
Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
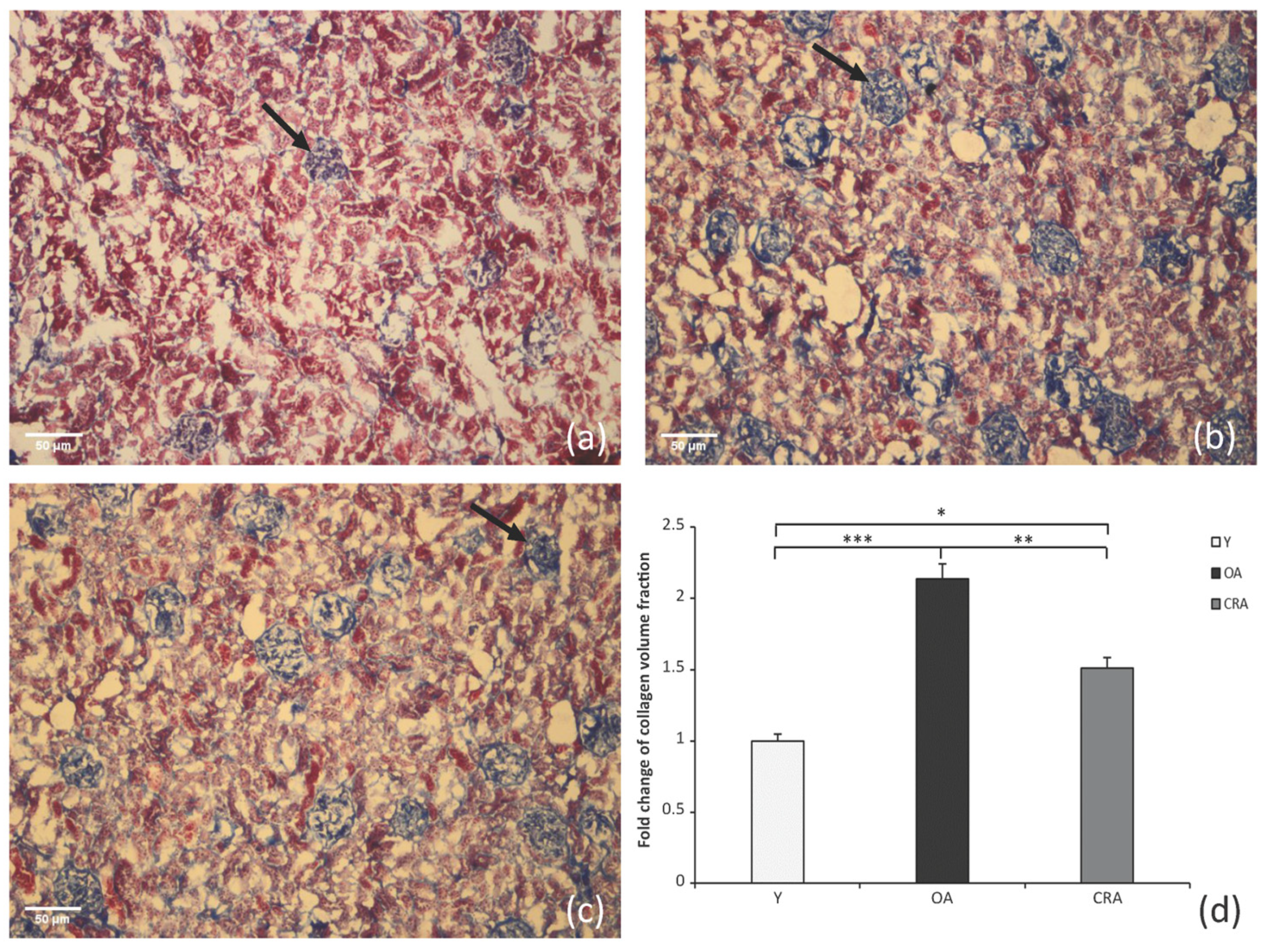
3.1. Caloric Restriction Decreases Interstitial Collagen Deposition
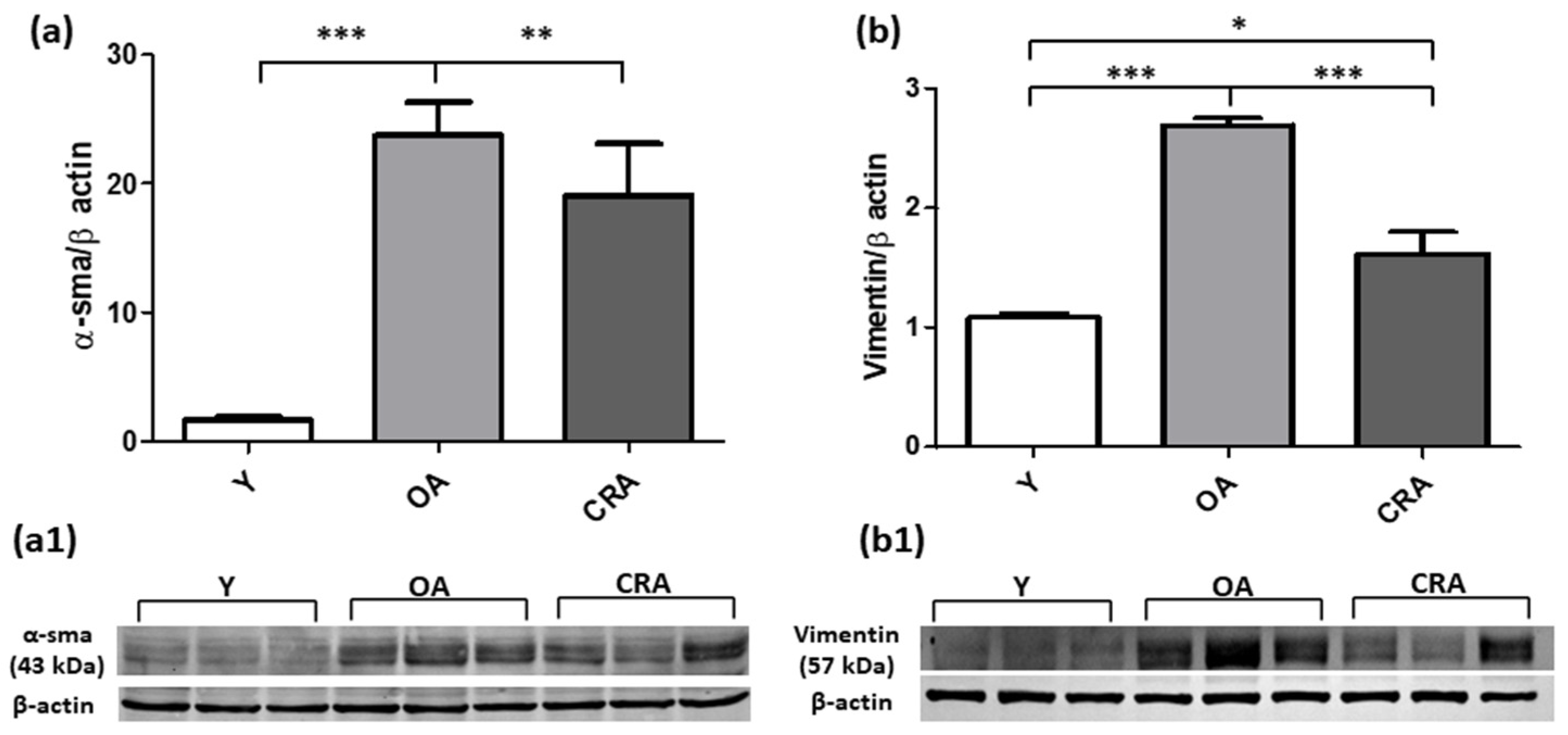
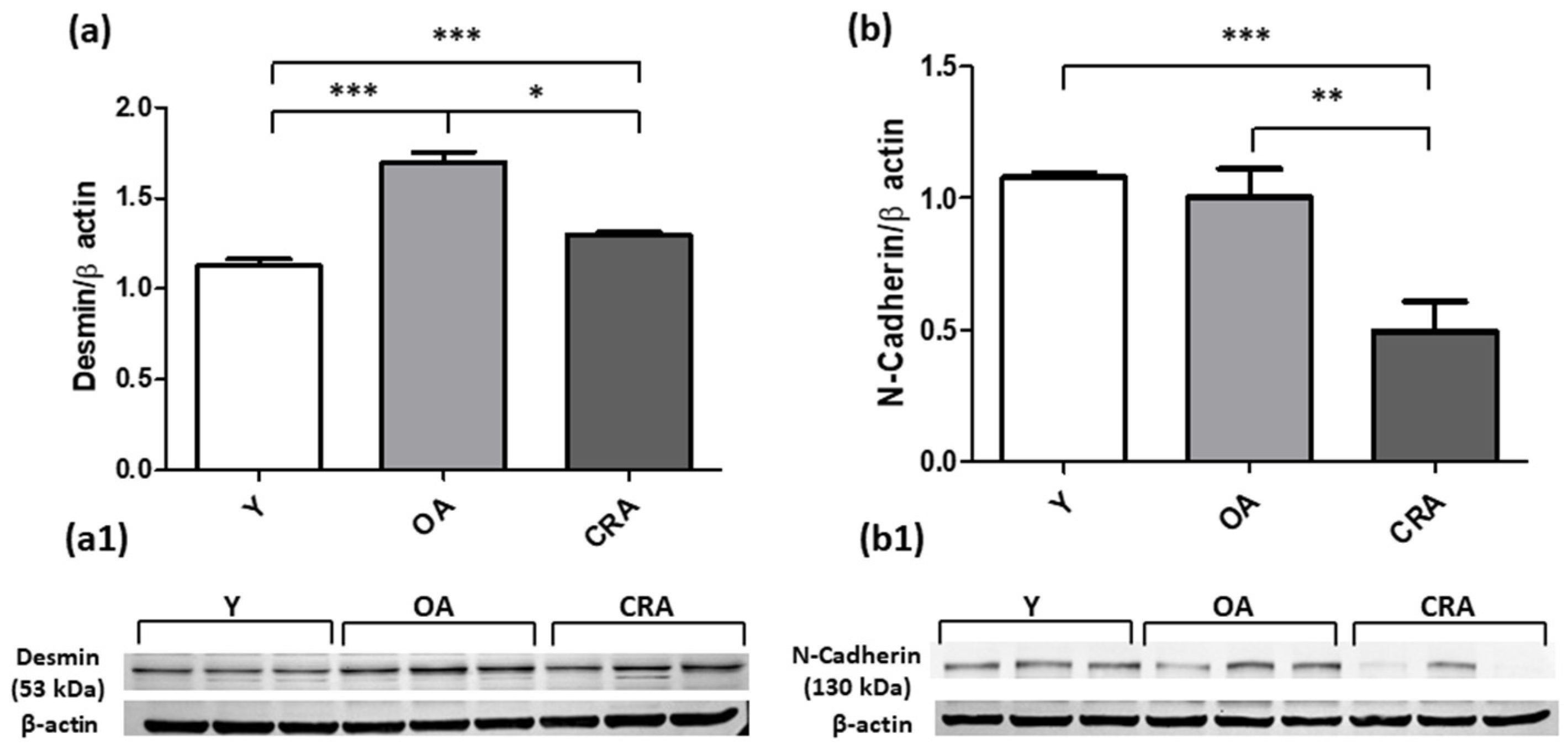
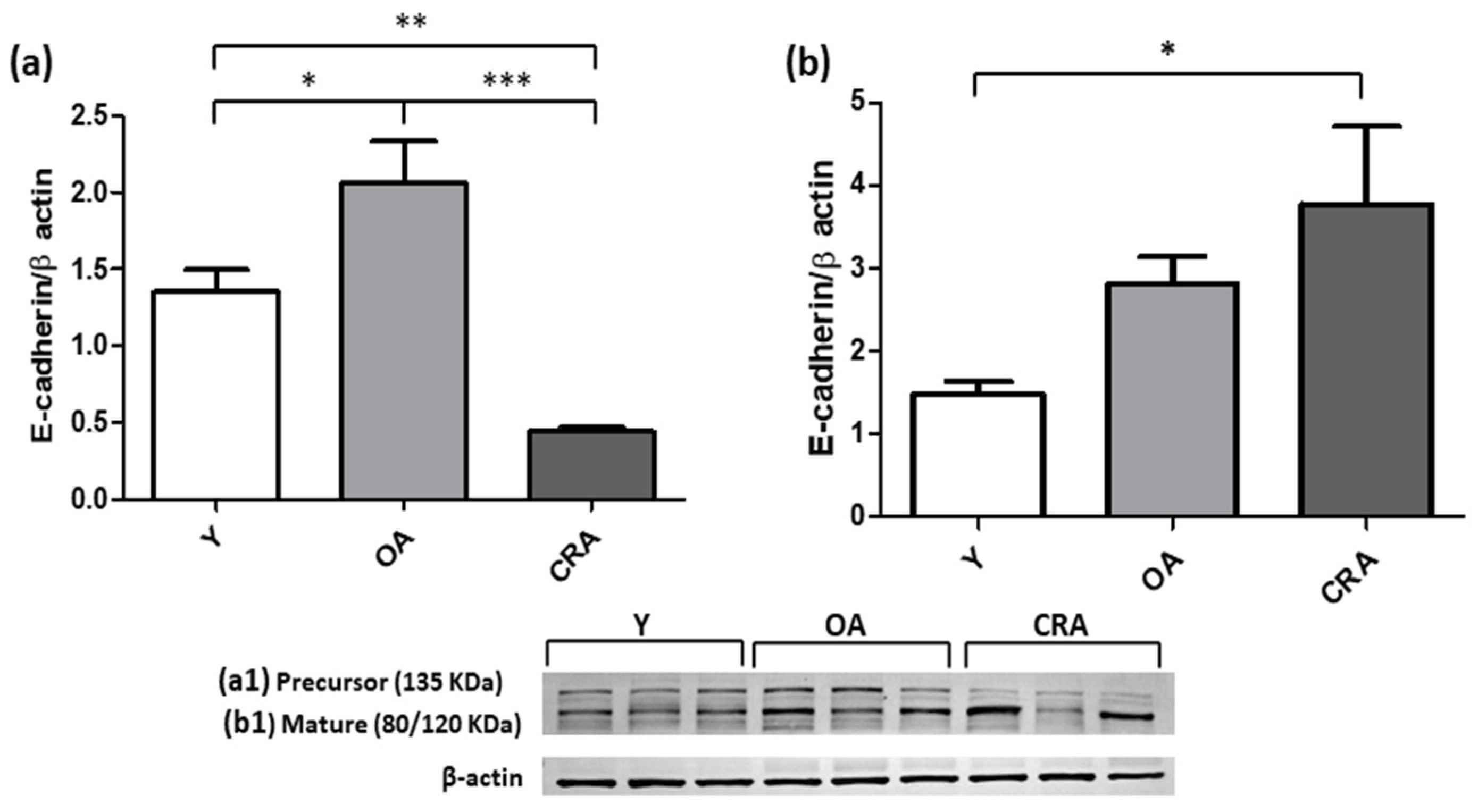
3.2. Caloric Restriction Downregulates Mesenchymal Markers and Restores E-Cadherin Expression
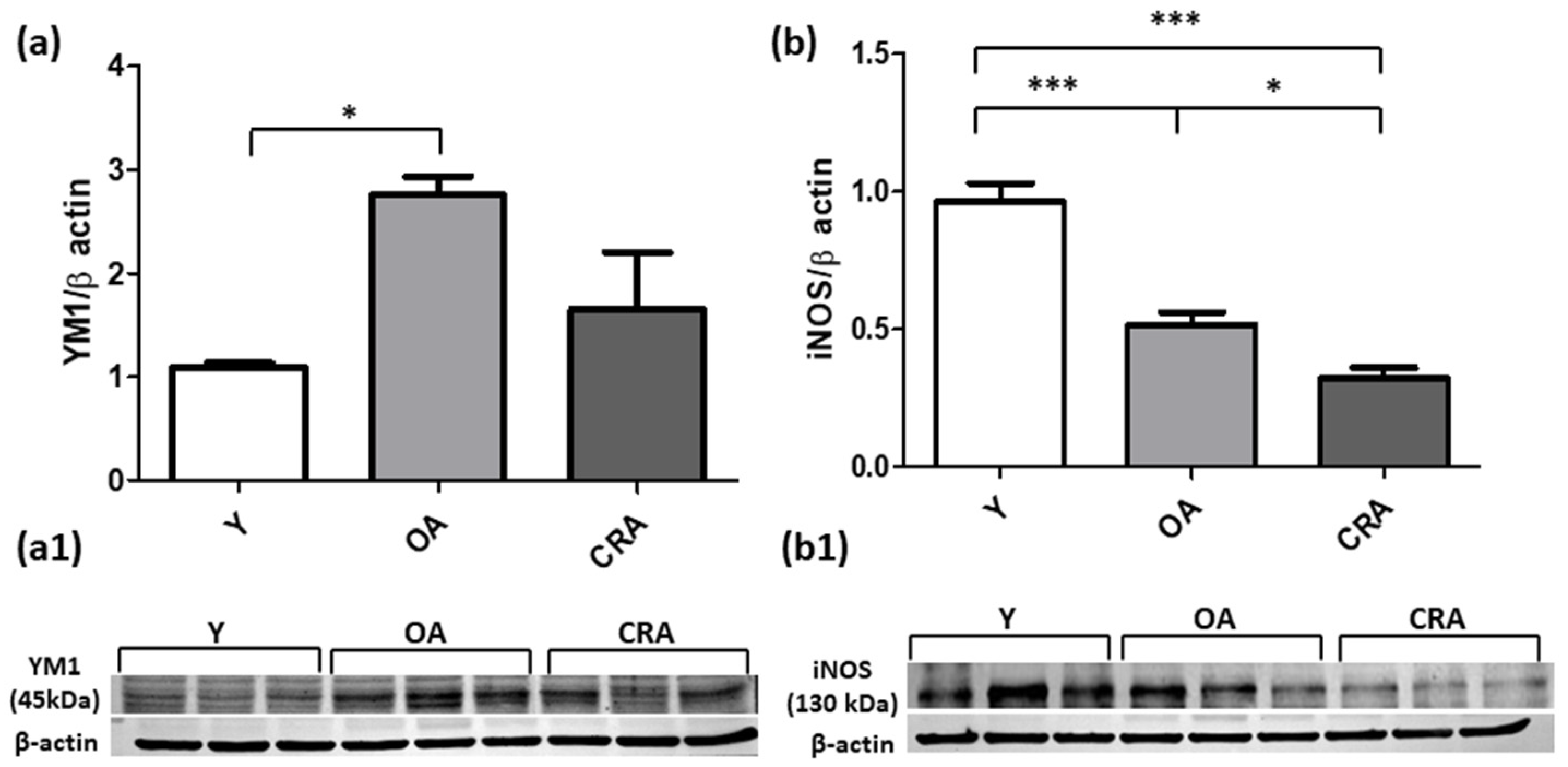
3.3. Caloric Restriction Affects Renal Inflammation
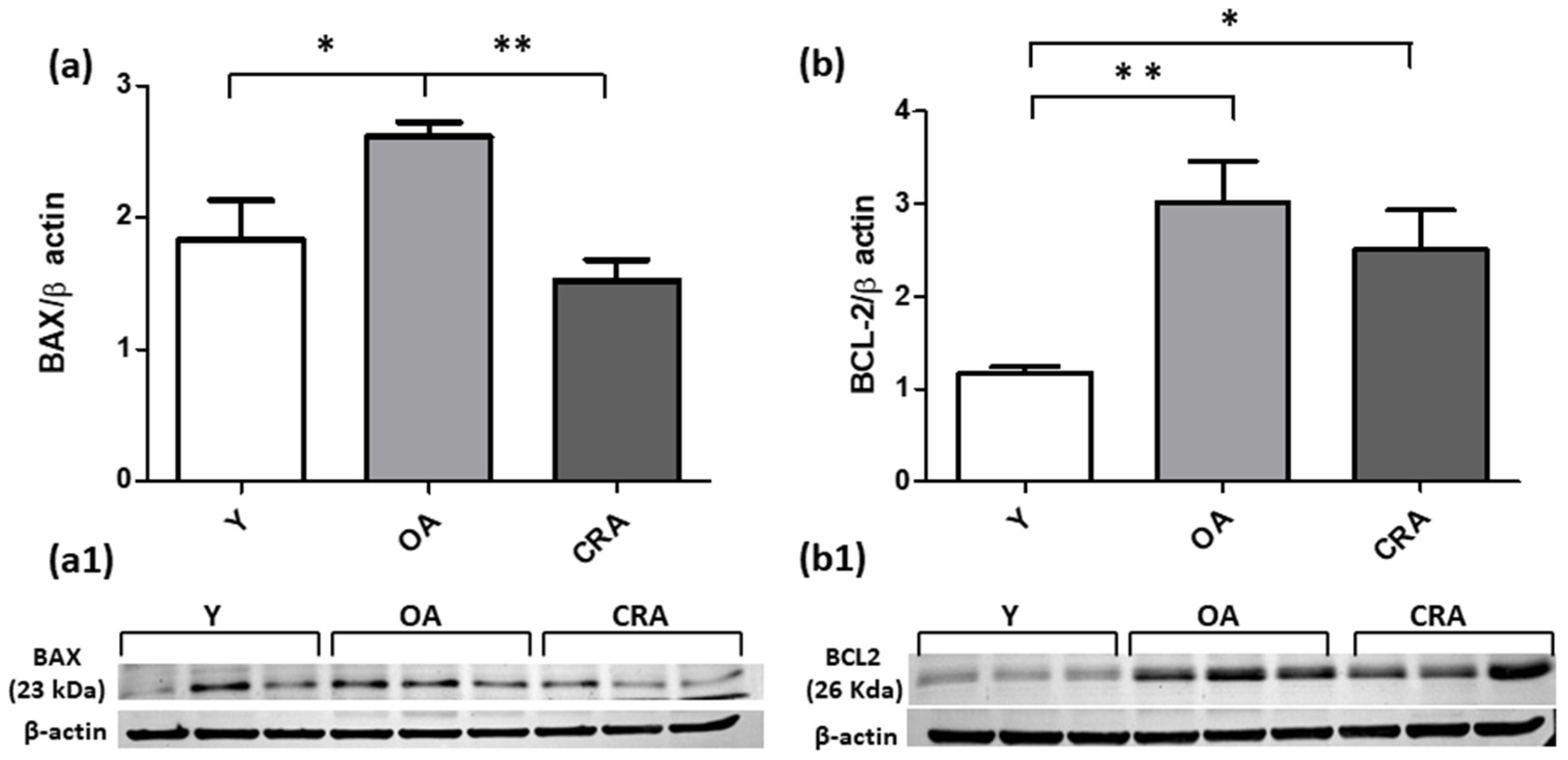
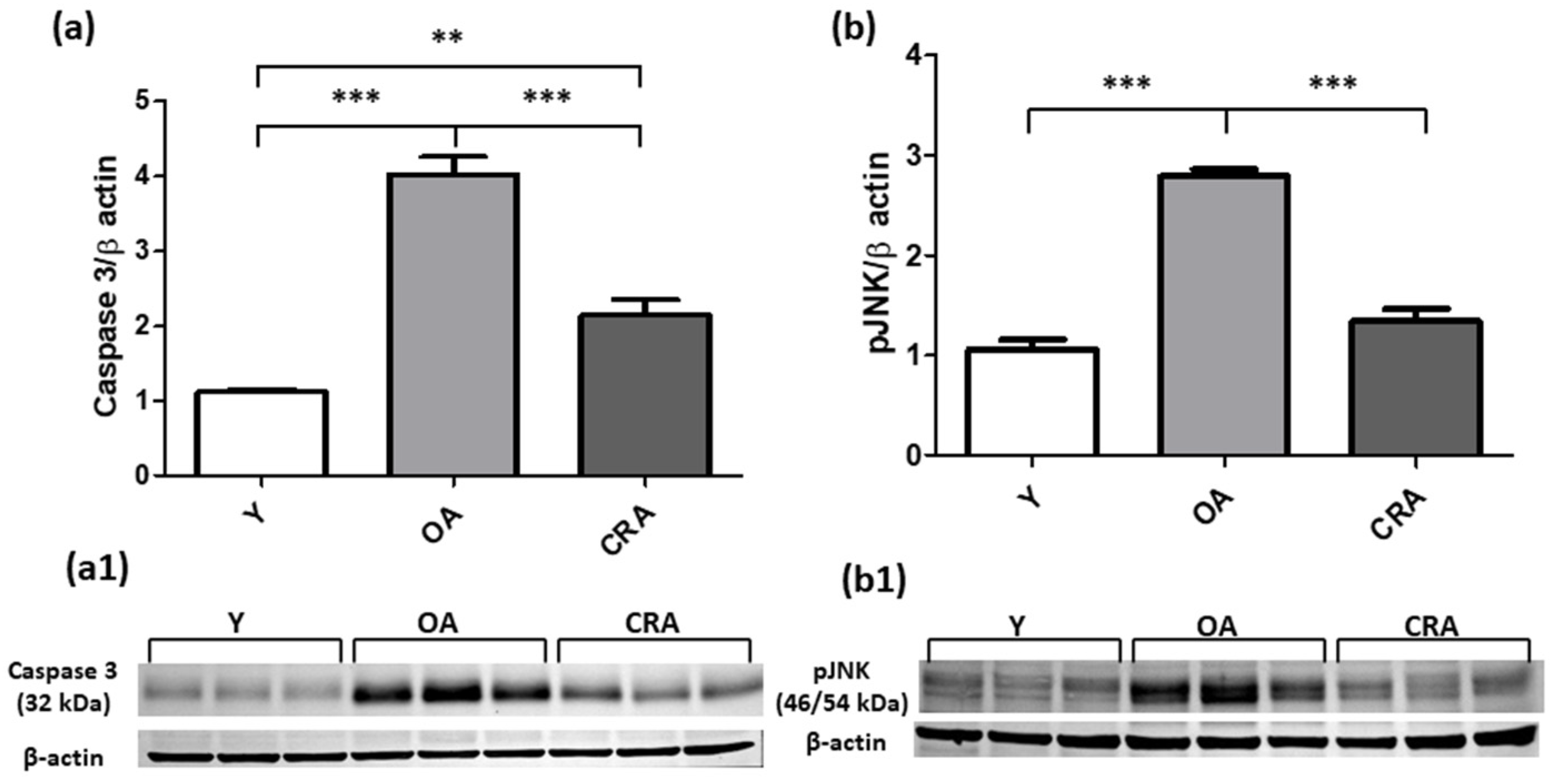
3.4. Caloric Restriction Protects from Renal Apoptotic Injury and Promotes Cell Division
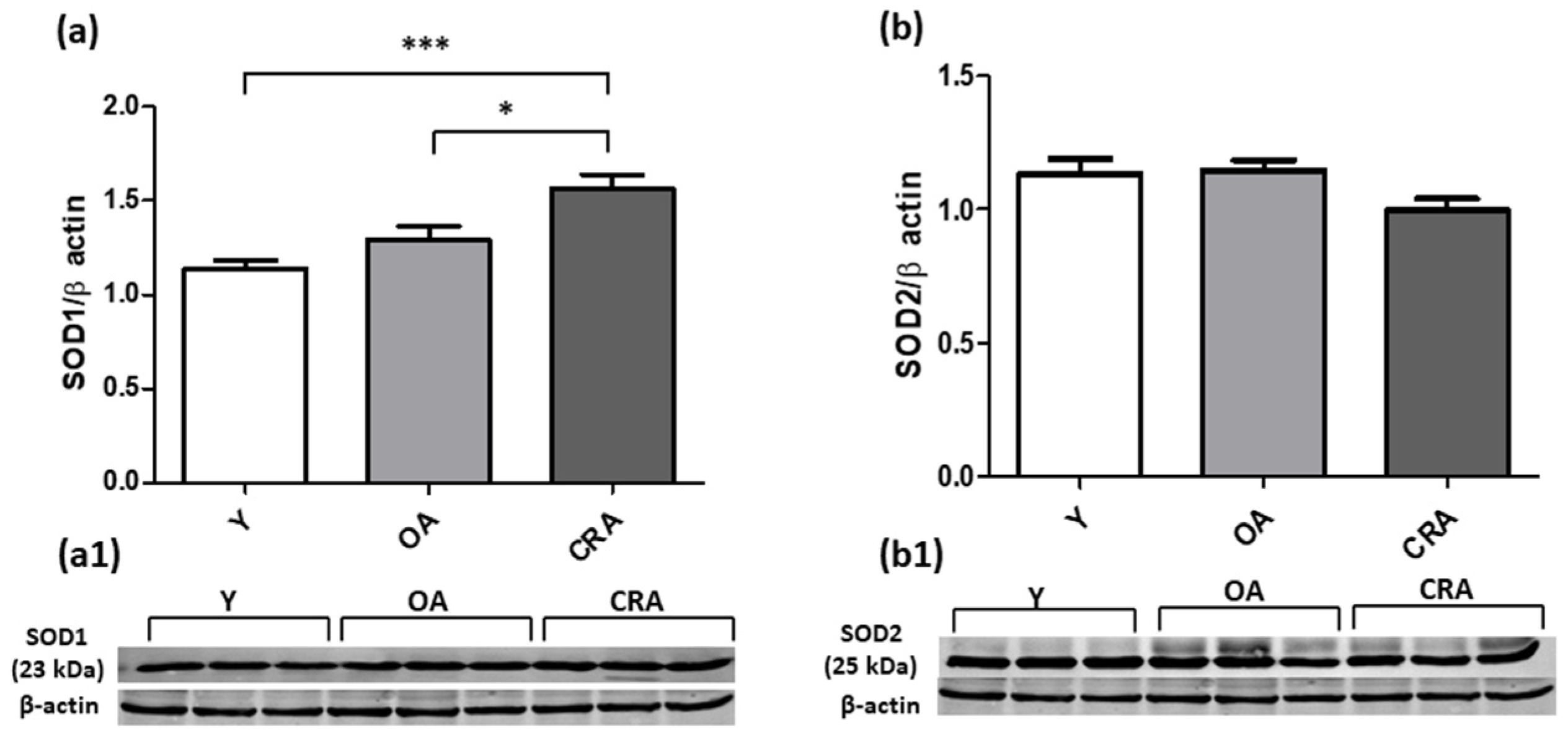
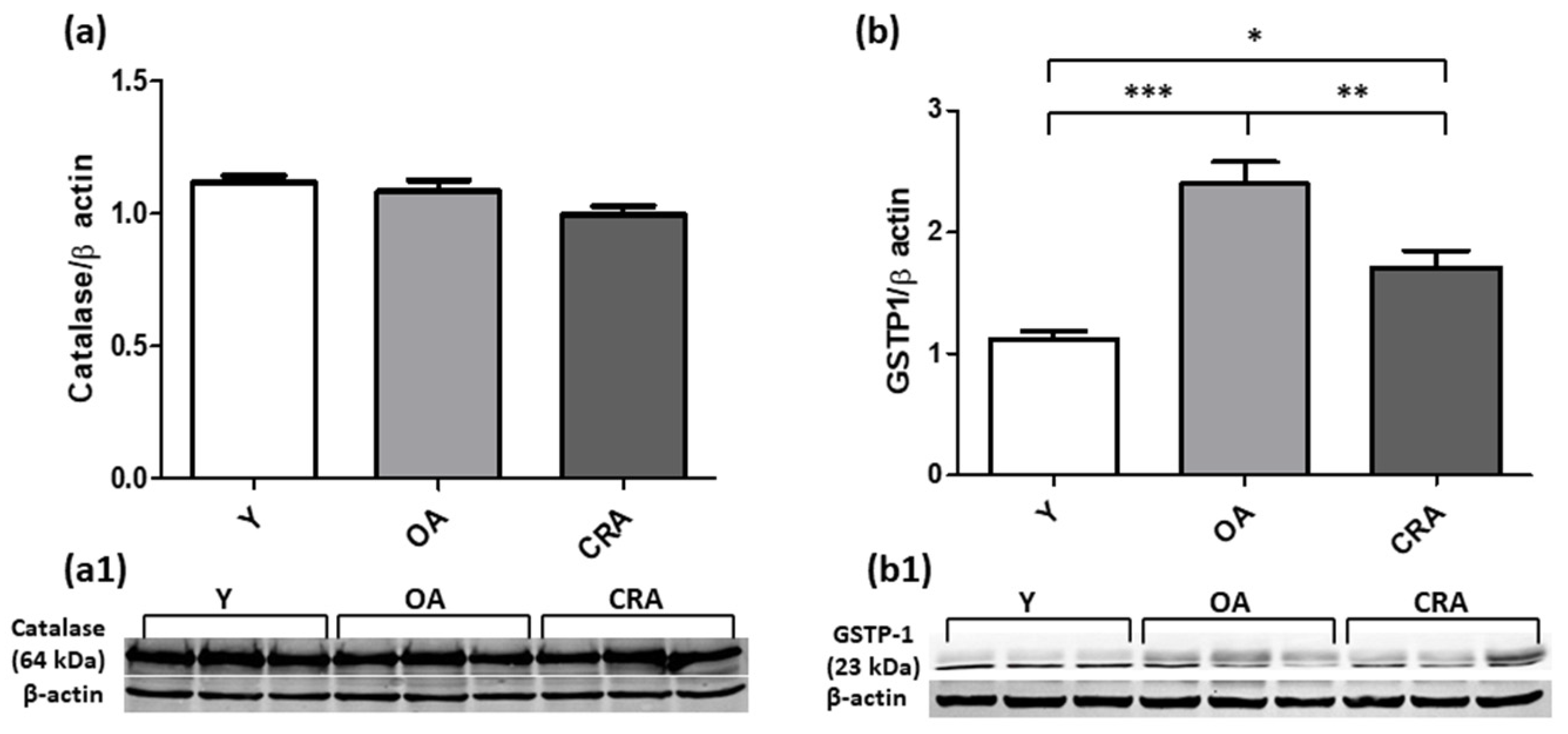
3.5. Caloric Restriction Enhances SOD1 Expression but Negatively Modulates GSTP1 Protein Level
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mather, K.A. Genetic and Environmental Factors in Ageing and Age-Related Disease. Genes 2022, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity May Accelerate the Aging Process. Front. Endocrinol. 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- James, W.P.T. Obesity: A Global Public Health Challenge. Clin. Chem. 2018, 64, 24–29. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Anjum, I.; Fayyaz, M.; Wajid, A.; Sohail, W.; Ali, A. Does Obesity Increase the Risk of Dementia: A Literature Review. Cureus 2018, 10, e2660. [Google Scholar] [CrossRef]
- Prasad, R.; Jha, R.K.; Keerti, A. Chronic Kidney Disease: Its Relationship with Obesity. Cureus 2022, 14, e30535. [Google Scholar] [CrossRef] [PubMed]
- La Russa, D.; Giordano, F.; Marrone, A.; Parafati, M.; Janda, E.; Pellegrino, D. Oxidative imbalance and kidney damage in cafeteria diet-induced rat model of metabolic syndrome: Effect of bergamot polyphenolic fraction. Antioxidants 2019, 8, 66. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Obesity, Senescence, and Senolytics. In From Obesity to Diabetes; Springer International Publishing: Cham, Switzerland, 2021; pp. 165–180. [Google Scholar]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Van der Hauwaert, C.; Savary, G.; Hennino, M.-F.; Pottier, N.; Glowacki, F.; Cauffiez, C. MicroRNAs in kidney fibrosis. Nephrol. Ther. 2015, 11, 474–482. [Google Scholar] [CrossRef]
- Leaf, I.A.; Duffield, J.S. What can target kidney fibrosis? Nephrol. Dial. Transplant. 2017, 32, i89–i97. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Su, H.; Wan, C.; Song, A.; Qiu, Y.; Xiong, W.; Zhang, C. Oxidative Stress and Renal Fibrosis: Mechanisms and Therapies. In Renal Fibrosis: Mechanisms and Therapies; Springer: Berlin/Heidelberg, Germany, 2019; pp. 585–604. [Google Scholar]
- Brunelli, E.; La Russa, D.; Pellegrino, D. Impaired Oxidative Status Is Strongly Associated with Cardiovascular Risk Factors. Oxidative Med. Cell. Longev. 2017, 2017, 6480145. [Google Scholar] [CrossRef] [PubMed]
- Russa, D.L.; Pellegrino, D.; Montesanto, A.; Gigliotti, P.; Perri, A.; Russa, A.L.; Bonofiglio, R. Oxidative Balance and Inflammation in Hemodialysis Patients: Biomarkers of Cardiovascular Risk? Oxidative Med. Cell. Longev. 2019, 2019, 8567275. [Google Scholar] [CrossRef]
- La Russa, D.; Montesano, D.; Pellegrino, D.; Frisina, M.; Bagetta, G.; Fallarino, F.; Amantea, D. Systemic administration of sunflower oil exerts neuroprotection in a mouse model of transient focal cerebral ischaemia. J. Pharm. Pharmacol. 2021, 74, 1776–1783. [Google Scholar] [CrossRef]
- Perri, A.; Lofaro, D.; La Russa, A.; Lupinacci, S.; Toteda, G.; Curti, A.; Urso, A.; Bonofiglio, R.; La Russa, D.; Pellegrino, D.; et al. Proinflammatory profile of visceral adipose tissue and oxidative stress in severe obese patients carrying the variant rs4612666 C of NLRP3 gene. Minerva Endocrinol. 2021, 46, 309–316. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Straface, E.; Gambardella, L.; Caruso, D.; Falliti, G.; Dossena, S.; Marino, A.; Morabito, R. Açaì (Euterpe oleracea) Extract Protects Human Erythrocytes from Age-Related Oxidative Stress. Cells 2022, 11, 2391. [Google Scholar] [CrossRef]
- Akki, R.; Siracusa, R.; Morabito, R.; Remigante, A.; Campolo, M.; Errami, M.; La Spada, G.; Cuzzocrea, S.; Marino, A. Neuronal-like differentiated SH-SY5Y cells adaptation to a mild and transient H2O2-induced oxidative stress. Cell Biochem. Funct. 2018, 36, 56–64. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M. V Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Harris, D.C.H.; Wang, Y. Macrophages in Kidney Injury, Inflammation, and Fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef] [PubMed]
- La Russa, D.; Marrone, A.; Mandalà, M.; Macirella, R.; Pellegrino, D. Antioxidant/Anti-Inflammatory Effects of Caloric Restriction in an Aged and Obese Rat Model: The Role of Adiponectin. Biomedicines 2020, 8, 532. [Google Scholar] [CrossRef]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R.; Sohal, R.S. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N. Engl. J. Med. 1997, 337, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.C.; Cai, G.Y.; Zhuo, L.; Gao, J.J.; Dong, D.; Cui, S.; Feng, Z.; Shi, S.Z.; Bai, X.Y.; Sun, X.F.; et al. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech. Ageing Dev. 2013, 134, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.X.; Dhahbi, J.M.; Mote, P.L.; Spindler, S.R. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10630–10635. [Google Scholar] [CrossRef]
- Dhahbi, J.M.; Kim, H.-J.; Mote, P.L.; Beaver, R.J.; Spindler, S.R. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 5524–5529. [Google Scholar] [CrossRef]
- Kuk, J.L.; Saunders, T.J.; Davidson, L.E.; Ross, R. Age-related changes in total and regional fat distribution. Ageing Res. Rev. 2009, 8, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Barzilai, N. Role of visceral adipose tissue in aging. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1117–1123. [Google Scholar] [CrossRef]
- Novelli, E.L.B.; Diniz, Y.S.; Galhardi, C.M.; Ebaid, G.M.X.; Rodrigues, H.G.; Mani, F.; Fernandes, A.A.H.; Cicogna, A.C.; Novelli Filho, J.L.V.B. Anthropometrical parameters and markers of obesity in rats. Lab. Anim. 2007, 41, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Jura, M.; Kozak, L.P. Obesity and related consequences to ageing. Age 2016, 38, 23. [Google Scholar] [CrossRef] [PubMed]
- Melk, A. Senescence of renal cells: Molecular basis and clinical implications. Nephrol. Dial. Transplant. 2003, 18, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Rakheja, D.; Yu, X.; Saxena, R.; Vaziri, N.D.; Silva, F.G. The aging kidney. Kidney Int. 2008, 74, 710–720. [Google Scholar] [CrossRef]
- Martin, J.E.; Sheaff, M.T. Renal ageing. J. Pathol. 2007, 211, 198–205. [Google Scholar] [CrossRef]
- Wiggins, J.E.; Goyal, M.; Sanden, S.K.; Wharram, B.L.; Shedden, K.A.; Misek, D.E.; Kuick, R.D.; Wiggins, R.C. Podocyte hypertrophy, “adaptation” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J. Am. Soc. Nephrol. 2005, 16, 2953–2966. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.I.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef]
- Calvo-Rubio, M.; Burón, M.I.; López-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Villalba, J.M.; González-Reyes, J.A. Dietary fat composition influences glomerular and proximal convoluted tubule cell structure and autophagic processes in kidneys from calorie-restricted mice. Aging Cell 2016, 15, 477–487. [Google Scholar] [CrossRef]
- Zha, Y.; Taguchi, T.; Nazneen, A.; Shimokawa, I.; Higami, Y.; Razzaque, M.S. Genetic suppression of GH-IGF-1 activity, combined with lifelong caloric restriction, prevents age-related renal damage and prolongs the life span in rats. Am. J. Nephrol. 2008, 28, 755–764. [Google Scholar] [CrossRef]
- Ivanova, L.; Butt, M.J.; Matsell, D.G. Mesenchymal transition in kidney collecting duct epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F1238–F1248. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to Mesenchymal Transition in Renal Fibrogenesis: Pathologic Significance, Molecular Mechanism, and Therapeutic Intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Lyons, J.G.; Thian, K.T.; Wang, Y.; Hsu, T.T.; Min, D.; Succar, L.; Rangan, G.K.; Hu, M.; Henderson, B.R.; et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-β1 in renal tubular epithelial cells. Am. J. Pathol. 2009, 175, 580–591. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages Regulate Renal Fibrosis Through Modulating TGFβ Superfamily Signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef]
- Anders, H.-J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr. Nephrol. 2015, 30, 199–209. [Google Scholar] [CrossRef]
- Klahr, S. The role of nitric oxide in hypertension and renal disease progression. Nephrol. Dial. Transplant. 2001, 16, 60–62. [Google Scholar] [CrossRef]
- Martin, B.; Caron, N.; Jadot, I.; Colombaro, V.; Federici, G.; Depommier, C.; Declèves, A.-É. Evaluation of inducible nitric oxide synthase inhibition on kidney function and structure in high-fat diet-induced kidney disease. Exp. Physiol. 2018, 103, 125–140. [Google Scholar] [CrossRef]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef]
- Kuloglu, T.; Aydin, S. Immunohistochemical expressions of adropin and ınducible nitric oxide synthase in renal tissues of rats with streptozotocin-ınduced experimental diabetes. Biotech. Histochem. 2014, 89, 104–110. [Google Scholar] [CrossRef]
- Inoue, T. M1 macrophage triggered by Mincle leads to a deterioration of acute kidney injury. Kidney Int. 2017, 91, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, Z.; Kadkhodaee, M.; Seifi, B.; Salehi, E. Inducible and endothelial nitric oxide synthase distribution and expression with hind limb per-conditioning of the rat kidney. Arch. Med. Sci. 2019, 15, 1081–1091. [Google Scholar] [CrossRef]
- Liu, X.; Chen, H.; Zhan, B.; Xing, B.; Zhou, J.; Zhu, H.; Chen, Z. Attenuation of reperfusion injury by renal ischemic postconditioning: The role of NO. Biochem. Biophys. Res. Commun. 2007, 359, 628–634. [Google Scholar] [CrossRef]
- Pak, E.S.; Jeong, L.S.; Hou, X.; Tripathi, S.K.; Lee, J.; Ha, H. Dual Actions of A2A and A3 Adenosine Receptor Ligand Prevents Obstruction-Induced Kidney Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 5667. [Google Scholar] [CrossRef]
- Song, L.; Shi, S.; Jiang, W.; Liu, X.; He, Y. Protective role of propofol on the kidney during early unilateral ureteral obstruction through inhibition of epithelial-mesenchymal transition. Am. J. Transl. Res. 2016, 8, 460–472. [Google Scholar] [PubMed]
- Meng, X.-M.; Mak, T.S.-K.; Lan, H.-Y. Macrophages in Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies; Springer: Berlin/Heidelberg, Germany, 2019; pp. 285–303. [Google Scholar]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. Physiol. 2010, 298, F1078–F1094. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Oldroyd, S.D.; Huang, L.H.; Yang, B.; Li, Y.; Ye, R.; El Nahas, A.M. Role of Apoptosis and Bcl-2/Bax in the Development of Tubulointerstitial Fibrosis during Experimental Obstructive Nephropathy. Nephron Exp. Nephrol. 2001, 9, 71–80. [Google Scholar] [CrossRef]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.-C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef]
- Janumyan, Y.M.; Sansam, C.G.; Chattopadhyay, A.; Cheng, N.; Soucie, E.L.; Penn, L.Z.; Andrews, D.; Knudson, C.M.; Yang, E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003, 22, 5459–5470. [Google Scholar] [CrossRef]
- Janumyan, Y.; Cui, Q.; Yan, L.; Sansam, C.G.; Valentin, M.; Yang, E. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27 phosphorylation by Mirk, revealing a novel role of BAX and BAK in quiescence regulation. J. Biol. Chem. 2008, 283, 34108–34120. [Google Scholar] [CrossRef]
- DU, X.; Xiao, J.; Fu, X.; Xu, B.; Han, H.; Wang, Y.; Pei, X. A proteomic analysis of Bcl-2 regulation of cell cycle arrest: Insight into the mechanisms. J. Zhejiang Univ. Sci. B 2021, 22, 839–855. [Google Scholar] [CrossRef]
- Lv, W.; Booz, G.W.; Fan, F.; Wang, Y.; Roman, R.J. Oxidative Stress and Renal Fibrosis: Recent Insights for the Development of Novel Therapeutic Strategies. Front. Physiol. 2018, 9, 105. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; de Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the Role of Mitochondria in Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Acuña-Castroviejo, D.; Carretero, M.; Doerrier, C.; López, L.C.; García-Corzo, L.; Tresguerres, J.A.; Escames, G. Melatonin protects lung mitochondria from aging. Age 2012, 34, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Choksi, K.B.; Nuss, J.E.; DeFord, J.H.; Papaconstantinou, J. Mitochondrial electron transport chain functions in long-lived Ames dwarf mice. Aging 2011, 3, 754–767. [Google Scholar] [CrossRef]
- Ojaimi, J.; Masters, C.L.; Opeskin, K.; McKelvie, P.; Byrne, E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 1999, 111, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Xiao, S.; Liu, J.; Guo, K.; Wu, F.; Yew, D.T.; Xu, J. Ginkgo biloba extract EGb761 protects against aging-associated mitochondrial dysfunction in platelets and hippocampi of SAMP8 mice. Platelets 2010, 21, 373–379. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Pich, M.M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species in Relation to Aging and Pathology. In Mitochondrial Pathogenesis; Springer: Berlin/Heidelberg, Germany, 2004; pp. 86–100. [Google Scholar]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 961–967. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef]
- Valerio, A.; Nisoli, E. Nitric oxide, interorganelle communication, and energy flow: A novel route to slow aging. Front. Cell Dev. Biol. 2015, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Simão, S.; Silva, E.; Pinto, V.; Amaral, J.S.; Afonso, J.; Serrão, M.P.; Pinho, M.J.; Soares-da-Silva, P. Aging increases Oxidative Stress and Renal Expression of Oxidant and Antioxidant Enzymes that Are Associated with an Increased Trend in Systolic Blood Pressure. Oxidative Med. Cell. Longev. 2009, 2, 138–145. [Google Scholar] [CrossRef]
- Enns, L.C.; Wiley, J.C.; Ladiges, W.C. Clinical Relevance of Transgenic Mouse Models for Aging Research. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Treuting, P.M.; Hopkins, H.C.; Ware, C.A.; Rabinovitch, P.R.; Ladiges, W.C. Generation of Genetically Altered Mouse Models for Aging Studies. Exp. Mol. Pathol. 2002, 72, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 634–644. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Health 2014, 3, 6. [Google Scholar] [CrossRef]
- Romi, M.M.; Arefian, N.; Wahyu Setyaningsih, W.A.; Perdana Putri, R.G.; Juffrie, M.; Ratna Sari, D.C. Calcitriol Treatment Attenuates Uric Acid-Induced Kidney Injury via Super Oxide Dismutase-1 (SOD-1) Upregulation and Fibrosis Reduction. Iran. Biomed. J. 2021, 25, 417–425. [Google Scholar] [CrossRef]
- Fujita, H.; Fujishima, H.; Takahashi, K.; Sato, T.; Shimizu, T.; Morii, T.; Shimizu, T.; Shirasawa, T.; Qi, Z.; Breyer, M.D.; et al. SOD1, but not SOD3, deficiency accelerates diabetic renal injury in C57BL/6-Ins2(Akita) diabetic mice. Metabolism 2012, 61, 1714–1724. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1—Mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef]
- Mura, C.V.; Gong, X.; Taylor, A.; Villalobos-Molina, R.; Scrofano, M.M. Effects of calorie restriction and aging on the expression of antioxidant enzymes and ubiquitin in the liver of Emory mice. Mech. Ageing Dev. 1996, 91, 115–129. [Google Scholar] [CrossRef]
- Walsh, M.E.; Shi, Y.; Van Remmen, H. The effects of dietary restriction on oxidative stress in rodents. Free Radic. Biol. Med. 2014, 66, 88–99. [Google Scholar] [CrossRef] [PubMed]
- La Russa, D.; Brunelli, E.; Pellegrino, D. Oxidative imbalance and kidney damage in spontaneously hypertensive rats: Activation of extrinsic apoptotic pathways. Clin. Sci. 2017, 131, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Dodia, C.; Feinstein, S.I.; Jain, M.K.; Fisher, A.B. 1-Cys Peroxiredoxin, a Bifunctional Enzyme with Glutathione Peroxidase and Phospholipase A2 Activities. J. Biol. Chem. 2000, 275, 28421–28427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 as an antioxidant enzyme: Protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. J. Cell. Biochem. 2008, 104, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grek, C.; Ye, Z.-W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic Functions of Glutathione S-Transferase P. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2014; pp. 143–175. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Russa, D.; Barberio, L.; Marrone, A.; Perri, A.; Pellegrino, D. Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model. Antioxidants 2023, 12, 1778. https://doi.org/10.3390/antiox12091778
La Russa D, Barberio L, Marrone A, Perri A, Pellegrino D. Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model. Antioxidants. 2023; 12(9):1778. https://doi.org/10.3390/antiox12091778
Chicago/Turabian StyleLa Russa, Daniele, Laura Barberio, Alessandro Marrone, Anna Perri, and Daniela Pellegrino. 2023. "Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model" Antioxidants 12, no. 9: 1778. https://doi.org/10.3390/antiox12091778
APA StyleLa Russa, D., Barberio, L., Marrone, A., Perri, A., & Pellegrino, D. (2023). Caloric Restriction Mitigates Kidney Fibrosis in an Aged and Obese Rat Model. Antioxidants, 12(9), 1778. https://doi.org/10.3390/antiox12091778