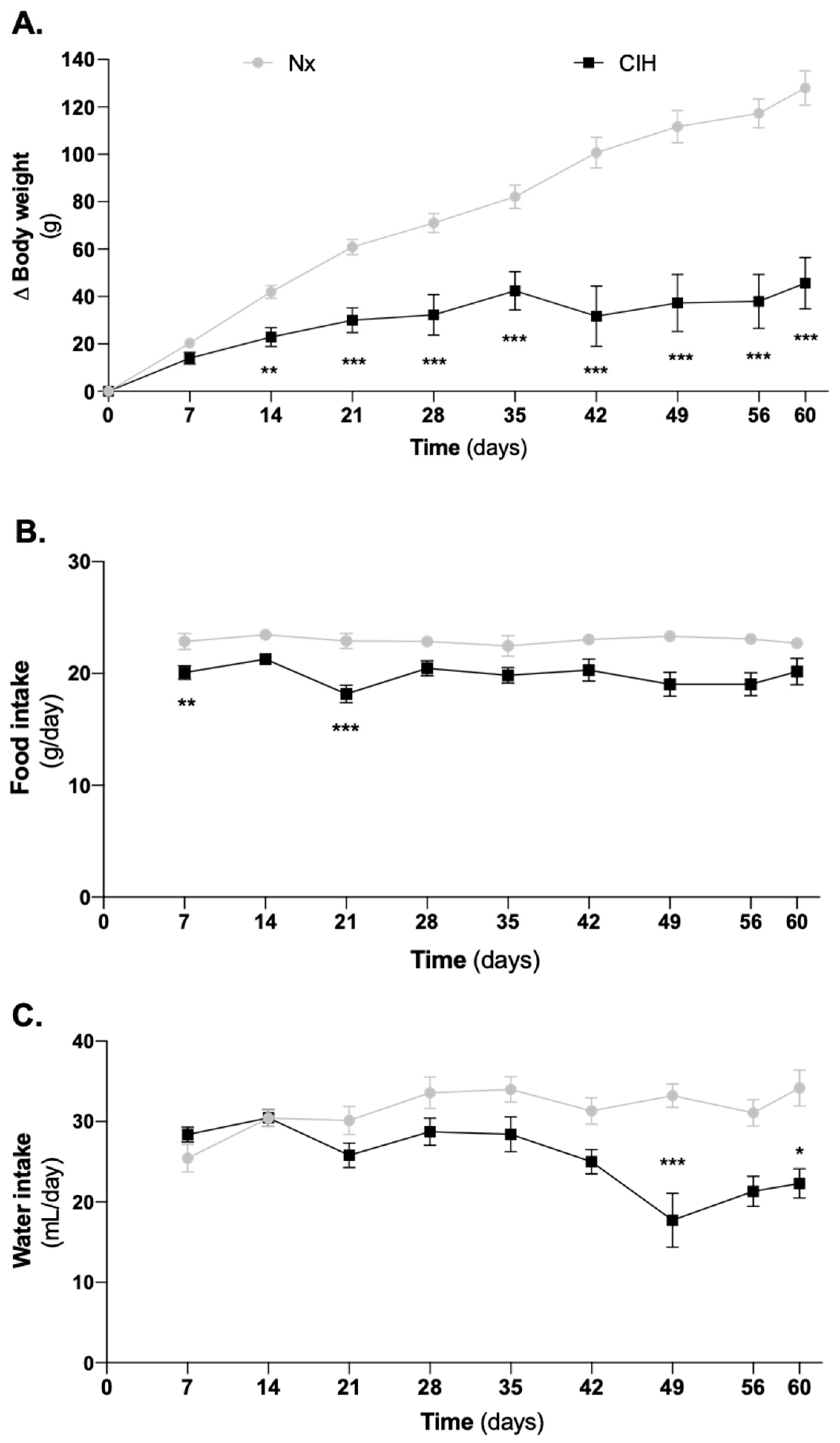
3.1. Impact of Intermittent Hypoxia Chronicity in Animals Body Weight, Food and Water Intake
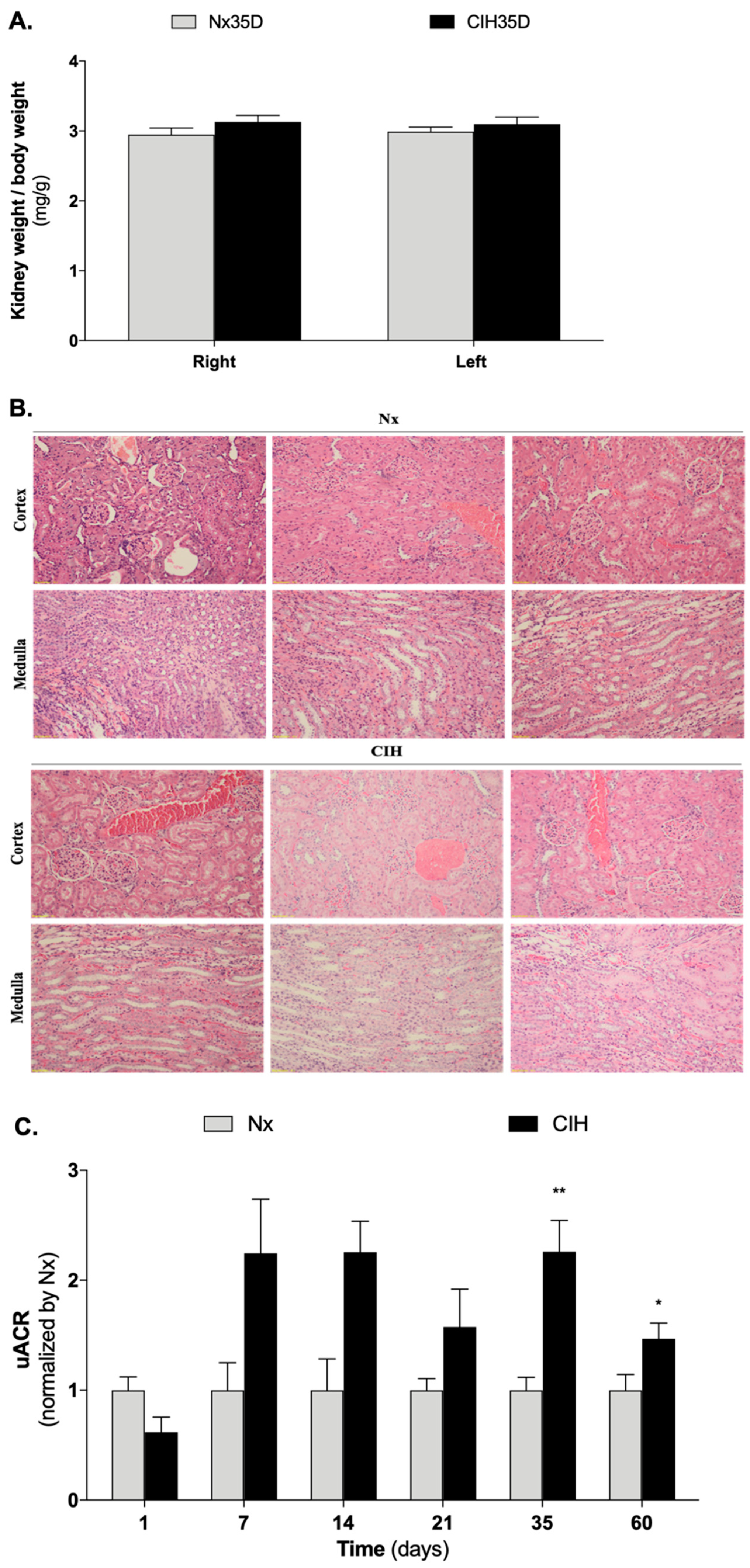
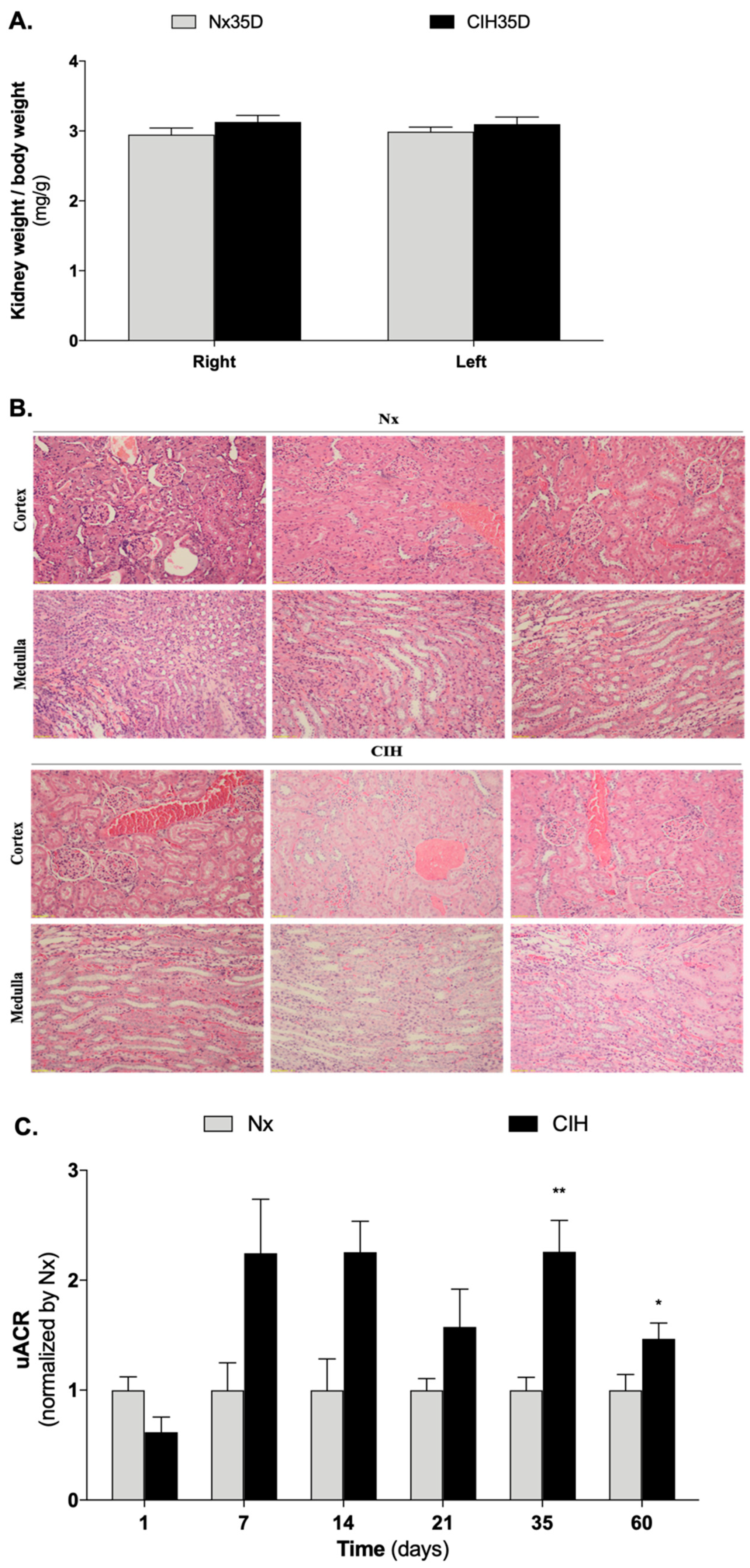
3.2. Impact of Intermittent Hypoxia Chronicity in Kidney Parameters
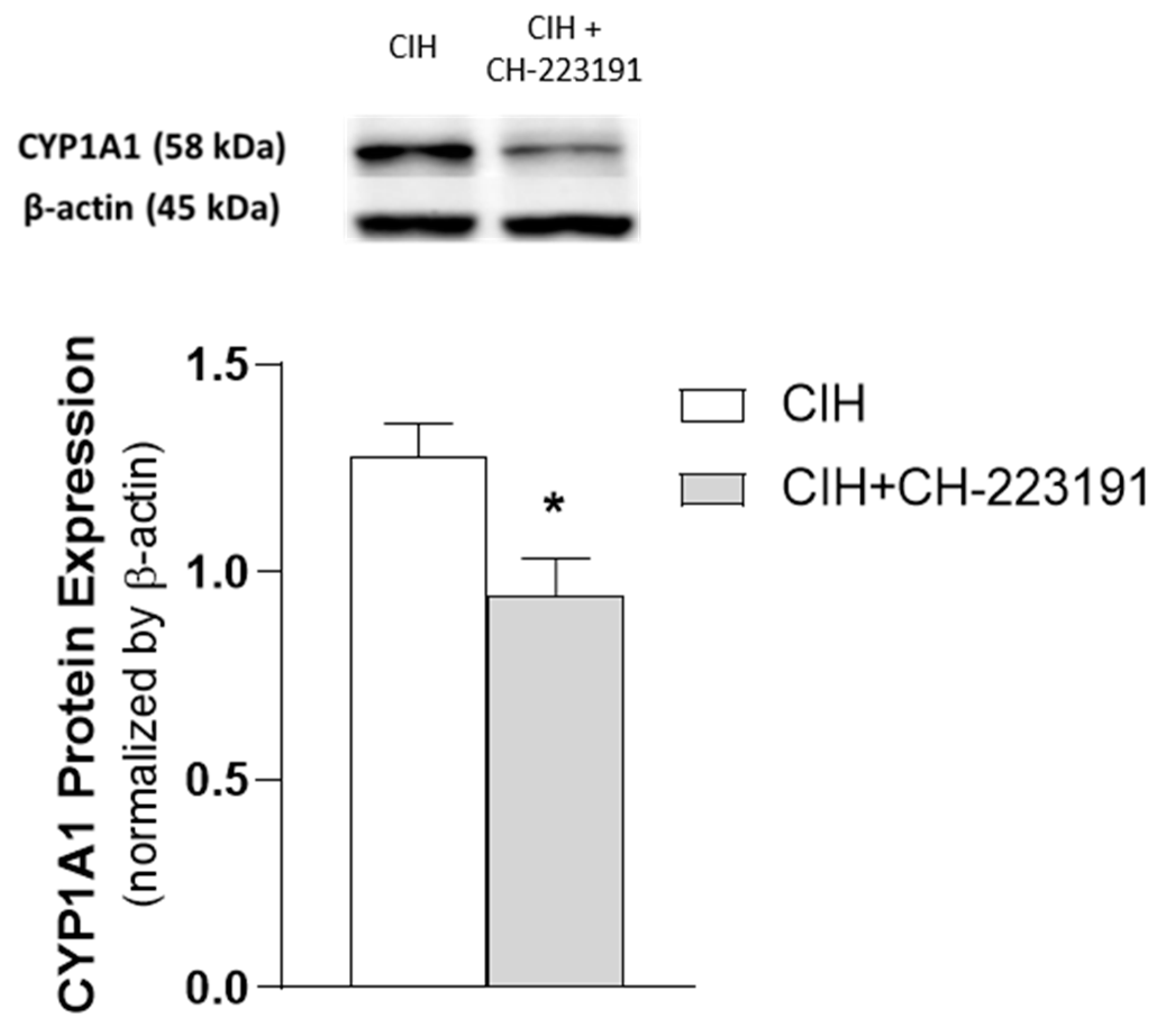
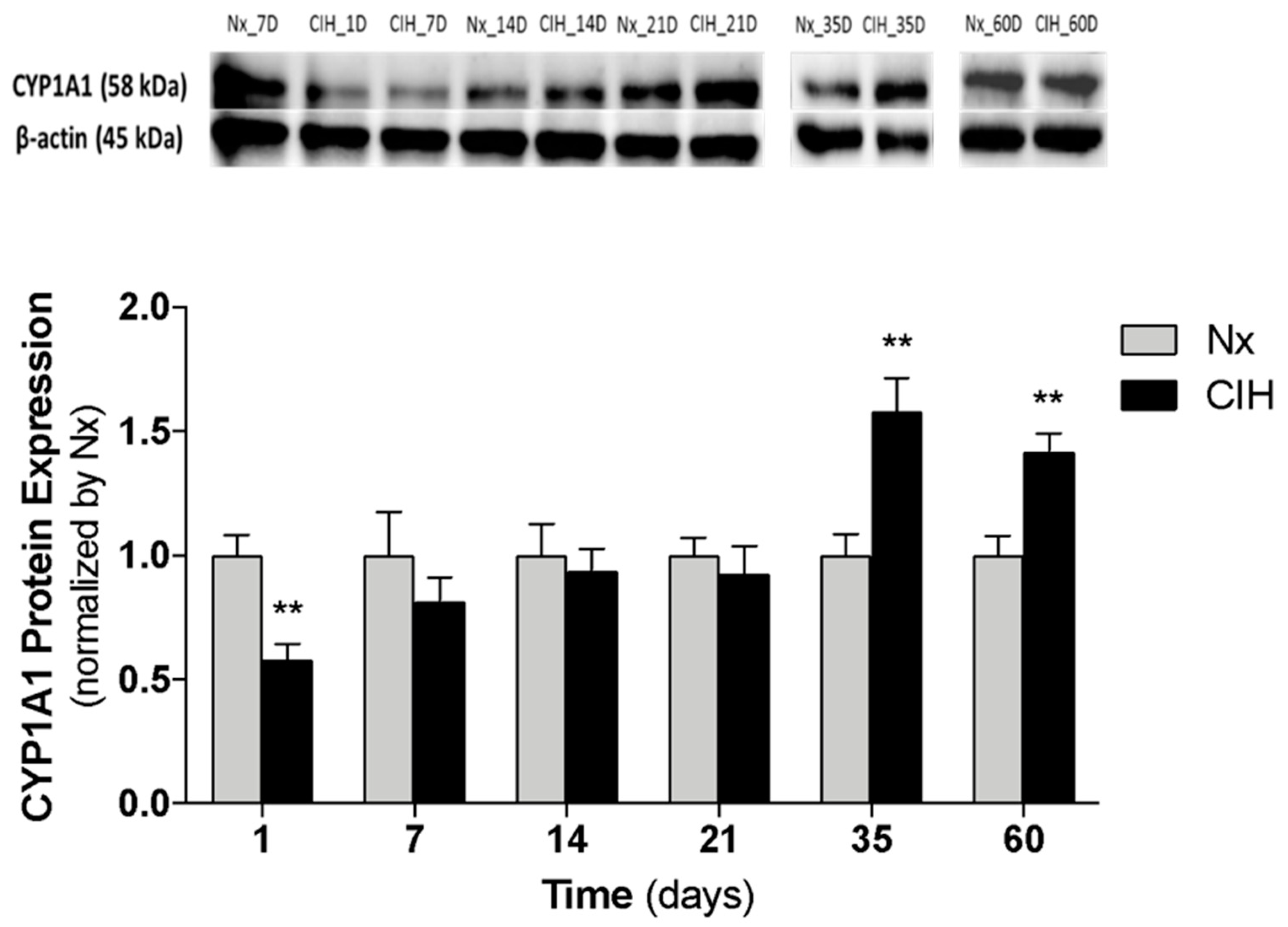
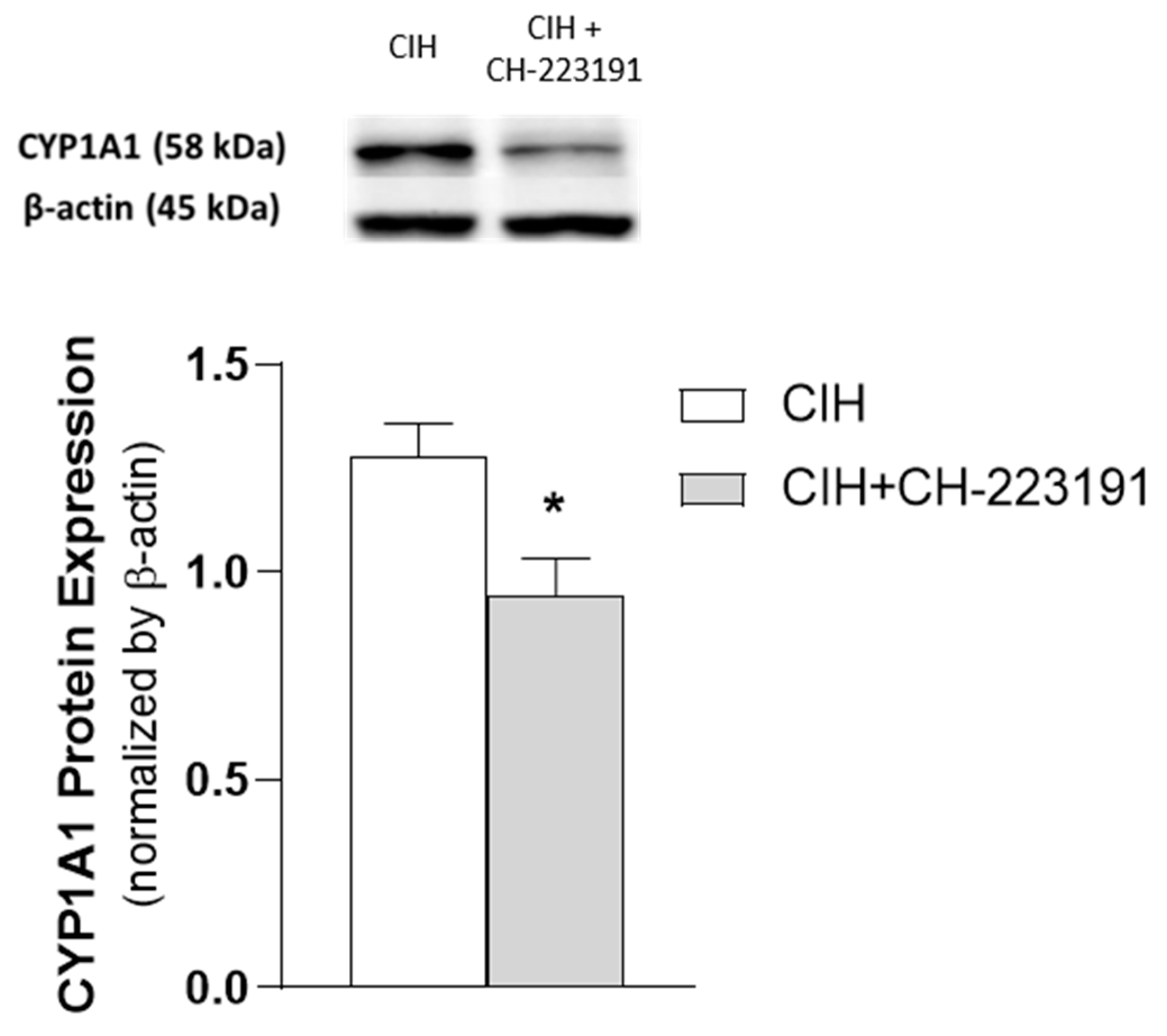
3.3. Impact of the Chronicity of Intermittent Hypoxia in AhR Activation at Kidney Cortex
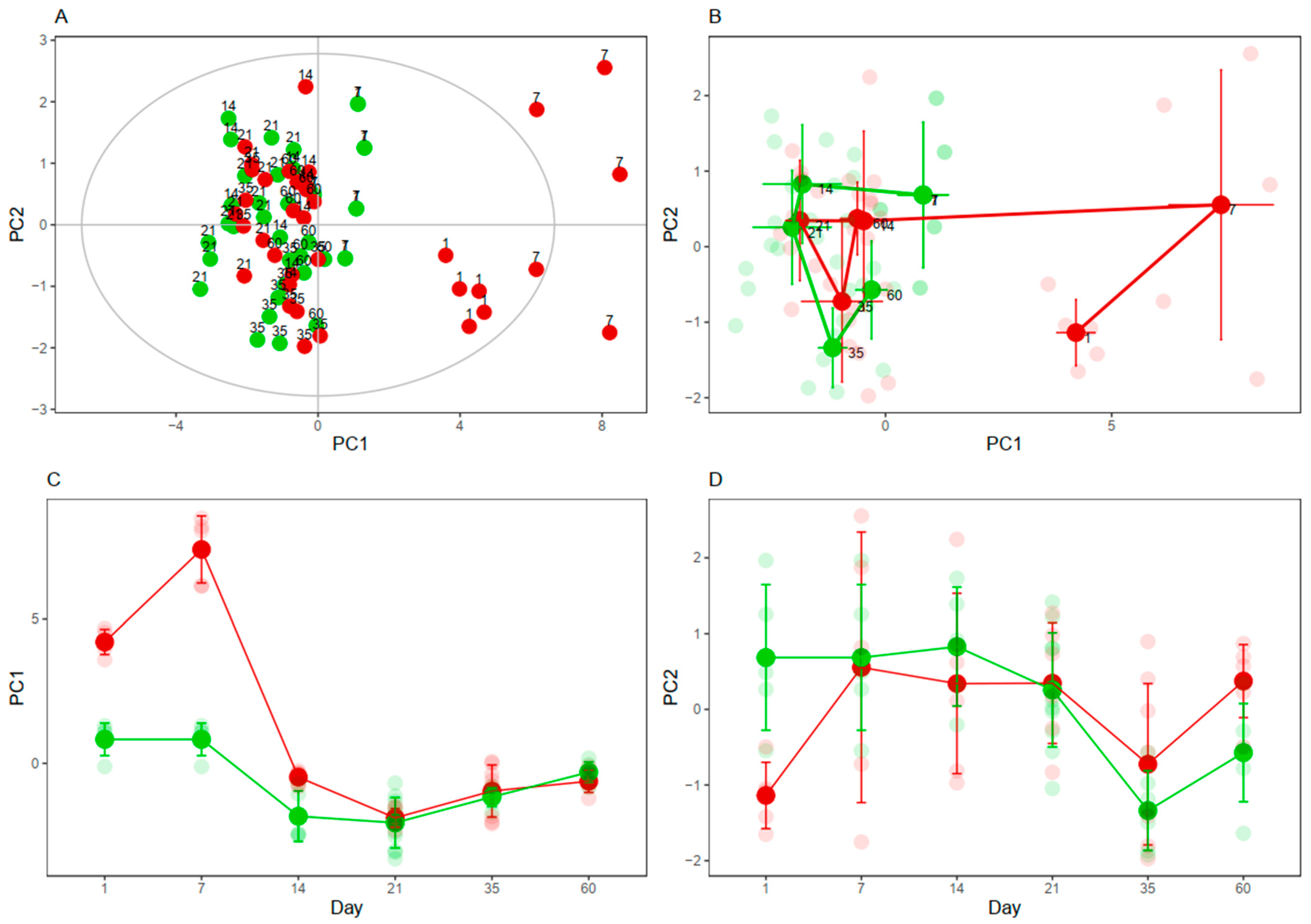
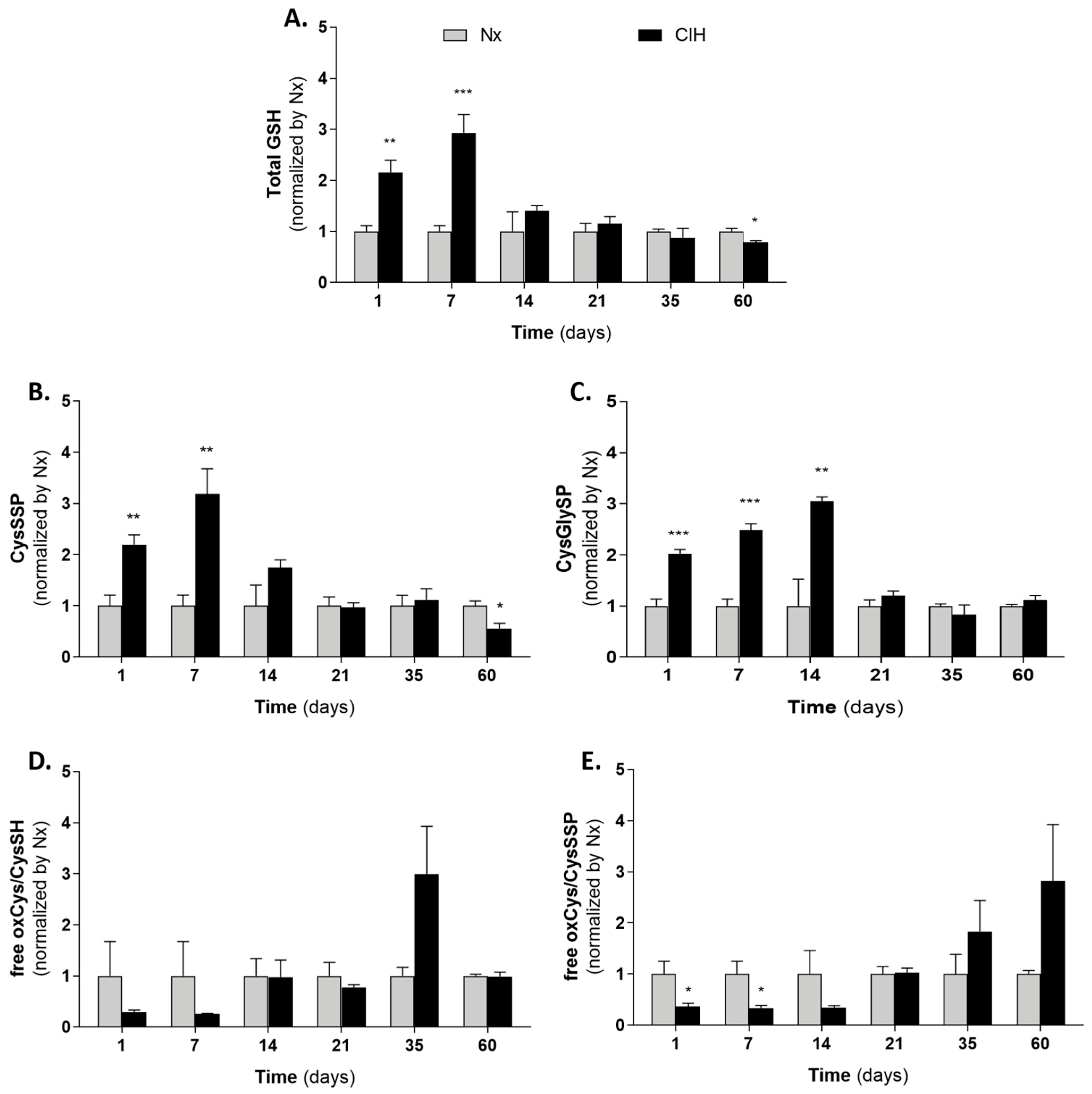
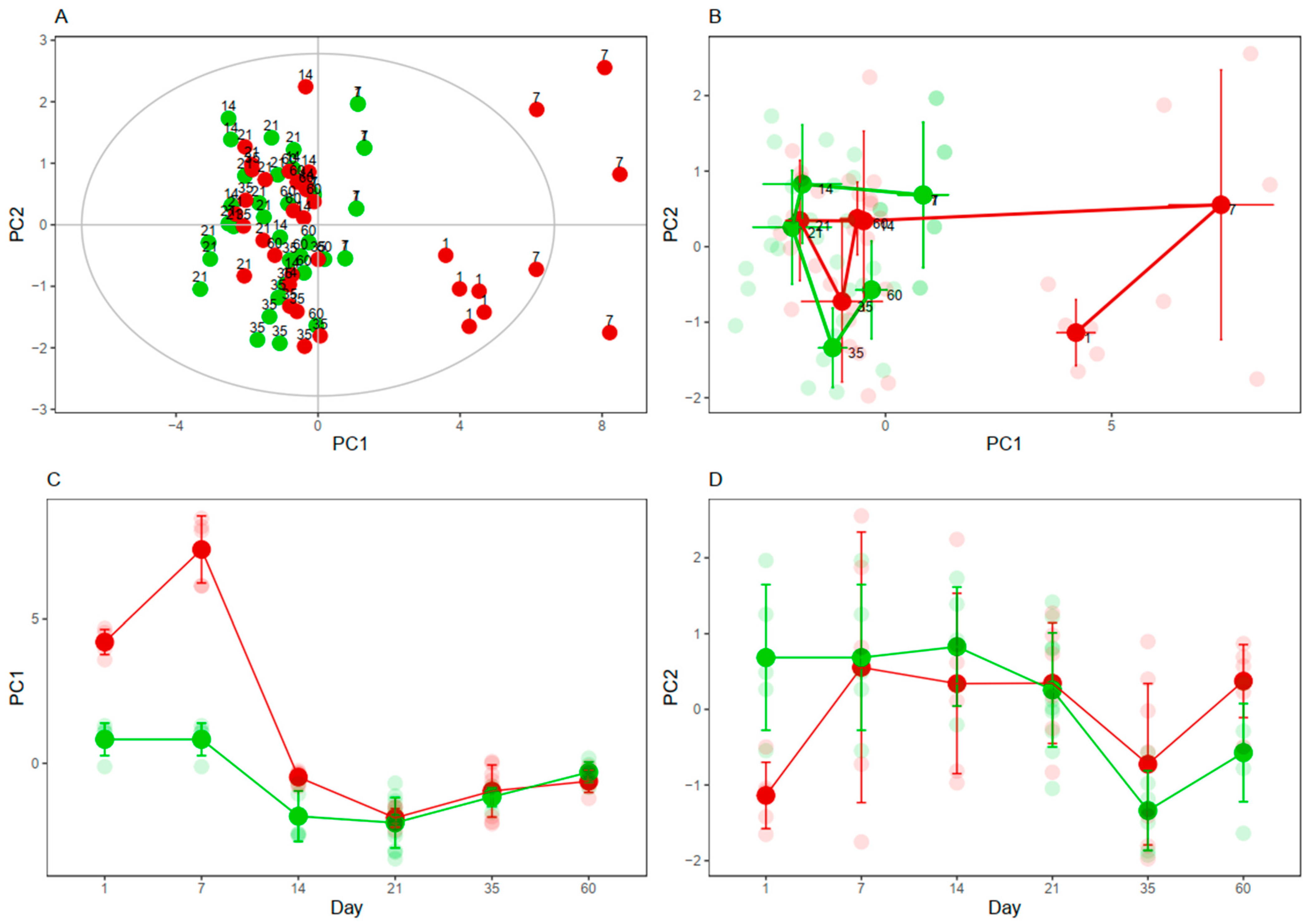
3.4. Cysteine-Related Thiolomic Profile
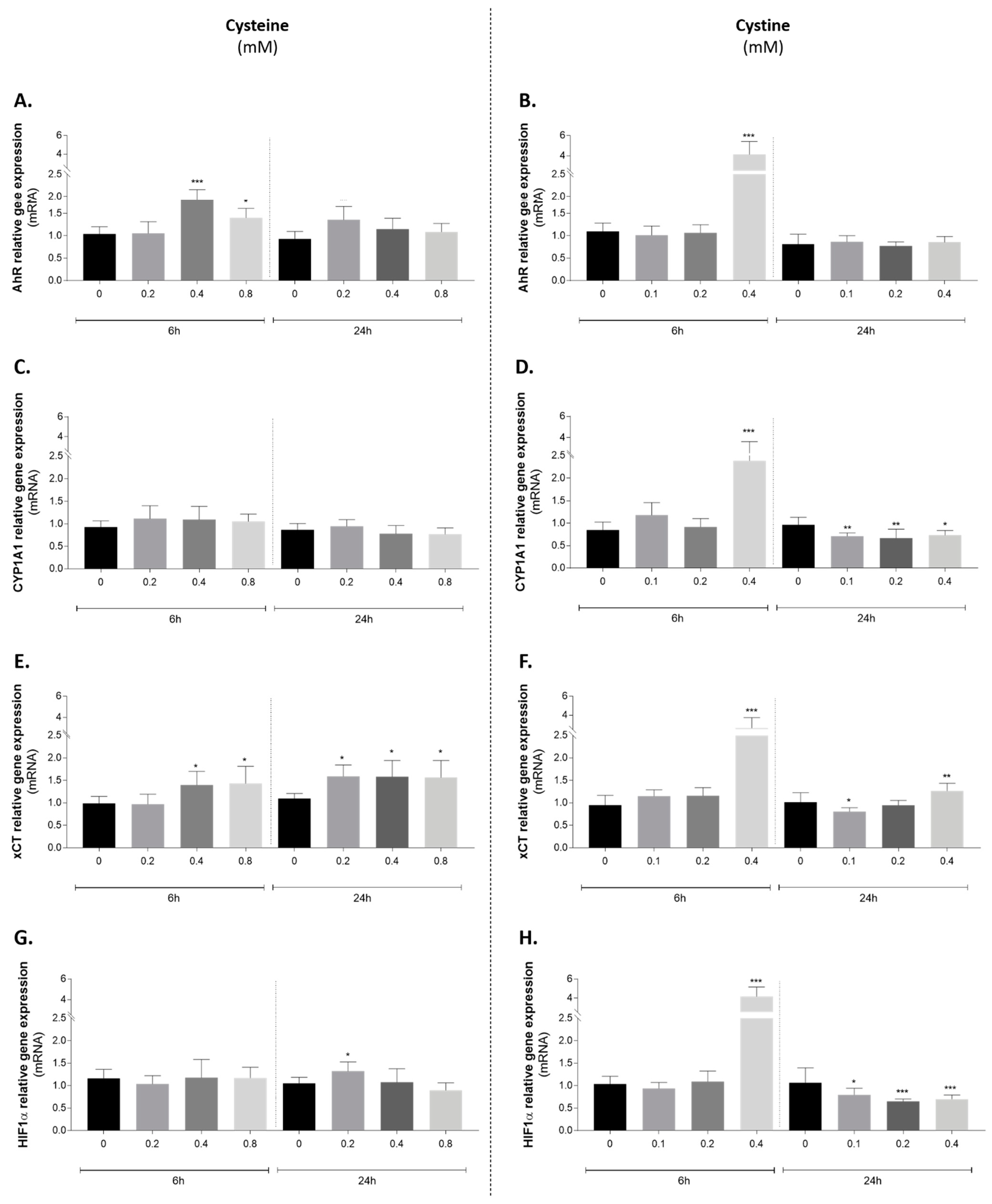
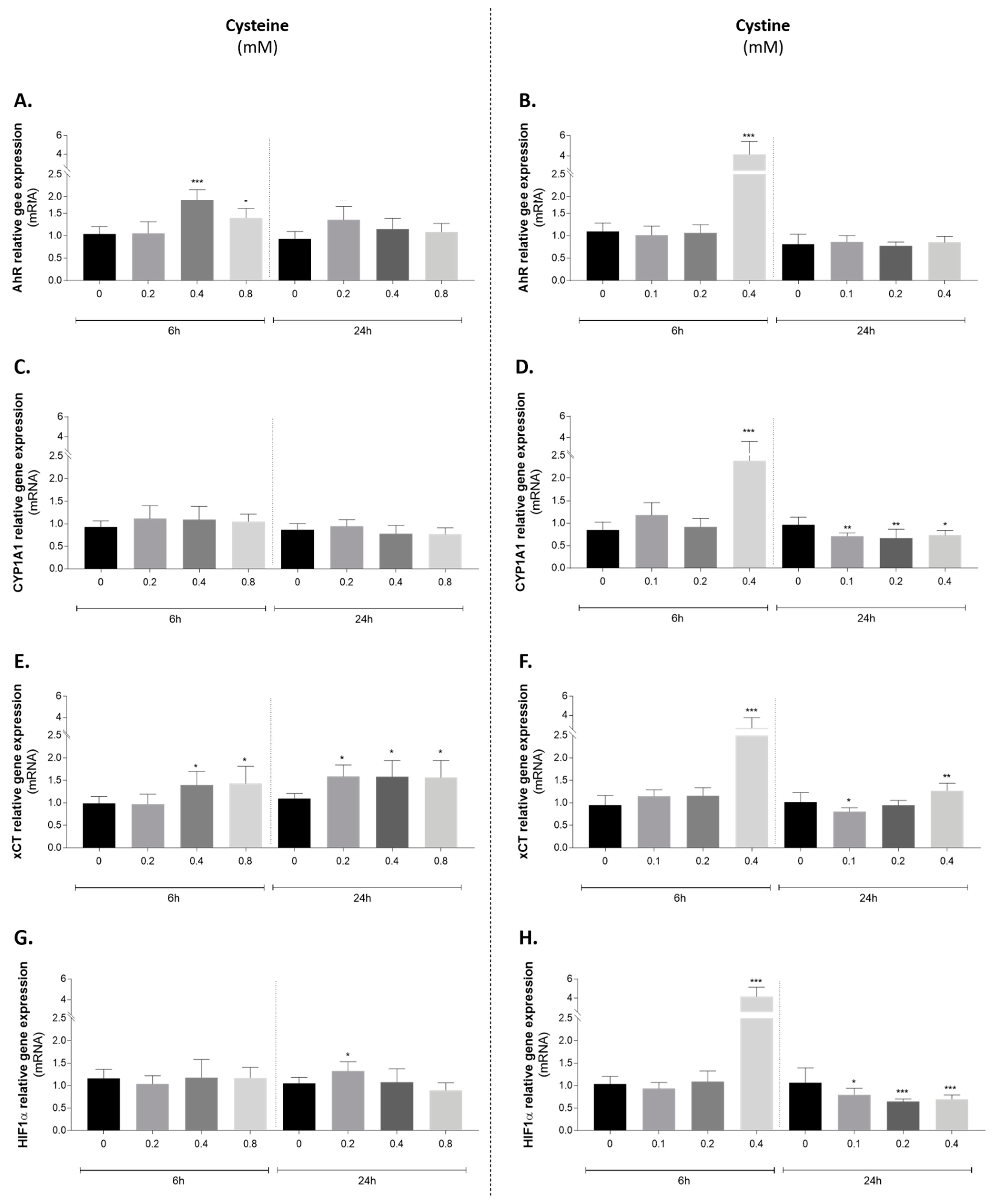
3.5. Effect of Cysteine and Cystine on AhR Canonical Pathway Activation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability Statement
Acknowledgments
Conflicts of Interest
References
- Senaratna, C.V.; Perret, J.L.; Lodge, C.J.; Lowe, A.J.; Campbell, B.E.; Matheson, M.C.; Hamilton, G.S.; Dharmage, S.C. Prevalence of obstructive sleep apnea in the general population: A systematic review. Sleep Med. Rev. 2017, 34, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, C.D. Intermittent hypoxia, cardiovascular disease and obstructive sleep apnoea. J. Thorac. Dis. 2018, 10, S33. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Cai, Y.; Wang, W.; Ji, L.; Dong, Y.; Zhang, X.; Su, M.; Liu, J.; Lu, G.; Zhang, X. Adiponectin protects the kidney against chronic intermittent hypoxia-induced injury through inhibiting endoplasmic reticulum stress. Sleep Breath. 2016, 20, 1069–1074. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yin, X.; Wang, Y.; Tan, Y.; Cai, L.; Wang, B.; Cai, J.; Fu, Y. Intermittent hypoxia-induced renal antioxidants and oxidative damage in male mice: Hormetic dose response. Dose Response 2012, 11, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, J.; Hu, K.; Tang, S.; Zhou, X.; Yu, S.; Xu, L. Angiotensin-(1-7) relieved renal injury induced by chronic intermittent hypoxia in rats by reducing inflammation, oxidative stress and fibrosis. Braz. J. Med. Biol. Res. 2017, 50, e5594. [Google Scholar] [CrossRef] [Green Version]
- Coelho, N.R.; Dias, C.G.; João Correia, M.; Grácio, P.; Serpa, J.; Monteiro, E.C.; Diogo, L.N.; Pereira, S.A. Cysteine Oxidative Dynamics Underlies Hypertension and Kidney Dysfunction Induced by Chronic Intermittent Hypoxia. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; Volume 1071, pp. 83–88. [Google Scholar]
- AlMarabeh, S.; Abdulla, M.H.; O’Halloran, K.D. Is aberrant reno-renal reflex control of blood pressure a contributor to chronic intermittent hypoxia-induced hypertension? Front. Physiol. 2019, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. J. Am. Med. Assoc. 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The physicians’ health study II randomized controlled trial. JAMA—J. Am. Med. Assoc. 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.S.; Ghasemzadeh, N.; Eapen, D.J.; Sher, S.; Arshad, S.; Ko, Y.A.; Veledar, E.; Samady, H.; Zafari, A.M.; Sperling, L.; et al. Novel Biomarker of Oxidative Stress Is Associated with Risk of Death in Patients with Coronary Artery Disease. Circulation 2016, 133, 361–369. [Google Scholar] [CrossRef]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Lorenzini, S.; Milzani, A.; Rossi, R. Age-related influence on thiol, disulfide, and protein-mixed disulfide levels in human plasma. J. Gerontol.—Ser. A Biol. Sci. Med. Sci. 2006, 61, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, R.; Giustarini, D.; Milzani, A.; Dalle-Donne, I. Cysteinylation and homocysteinylation of plasma protein thiols during ageing of healthy human beings. J. Cell. Mol. Med. 2009, 13, 3131–3140. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.V.S.; Laurindo, F.R.M. Implications of plasma thiol redox in disease. Clin. Sci. 2018, 132, 1257–1280. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2020, 124, 862–879. [Google Scholar] [CrossRef]
- Fu, X.; Cate, S.A.; Dominguez, M.; Osborn, W.; Özpolat, T.; Konkle, B.A.; Chen, J.; López, J.A. Cysteine Disulfides (Cys-ss-X) as Sensitive Plasma Biomarkers of Oxidative Stress. Sci. Rep. 2019, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves-Dias, C.; Sequeira, C.O.; Vicente, J.B.; Correia, M.J.; Coelho, N.R.; Morello, J.; Antunes, A.M.M.; Soto, K.; Monteiro, E.C.; Pereira, S.A. A Mechanistic-Based and Non-invasive Approach to Quantify the Capability of Kidney to Detoxify Cysteine-Disulfides. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2021; Volume 1306, pp. 109–120. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Grilo, N.M.; João Correia, M.; Miranda, J.P.; Cipriano, M.; Serpa, J.; Matilde Marques, M.; Monteiro, E.C.; Antunes, A.M.M.; Diogo, L.N.; Pereira, S.A. Unmasking efavirenz neurotoxicity: Time matters to the underlying mechanisms. Eur. J. Pharm. Sci. 2017, 105, 47–54. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [CrossRef]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barouki, R.; Coumoul, X.; Fernandez-Salguero, P. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007, 581, 3608–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebert, D. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog. Lipid Res. 2017, 67, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sorrentino, C.; Denison, M.S.; Kolaja, K.; Fielden, M.R. Induction of Cyp1a1 Is a Nonspecific Biomarker of Aryl Hydrocarbon Receptor Activation: Results of Large Scale Screening of Pharmaceuticals and Toxicants in Vivo and in Vitro. Mol. Pharmacol. 2007, 71, 1475–1486. [Google Scholar] [CrossRef]
- Coelho, N.R.; Tomkiewicz, C.; Correia, M.J.; Gonçalves-Dias, C.; Barouki, R.; Pereira, S.A.; Coumoul, X.; Monteiro, E.C. First evidence of aryl hydrocarbon receptor as a druggable target in hypertension induced by chronic intermittent hypoxia. Pharmacol. Res. 2020, 159, 104869. [Google Scholar] [CrossRef]
- Coelho, N.R.; Matos, C.; Pimpão, A.B.; Correia, M.J.; Sequeira, C.O.; Morello, J.; Pereira, S.A.; Monteiro, E.C. AHR canonical pathway: In vivo findings to support novel antihypertensive strategies. Pharmacol. Res. 2021, 165, 105407. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C. Antioxidant Functions of the Aryl Hydrocarbon Receptor. Stem Cells Int. 2016, 2016, 7943495. [Google Scholar] [CrossRef] [Green Version]
- States, B.; Foreman, J.W.; Segal, S. Cysteine and glutathione levels in developing rat kidney and liver. Pediatr. Res. 1987, 22, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic. Biol. Med. 2011, 50, 495–509. [Google Scholar] [CrossRef] [Green Version]
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased Prevalence of Sleep-Disordered Breathing in Adults. Am. J. Epidemiol. 2013, 177, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Mähler, M.; Berar, M.; Feinstein, R.; Gallagher, A.; Illgen-Wilcke, B.; Pritchett-Corning, K.; Raspa, M. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab. Anim. 2014, 48, 178–192. [Google Scholar] [CrossRef]
- Diogo, L.N.; Pereira, S.A.; Nunes, A.R.; Afonso, R.A.; Santos, A.I.; Monteiro, E.C. Efficacy of carvedilol in reversing hypertension induced by chronic intermittent hypoxia in rats. Eur. J. Pharmacol. 2015, 765, 58–67. [Google Scholar] [CrossRef]
- O’Neill, J.; Jasionek, G.; Drummond, S.E.; Brett, O.; Lucking, E.F.; Abdulla, M.A.; O’Halloran, K.D. Renal cortical oxygen tension is decreased following exposure to long-term but not short-term intermittent hypoxia in the rat. Am. J. Physiol. Ren. Physiol. 2019, 316, 635–645. [Google Scholar] [CrossRef] [PubMed]
- George, S.K.; Dipu, M.T.; Mehra, U.R.; Singh, P.; Verma, A.K.; Ramgaokar, J.S. Improved HPLC method for the simultaneous determination of allantoin, uric acid and creatinine in cattle urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 832, 134–137. [Google Scholar] [CrossRef]
- Przemysław, W.; Piotr, K.; Grażyna, C.; Danuta, K.-P.; Małgorzata, I.; Bernadeta, M.; Małgorzata, S.; Witold, S. Total, free, and protein-bound thiols in plasma of peritoneal dialysis and predialysis patients. Int. Urol. Nephrol. 2011, 43, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Godinho-Pereira, J.; Oudot, C.; Sequeira, C.O.; Macià, A.; Carvalho, F.; Motilva, M.J.; Pereira, S.A.; Matzapetakis, M.; Brenner, C.; et al. Berry fruits modulate kidney dysfunction and urine metabolome in Dahl salt-sensitive rats. Free Radic. Biol. Med. 2020, 154, 119–131. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Martins, F.; Hipólito, A.; Mendes, C.; Sequeira, C.O.; Pires, R.F.; Almeida, A.M.; Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J. The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy. Front. Oncol. 2021, 11, 656229. [Google Scholar] [CrossRef]
- Miura, K.; Ishii, T.; Sugita, Y.; Bannai, S. Cystine uptake and glutathione level in endothelial cells exposed to oxidative stress. Am. J. Physiol.—Cell Physiol. 1992, 262, 50–58. [Google Scholar] [CrossRef]
- Li, H.; Marshall, Z.M.; Whorton, A.R. Stimulation of cystine uptake by nitric oxide: Regulation of endothelial cell glutathione levels. Am. J. Physiol.—Cell Physiol. 1999, 276, C803–C811. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Coelho, F.; Silva, F.; Gouveia-Fernandes, S.; Martins, C.; Lopes, N.; Domingues, G.; Brito, C.; Almeida, A.M.; Pereira, S.A.; Serpa, J. Monocytes as Endothelial Progenitor Cells (EPCs), Another Brick in the Wall to Disentangle Tumor Angiogenesis. Cells 2020, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Keun, H.C.; Ebbels, T.M.D.; Bollard, M.E.; Beckonert, O.; Antti, H.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Geometric trajectory analysis of metabolic responses to toxicity can define treatment specific profiles. Chem. Res. Toxicol. 2004, 17, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Sumayao, R.; Newsholme, P.; McMorrow, T. The role of cystinosin in the intermediary thiol metabolism and redox homeostasis in kidney proximal tubular cells. Antioxidants 2018, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells. Circ. J. 2013, 77, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, C.; Lei, W.; Wang, K.; Zheng, J. Roles of aryl hydrocarbon receptor in endothelial angiogenic responses. Biol. Reprod. 2020, 103, 927–937. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 2005, 111, 2973–2980. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Park, H.; Koval, M.; Orr, M.; Reed, M.; Liang, Y.; Smith, D.; Pohl, J.; Jones, D.P. A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic. Biol. Med. 2010, 48, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Opazo, A.; Mitchell, G.S. Therapeutic potential of intermittent hypoxia: A matter of dose. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2014, 307, 1181–1197. [Google Scholar] [CrossRef] [Green Version]
- Nanduri, J.; Peng, Y.J.; Wang, N.; Khan, S.A.; Semenza, G.L.; Kumar, G.K.; Prabhakar, N.R. Epigenetic regulation of redox state mediates persistent cardiorespiratory abnormalities after long-term intermittent hypoxia. J. Physiol. 2017, 595, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.J.; Baumgärtl, H.; Lübbers, D.; Wilcox, C.S. Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney Int. 2001, 59, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.J.; Blau, J.; Xie, H.; Chabrashvili, T.; Wilcox, C.S. Angiotensin-induced defects in renal oxygenation: Role of oxidative stress. Am. J. Physiol. Circ. Physiol. 2005, 288, H22–H28. [Google Scholar] [CrossRef]
- Palm, F.; Nordquist, L. Renal oxidative stress, oxygenation, and hypertension. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R1229–R1241. [Google Scholar] [CrossRef] [Green Version]
- Adeseun, G.A.; Rosas, S.E. The Impact of Obstructive Sleep Apnea on Chronic Kidney Disease. Curr. Hypertens. Rep. 2010, 12, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Parati, G.; Ochoa, J.E.; Bilo, G.; Mattaliano, P.; Salvi, P.; Kario, K.; Lombardi, C. Obstructive sleep apnea syndrome as a cause of resistant hypertension. Hypertens. Res. 2014, 37, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473. [Google Scholar] [CrossRef]
- Lermant, A.; Murdoch, C.E. Cysteine glutathionylation acts as a redox switch in endothelial cells. Antioxidants 2019, 8, 315. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.-L.; Escobar, J.; Izquierdo-Álvarez, A.; Gil, A.; Pérez, S.; Pereda, J.; Zapico, I.; Vento, M.; Sabater, L.; Marina, A.; et al. Disulfide stress: A novel type of oxidative stress in acute pancreatitis. Free Radic. Biol. Med. 2014, 70, 265–277. [Google Scholar] [CrossRef]
- Parakh, S.; Atkin, J.D. Novel roles for protein disulphide isomerase in disease states: A double edged sword? Front. Cell Dev. Biol. 2015, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Serebrovskaya, T.V.; Manukhina, E.B.; Smith, M.L.; Downey, H.F.; Mallet, R.T. Intermittent hypoxia: Cause of or therapy for systemic hypertension? Exp. Biol. Med. 2008, 233, 627–650. [Google Scholar] [CrossRef]
- Abuyassin, B.; Badran, M.; Ayas, N.T.; Laher, I. Intermittent hypoxia causes histological kidney damage and increases growth factor expression in a mouse model of obstructive sleep apnea. PLoS ONE 2018, 13, e0192084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Wang, J.; Zhang, H.; Shan, Q.; Zhou, B. Renal denervation improves chronic intermittent hypoxia induced hypertension and cardiac fibrosis and balances gut microbiota. Life Sci. 2020, 262, 118500. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, S.; Kong, L.; Chen, J.; Feng, W.; Cai, J.; Miao, L.; Tan, Y. Metallothionein deletion exacerbates intermittent hypoxia-induced renal injury in mice. Toxicol. Lett. 2015, 232, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, W.; Lu, H.; Liu, C.; Yu, B.; Xu, H.; Tang, Y.; Li, S.; Zhou, J.; Shao, C. Soluble Receptor for Advanced Glycation End Product Ameliorates Chronic Intermittent Hypoxia Induced Renal Injury, Inflammation, and Apoptosis via P38/JNK Signaling Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 1015390. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, T.; Dou, Z.J.; Wang, M.T.; Hu, Z.X.; Wang, B. CB1 receptor antagonist rimonabant protects against chronic intermittent hypoxia-induced renal injury in rats. BMC Nephrol. 2021, 22, 153. [Google Scholar] [CrossRef]
- Poonit, N.D.; Zhang, Y.C.; Ye, C.Y.; Cai, H.L.; Yu, C.Y.; Li, T.; Cai, X.H. Chronic intermittent hypoxia exposure induces kidney injury in growing rats. Sleep Breath. 2018, 22, 453–461. [Google Scholar] [CrossRef]
- AlMarabeh, S.; O’Neill, J.; Cavers, J.; Lucking, E.F.; O’Halloran, K.D.; Abdulla, M.H. Chronic intermittent hypoxia impairs diuretic and natriuretic responses to volume expansion in rats with preserved low-pressure baroreflex control of the kidney. Am. J. Physiol.—Ren. Physiol. 2021, 320, F1–F16. [Google Scholar] [CrossRef]
- Faulx, M.D.; Storfer-Isser, A.; Kirchner, H.L.; Jenny, N.S.; Tracy, R.P.; Redline, S. Obstructive sleep apnea is associated with increased urinary albumin excretion. Sleep 2007, 30, 923–929. [Google Scholar] [CrossRef] [Green Version]
- Tsioufis, C.; Thomopoulos, C.; Dimitriadis, K.; Amfilochiou, A.; Tsiachris, D.; Selima, M.; Petras, D.; Kallikazaros, I.; Stefanadis, C. Association of Obstructive Sleep Apnea With Urinary Albumin Excretion in Essential Hypertension: A Cross-sectional Study. Am. J. Kidney Dis. 2008, 52, 285–293. [Google Scholar] [CrossRef]
- Teumer, A.; Li, Y.; Ghasemi, S.; Prins, B.P.; Wuttke, M.; Hermle, T.; Giri, A.; Sieber, K.B.; Qiu, C.; Kirsten, H.; et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat. Commun. 2019, 10, 4130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti, D.; Rao, A.; Gustafsson, S.; Assimes, T.L.; Montgomery, S.B.; Ingelsson, E. Identification of 22 novel loci associated with urinary biomarkers of albumin, sodium, and potassium excretion. Kidney Int. 2019, 95, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, A. Melatonin in humans. N. Engl. J. Med. 1997, 336, 186–195. [Google Scholar] [CrossRef]
- İlhan, S.; Ateşşahin, D.; Ateşşahin, A.; Mutlu, E.; Onat, E.; Şahna, E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced hypertension: The beneficial effects of melatonin. Toxicol. Ind. Health 2015, 31, 298–303. [Google Scholar] [CrossRef]
- Simko, F.; Paulis, L. Melatonin as a potential antihypertensive treatment. J. Pineal Res. 2007, 42, 319–322. [Google Scholar] [CrossRef]
- Borghi, C.; Cicero, A.F.G. Nutraceuticals with a clinically detectable blood pressure-lowering effect: A review of available randomized clinical trials and their meta-analyses. Br. J. Clin. Pharmacol. 2017, 83, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Coelho, F.; Martins, F.; Pereira, S.A.; Serpa, J. Anti-angiogenic therapy: Current challenges and future perspectives. Int. J. Mol. Sci. 2021, 22, 3765. [Google Scholar] [CrossRef]
- Wenzel, P. Monocytes as immune targets in arterial hypertension. Br. J. Pharmacol. 2019, 176, 1966–1977. [Google Scholar] [CrossRef]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Alexander, R.W.; Harrison, D.G.; Quyyumi, A.A. Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension 2008, 52, 80–85. [Google Scholar] [CrossRef]
- Boneberg, R.; Pardun, A.; Hannemann, L.; Hildebrandt, O.; Koehler, U.; Kinscherf, R.; Hildebrandt, W. High Plasma Cystine Levels Are Associated with Blood Pressure and Reversed by CPAP in Patients with Obstructive Sleep Apnea. J. Clin. Med. 2021, 10, 1387. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Juan, S.H.; Lee, J.L.; Ho, P.Y.; Lee, Y.H.; Lee, W. Sen Antiproliferative and antiangiogenic effects of 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, in human umbilical vascular endothelial cells. Eur. J. Pharmacol. 2006, 530, 1–8. [Google Scholar] [CrossRef]
- Kharait, S.; Haddad, D.J.; Springer, M.L. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem. Biophys. Res. Commun. 2011, 409, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Andersson, H.; Garscha, U.; Brittebo, E. Effects of PCB126 and 17β-oestradiol on endothelium-derived vasoactive factors in human endothelial cells. Toxicology 2011, 285, 46–56. [Google Scholar] [CrossRef]
- Kopf, P.G.; Huwe, J.K.; Walker, M.K. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin are associated with increased superoxide. Cardiovasc. Toxicol. 2008, 8, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Kopf, P.G.; Scott, J.A.; Agbor, L.N.; Boberg, J.R.; Elased, K.M.; Huwe, J.K.; Walker, M.K. Cytochrome P4501A1 is required for vascular dysfunction and hypertension induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2010, 117, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl Hydrocarbon Receptor Activation in Chronic Kidney Disease: Role of Uremic Toxins. Nephron 2017, 137, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Burtey, S. Accumulation of protein-bound uremic toxins: The kidney remains the leading culprit in the gut-liver-kidney axis. Kidney Int. 2020, 97, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.-Y.; Yisireyili, M.; Saito, S.; Lee, C.-T.; Adelibieke, Y.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Expression of Mas Receptor via OAT3/AhR/Stat3 Pathway in Proximal Tubular Cells. PLoS ONE 2014, 9, e91517. [Google Scholar] [CrossRef]
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Torano, J.S.; Zaal, E.A.; Berkers, C.R.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 16105–16110. [Google Scholar] [CrossRef] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.Y.; Bolati, W.; Lee, C.T.; Chien, Y.S.; Yisireyili, M.; Saito, S.; Pei, S.N.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Mas Receptor via Aryl Hydrocarbon Receptor/Nuclear Factor-kappa B, and Induces Cell Proliferation and Tissue Factor Expression in Vascular Smooth Muscle Cells. Nephron 2016, 133, 205–212. [Google Scholar] [CrossRef]
- Goncalves-Dias, C.; Morello, J.; Correia, M.J.; Coelho, N.R.; Antunes, A.M.M.; MacEdo, M.P.; Monteiro, E.C.; Soto, K.; Pereira, S.A. Mercapturate pathway in the tubulocentric perspective of diabetic kidney disease. Nephron 2019, 143, 17–23. [Google Scholar] [CrossRef]
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 963. [Google Scholar] [CrossRef]
- Nie, M.; Blankenship, A.L.; Giesy, J.P. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ. Toxicol. Pharmacol. 2001, 10, 17–27. [Google Scholar] [CrossRef]
- Wolff, M.; Jelkmann, W.; Dunst, J.; Depping, R.; Depping, N.R. The Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT/HIF-1β) is Influenced by Hypoxia and Hypoxia-Mimetics. Cell Physiol. Biochem. 2013, 32, 849–858. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Domann, F.E. Regulatory crosstalk and interference between the and hypoxia sensing pathways at the AhR-ARNT-HIF1α signaling node. Chem. Biol. Interact. 2014, 218, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Belaidi, E.; Moulin, S.; Horman, S.; Van Der Zon, G.C.; Viollet, B.; Levy, P.; Bertrand, L.; Pepin, J.L.; Godin-Ribuot, D.; et al. Chronic intermittent hypoxia impairs insulin sensitivity but improves whole-body glucose tolerance by activating skeletal muscle AMPK. Diabetes 2017, 66, 2942–2951. [Google Scholar] [CrossRef] [Green Version]
- Dematteis, M.; Julien, C.; Guillermet, C.; Sturm, N.; Lantuejoul, S.; Mallaret, M.; Lévy, P.; Gozal, E. Intermittent Hypoxia Induces Early Functional Cardiovascular Remodeling in Mice. Am. J. Respir. Crit. Care Med. 2008, 177, 227–235. [Google Scholar] [CrossRef]
- Fenik, V.B.; Singletary, T.; Branconi, J.L.; Davies, R.O.; Kubin, L. Glucoregulatory consequences and cardiorespiratory parameters in rats exposed to chronic-intermittent hypoxia: Effects of the duration of exposure and losartan. Front. Neurol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, A.; Kayali, F.; Zhang, J.; Hirotsu, C.; Wang, Y.; Gozal, D. Metabolic effects of intermittent hypoxia in mice: Steady versus high-frequency applied hypoxia daily during the rest period. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2012, 303, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, A.; Zhang, S.X.L.; Almendros, I.; Wang, Y.; Peris, E.; Qiao, Z.; Gozal, D. Resveratrol attenuates intermittent hypoxia-induced macrophage migration to visceral white adipose tissue and insulin resistance in male mice. Endocrinology 2015, 156, 437–443. [Google Scholar] [CrossRef]
- Xu, C.X.; Wang, C.; Zhang, Z.M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. 2015, 39, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshorbagy, A.K.; Church, C.; Valdivia-Garcia, M.; Smith, A.D.; Refsum, H.; Cox, R. Dietary cystine level affects metabolic rate and glycaemic control in adult mice. J. Nutr. Biochem. 2012, 23, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randerath, W.; Bassetti, C.L.; Bonsignore, M.R.; Farre, R.; Ferini-Strambi, L.; Grote, L.; Hedner, J.; Kohler, M.; Martinez-Garcia, M.A.; Mihaicuta, S.; et al. Challenges and perspectives in obstructive sleep apnoea. Eur. Respir. J. 2018, 52, 1702616. [Google Scholar] [CrossRef]
- Pragyan, P.; Kesharwani, S.S.; Nandekar, P.P.; Rathod, V.; Sangamwar, A.T. Predicting drug metabolism by CYP1A1, CYP1A2, and CYP1B1: Insights from MetaSite, molecular docking and quantum chemical calculations. Mol. Divers. 2014, 18, 865–878. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, M.J.; Pimpão, A.B.; Lopes-Coelho, F.; Sequeira, C.O.; Coelho, N.R.; Gonçalves-Dias, C.; Barouki, R.; Coumoul, X.; Serpa, J.; Morello, J.; et al. Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex. Antioxidants 2021, 10, 1484. https://doi.org/10.3390/antiox10091484
Correia MJ, Pimpão AB, Lopes-Coelho F, Sequeira CO, Coelho NR, Gonçalves-Dias C, Barouki R, Coumoul X, Serpa J, Morello J, et al. Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex. Antioxidants. 2021; 10(9):1484. https://doi.org/10.3390/antiox10091484
Chicago/Turabian StyleCorreia, Maria João, António B. Pimpão, Filipa Lopes-Coelho, Catarina O. Sequeira, Nuno R. Coelho, Clara Gonçalves-Dias, Robert Barouki, Xavier Coumoul, Jacinta Serpa, Judit Morello, and et al. 2021. "Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex" Antioxidants 10, no. 9: 1484. https://doi.org/10.3390/antiox10091484
APA StyleCorreia, M. J., Pimpão, A. B., Lopes-Coelho, F., Sequeira, C. O., Coelho, N. R., Gonçalves-Dias, C., Barouki, R., Coumoul, X., Serpa, J., Morello, J., Monteiro, E. C., & Pereira, S. A. (2021). Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex. Antioxidants, 10(9), 1484. https://doi.org/10.3390/antiox10091484