

α-Phenyl-N-tert-Butylnitrone and Analogous α-Aryl-N-alkylnitrones as Neuroprotective Antioxidant Agents for Stroke

Abstract
1. Introduction
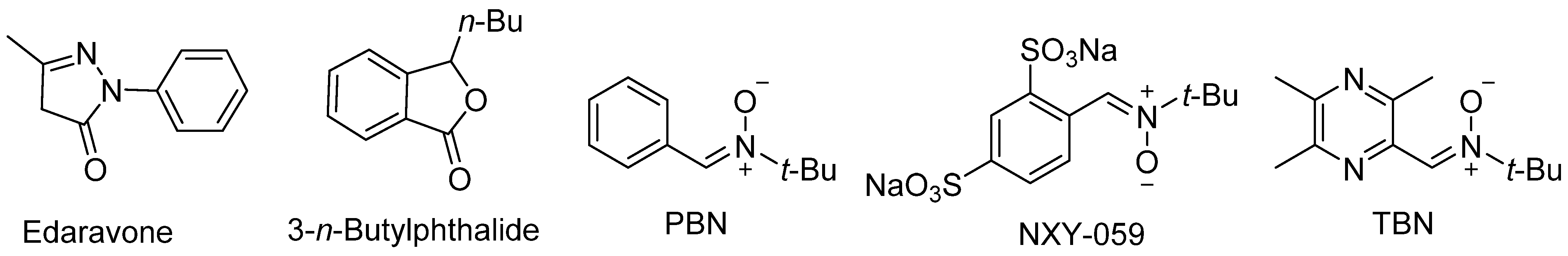
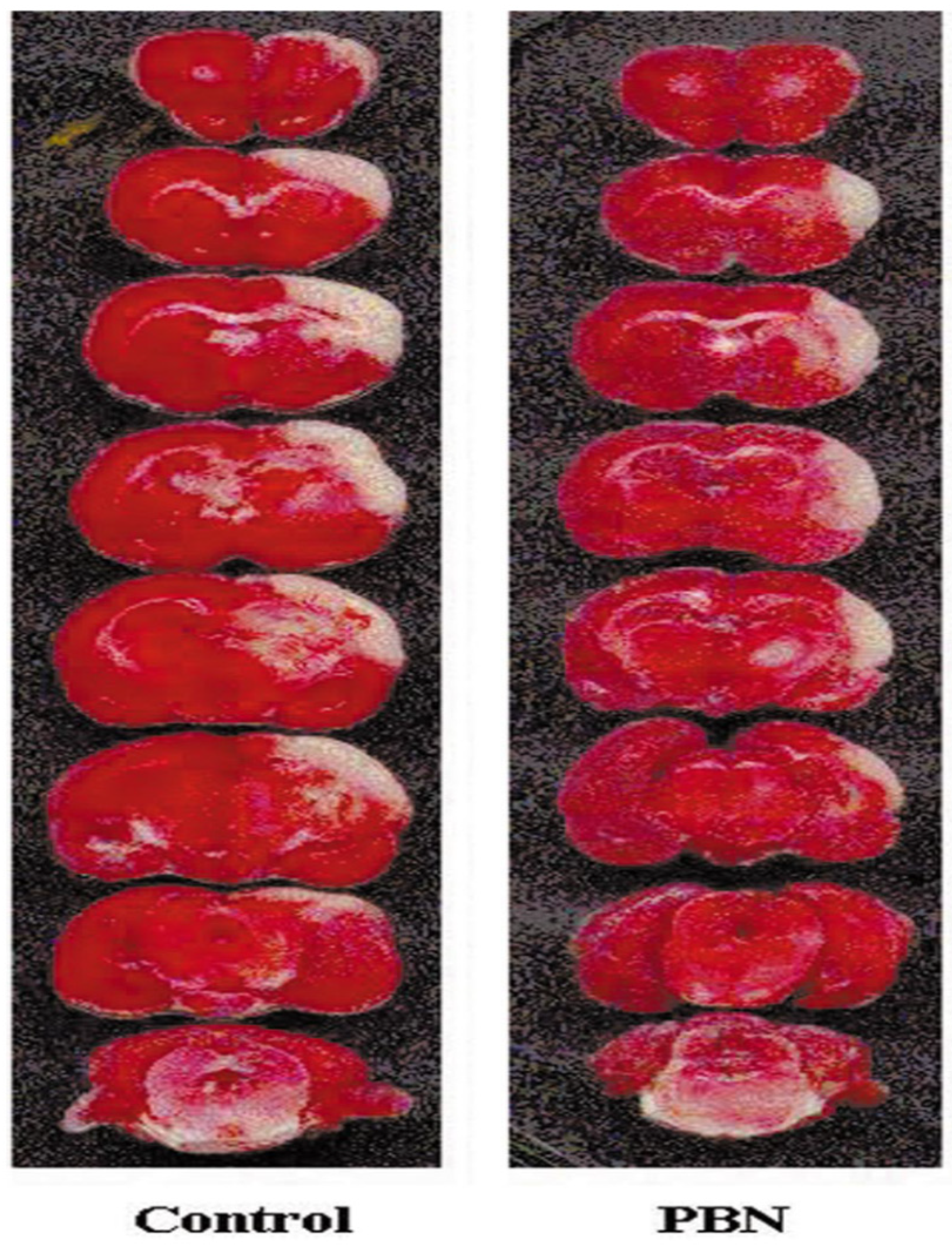
2. PBN, an Antioxidant and Neuroprotective Agent
3. Neuroprotection of PBN-Derived Nitrones from Author’s Laboratory
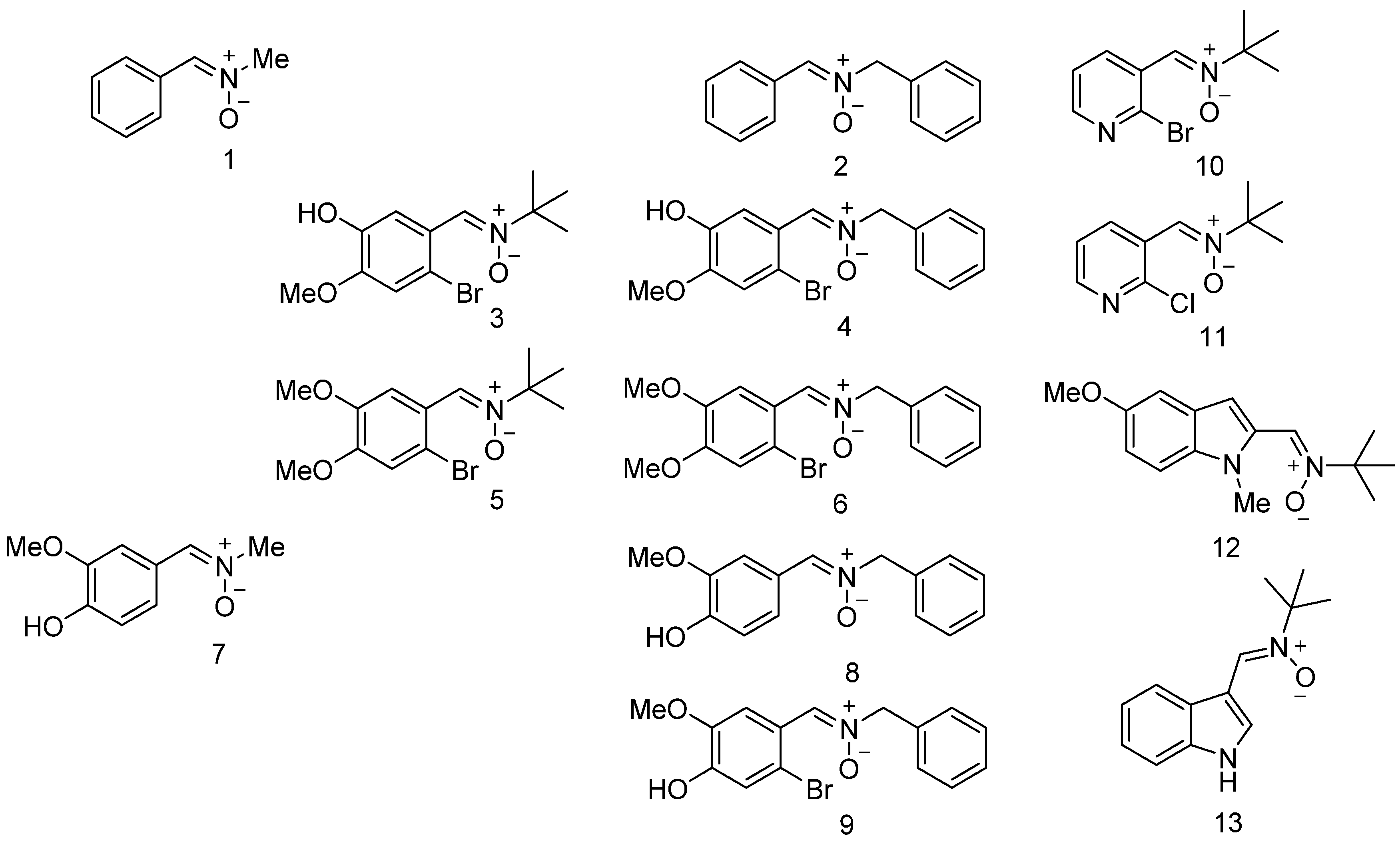
3.1. Neuroprotection of PBN-Derived Nitrones 1–13
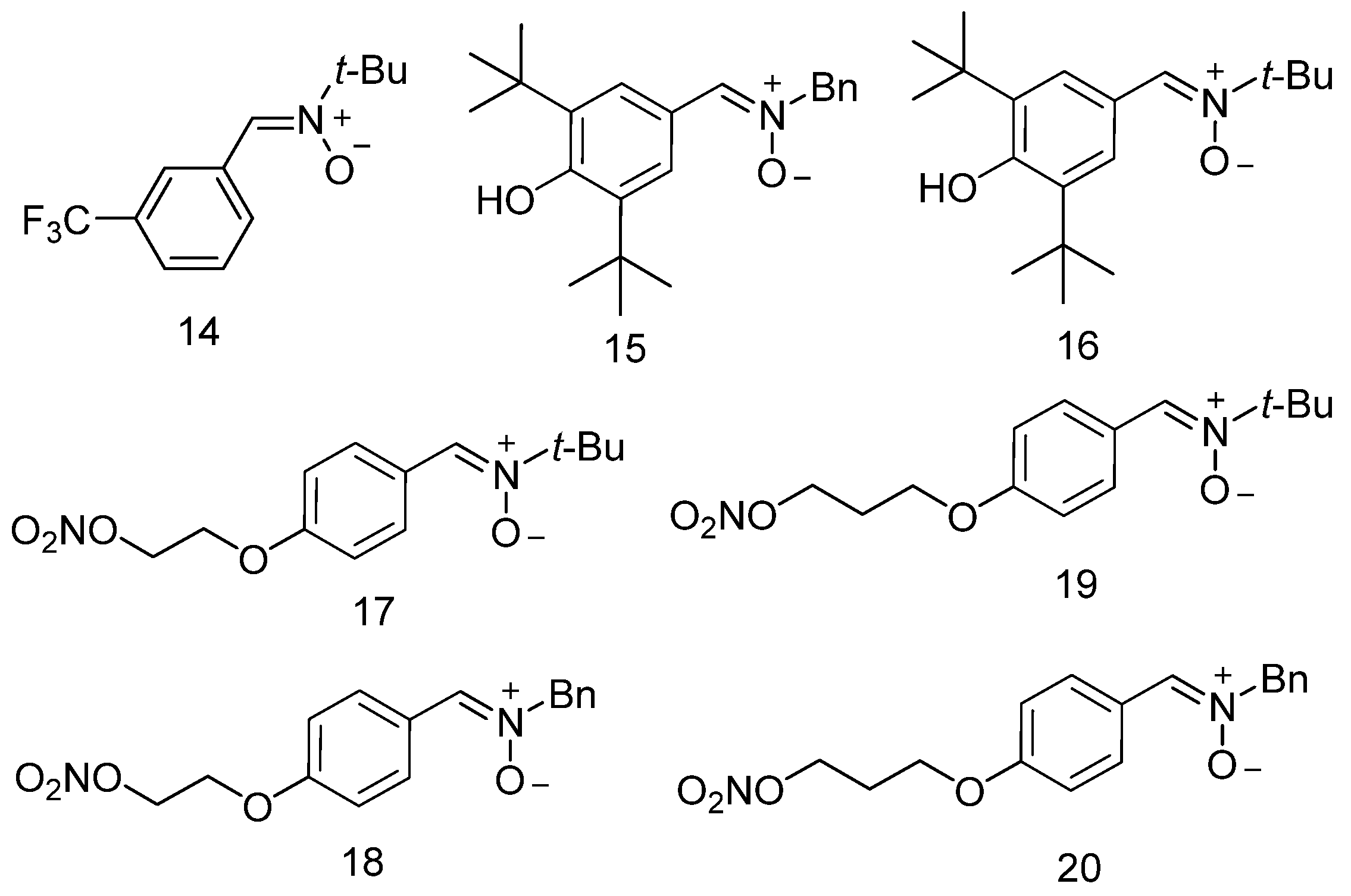
3.2. Neuroprotection of PBN-Derived Nitrones 14–20
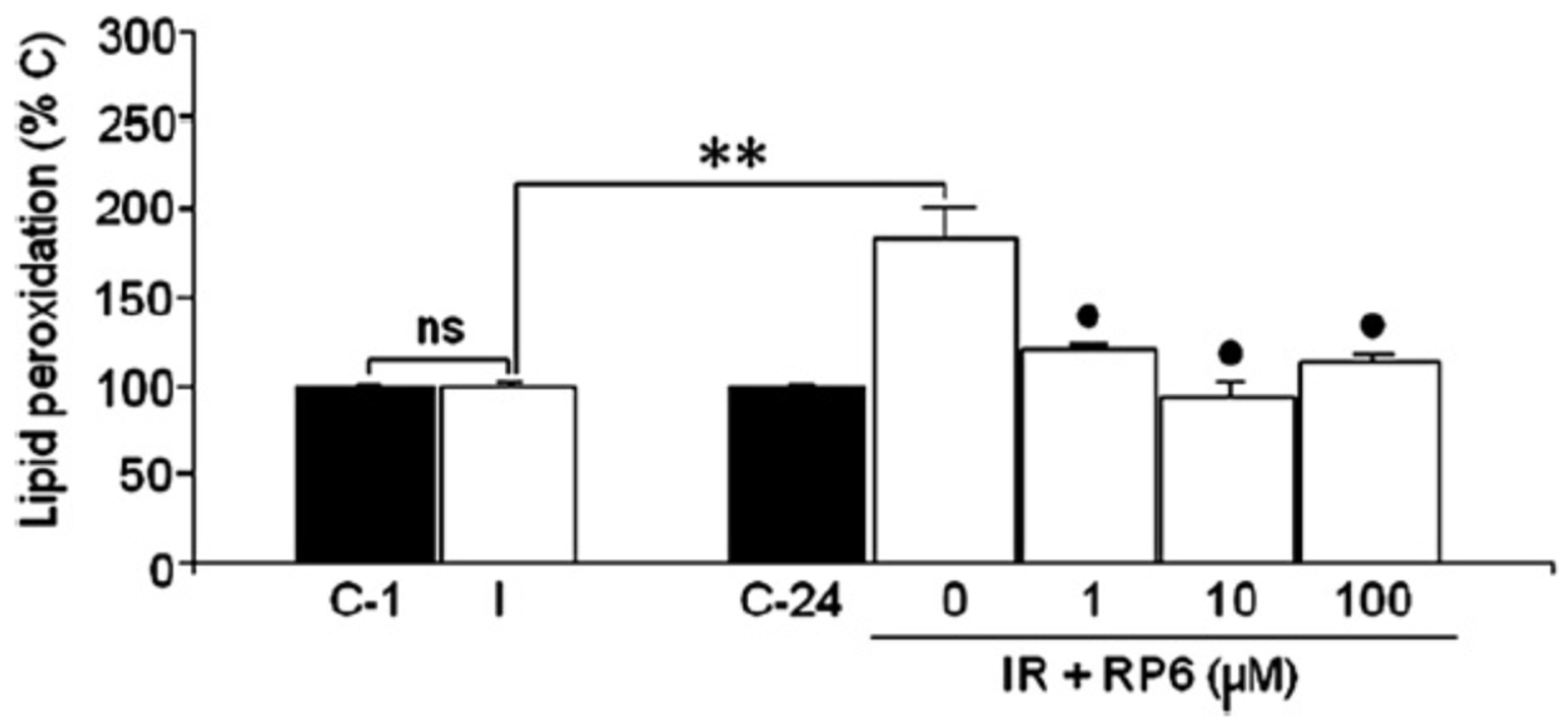
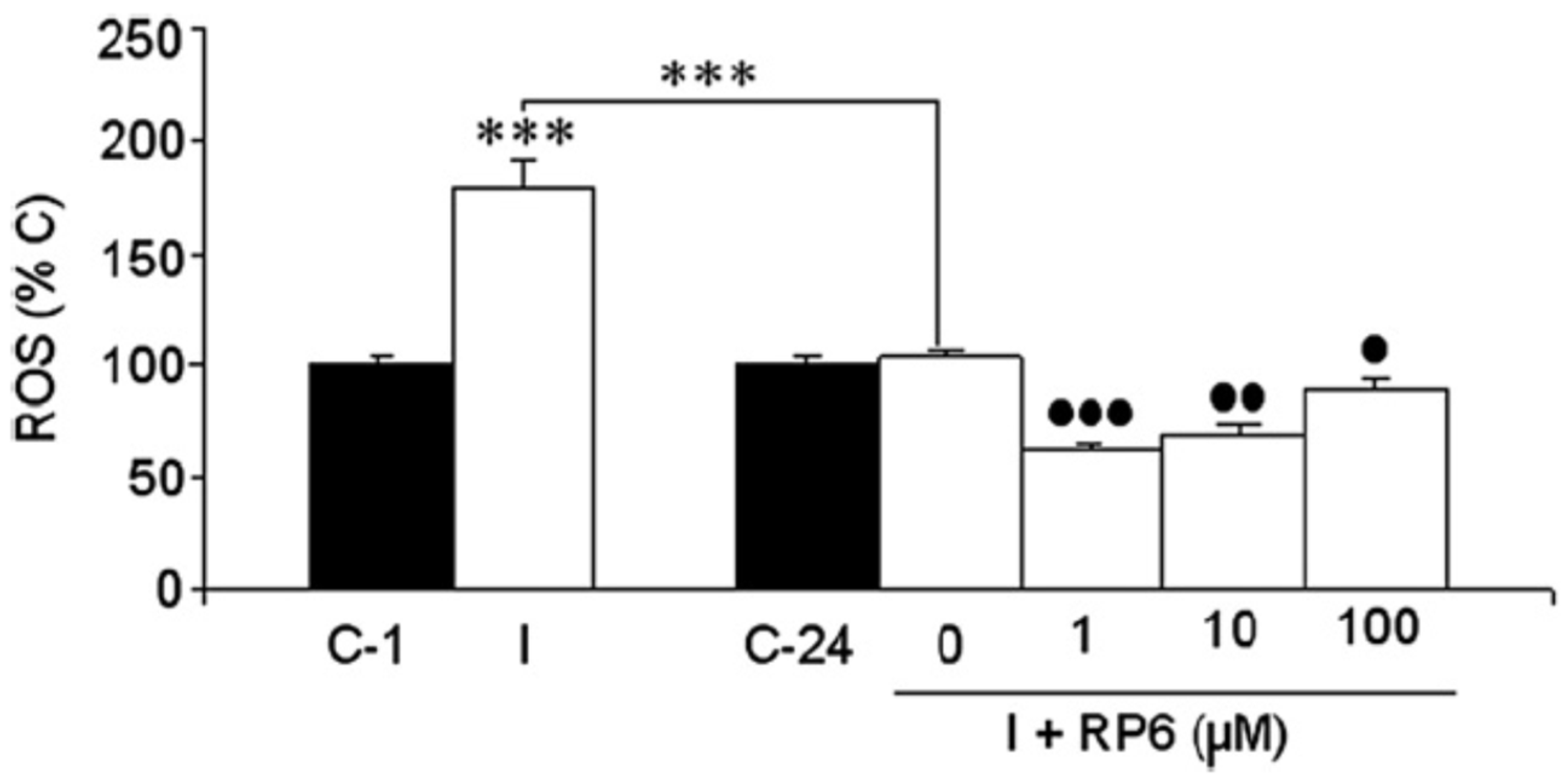
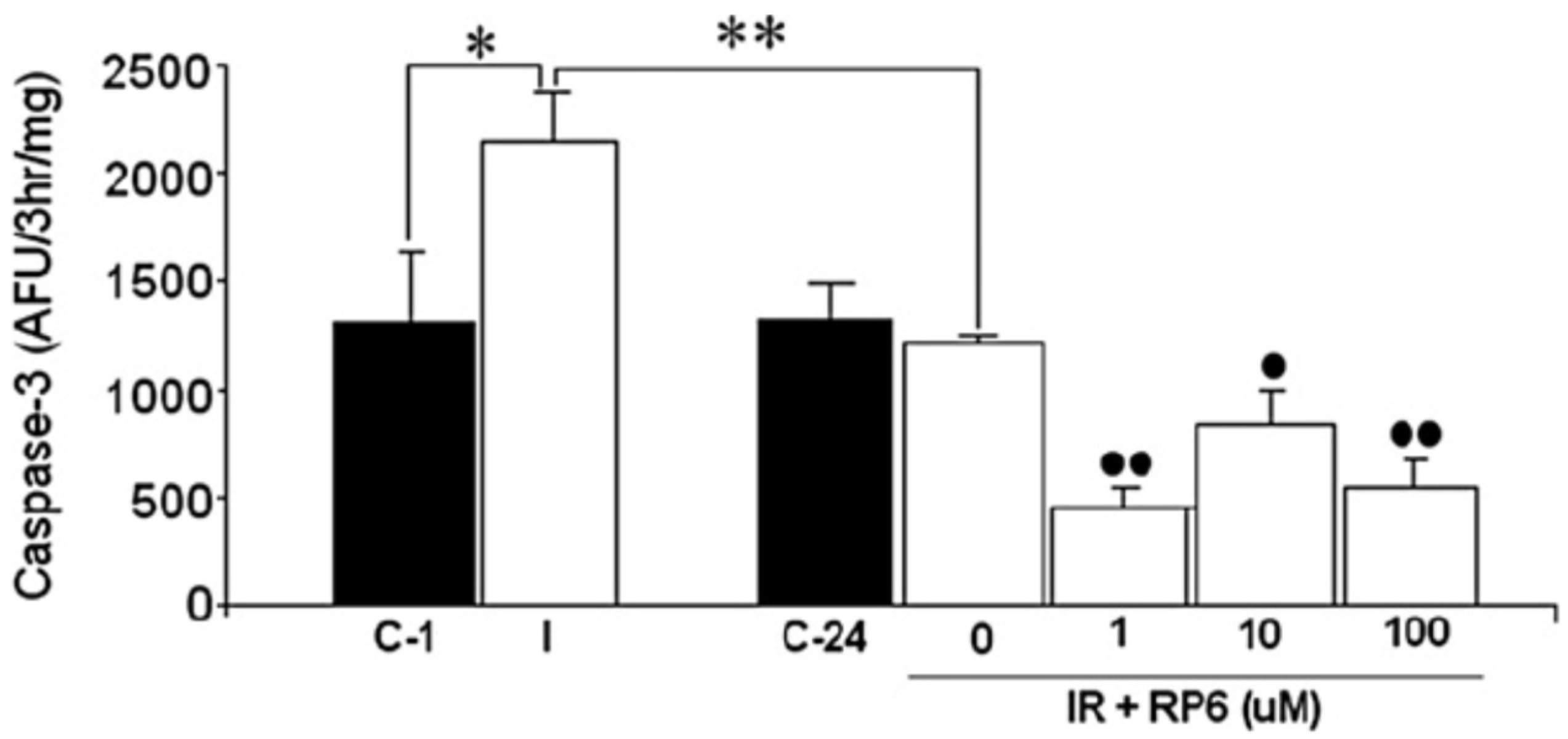
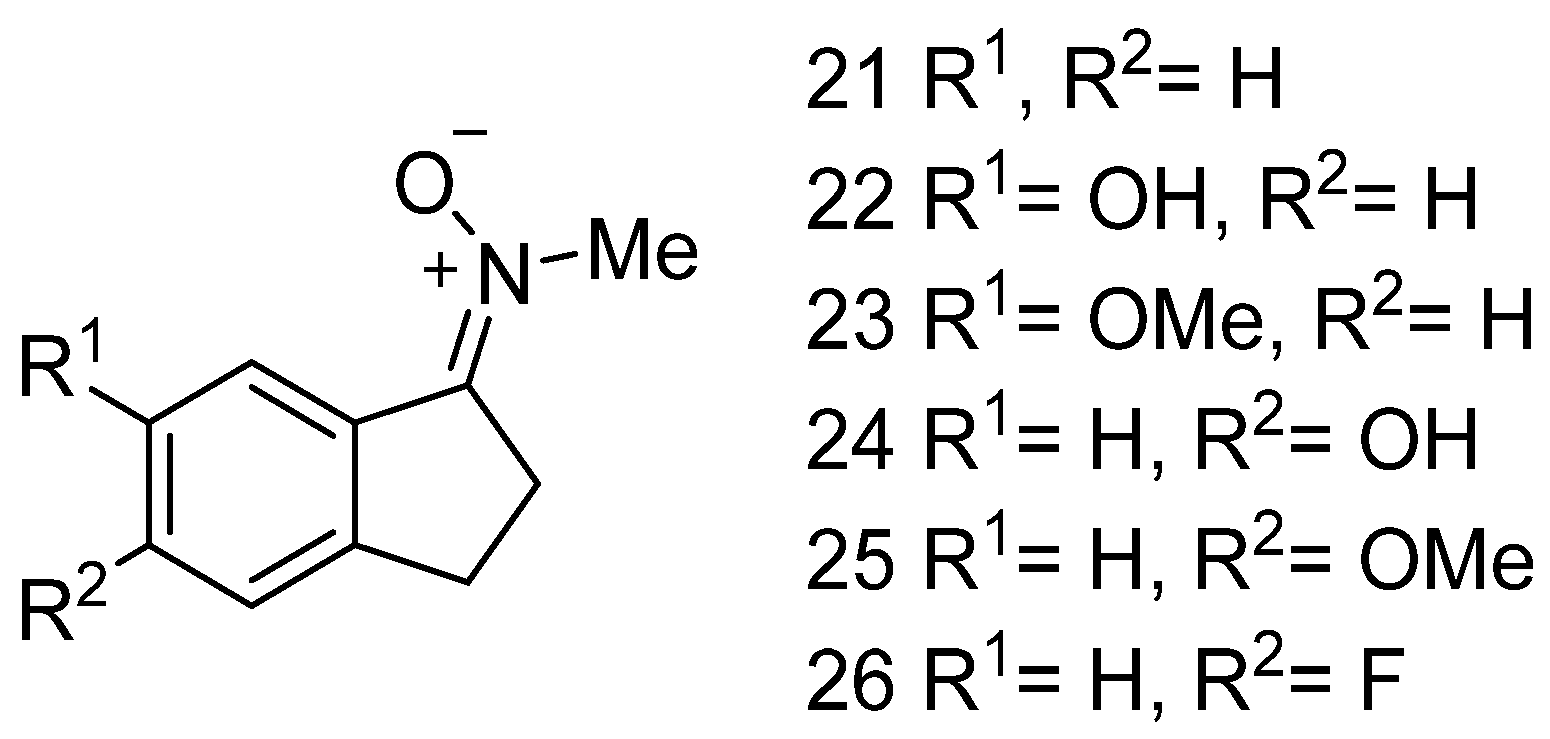
3.3. Neuroprotection of PBN-Indanonitrones 21–26
3.4. Neuroprotection of PBN-Derived Homo-Bis-Nitrones 27–35
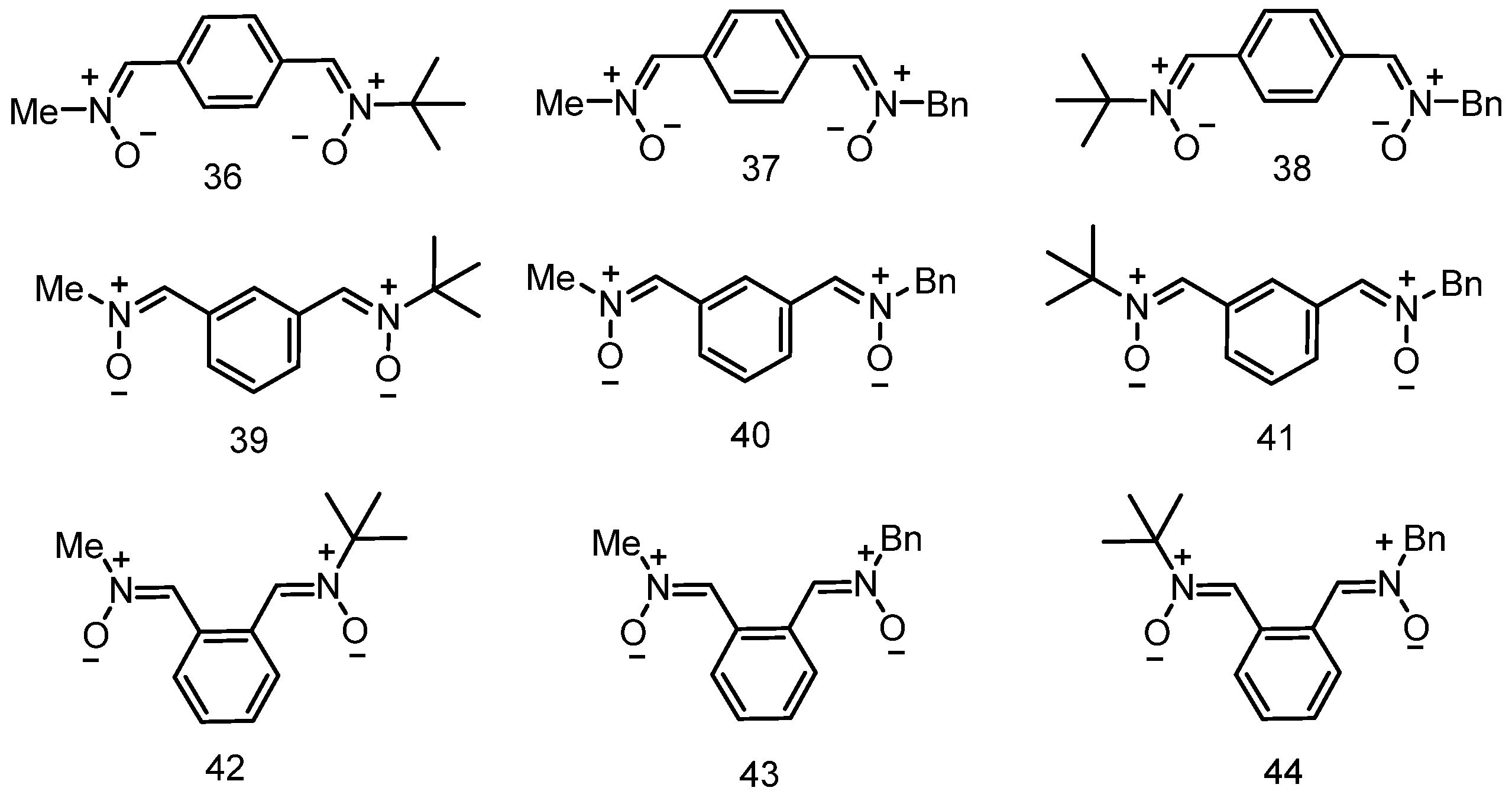
3.5. Neuroprotection of PBN-Derived Hetero-Bis-Nitrones 36–44
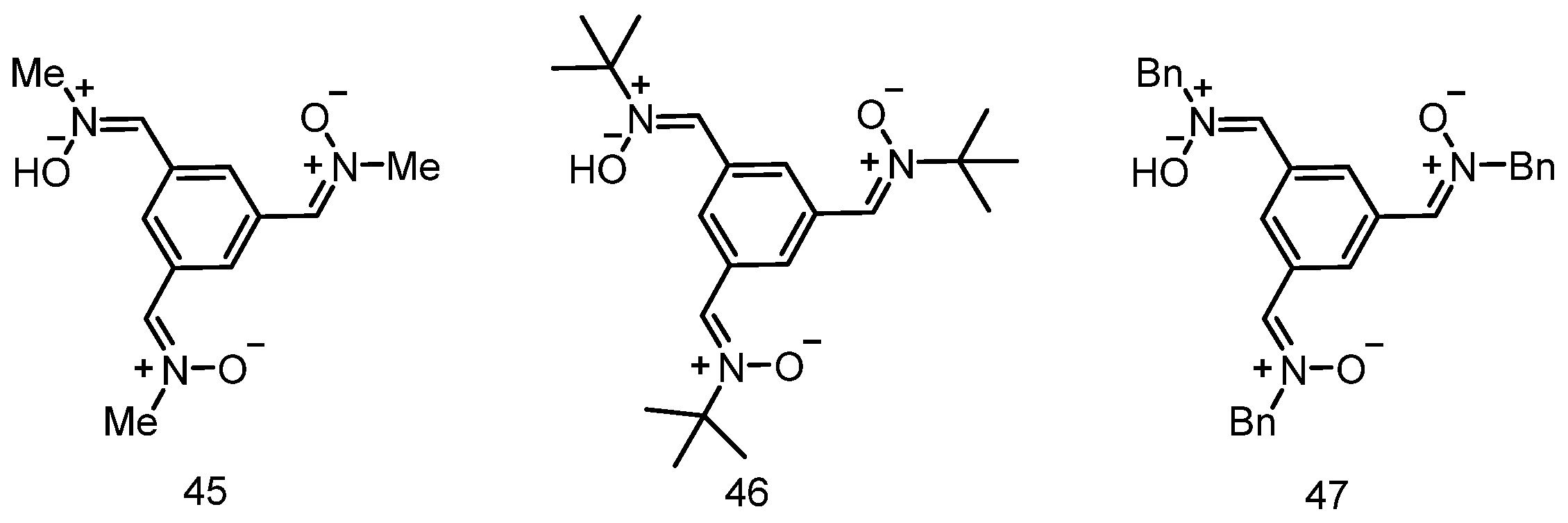
3.6. Neuroprotection of PBN-Derived Homo-Tris-Nitrones 45–47
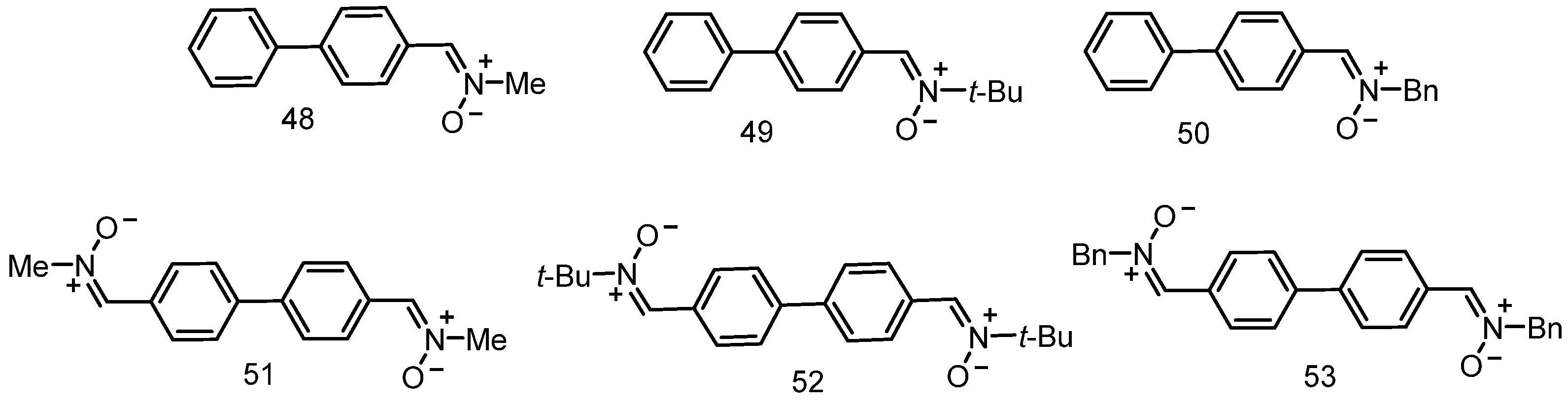
3.7. Neuroprotection of PBN-Derived 1,1′-Biphenylnitrones 48–53
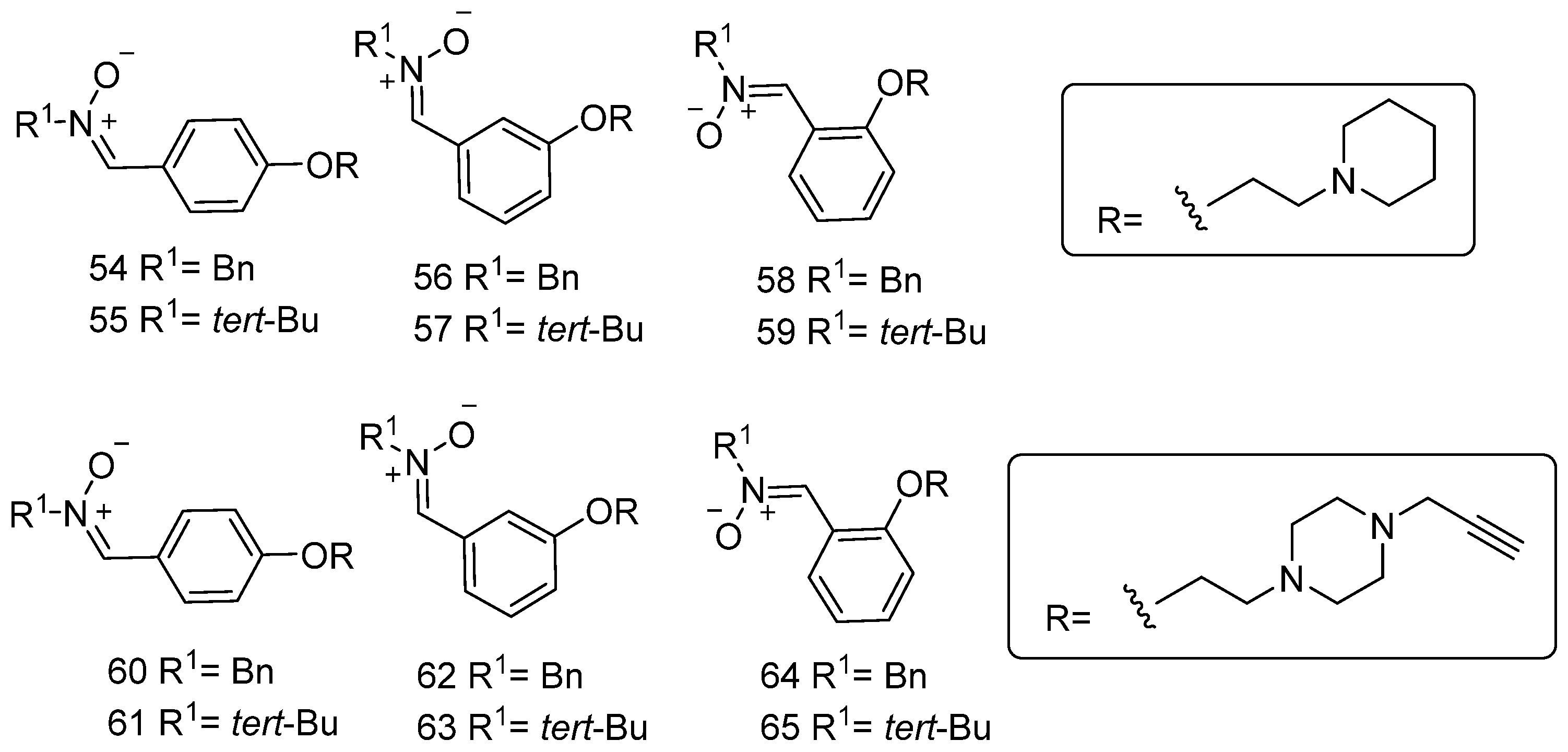
3.8. Neuroprotection of Polyfunctionalized PBN-Based Nitrones 54–65
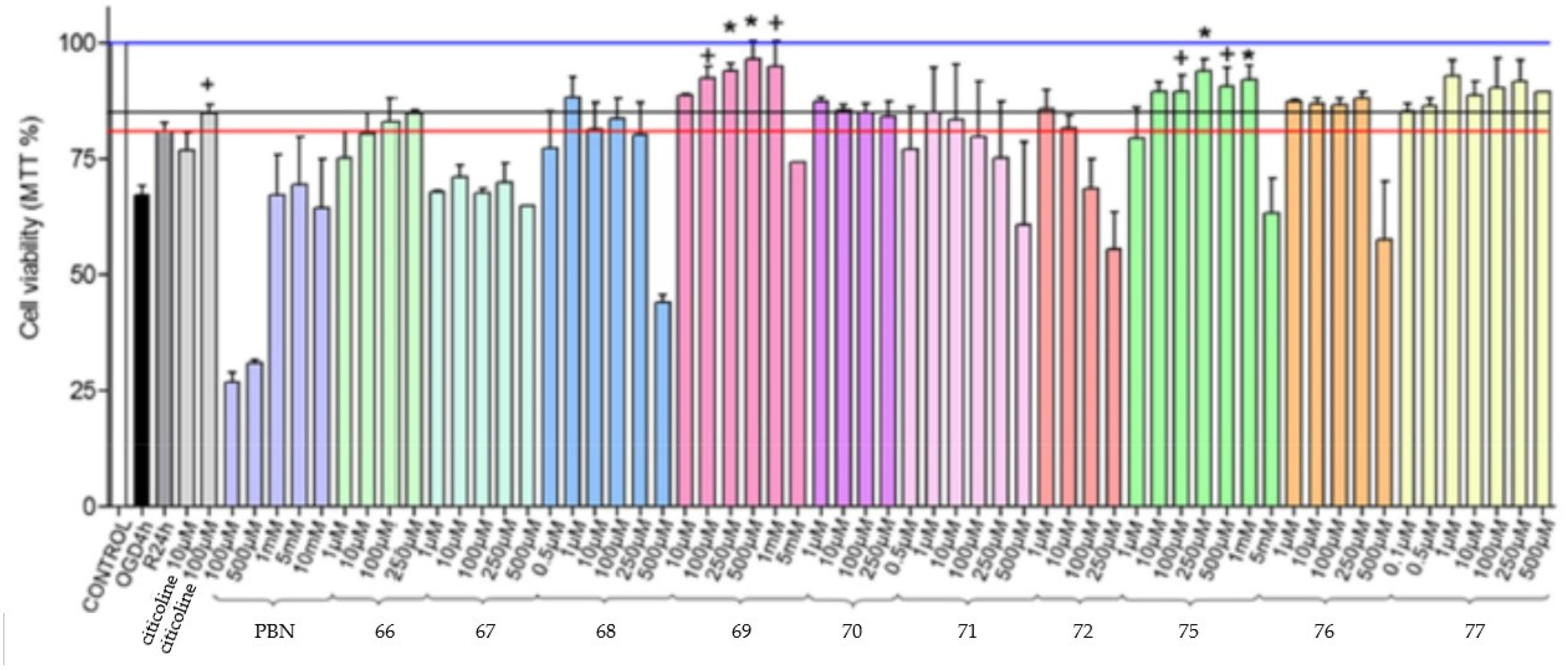
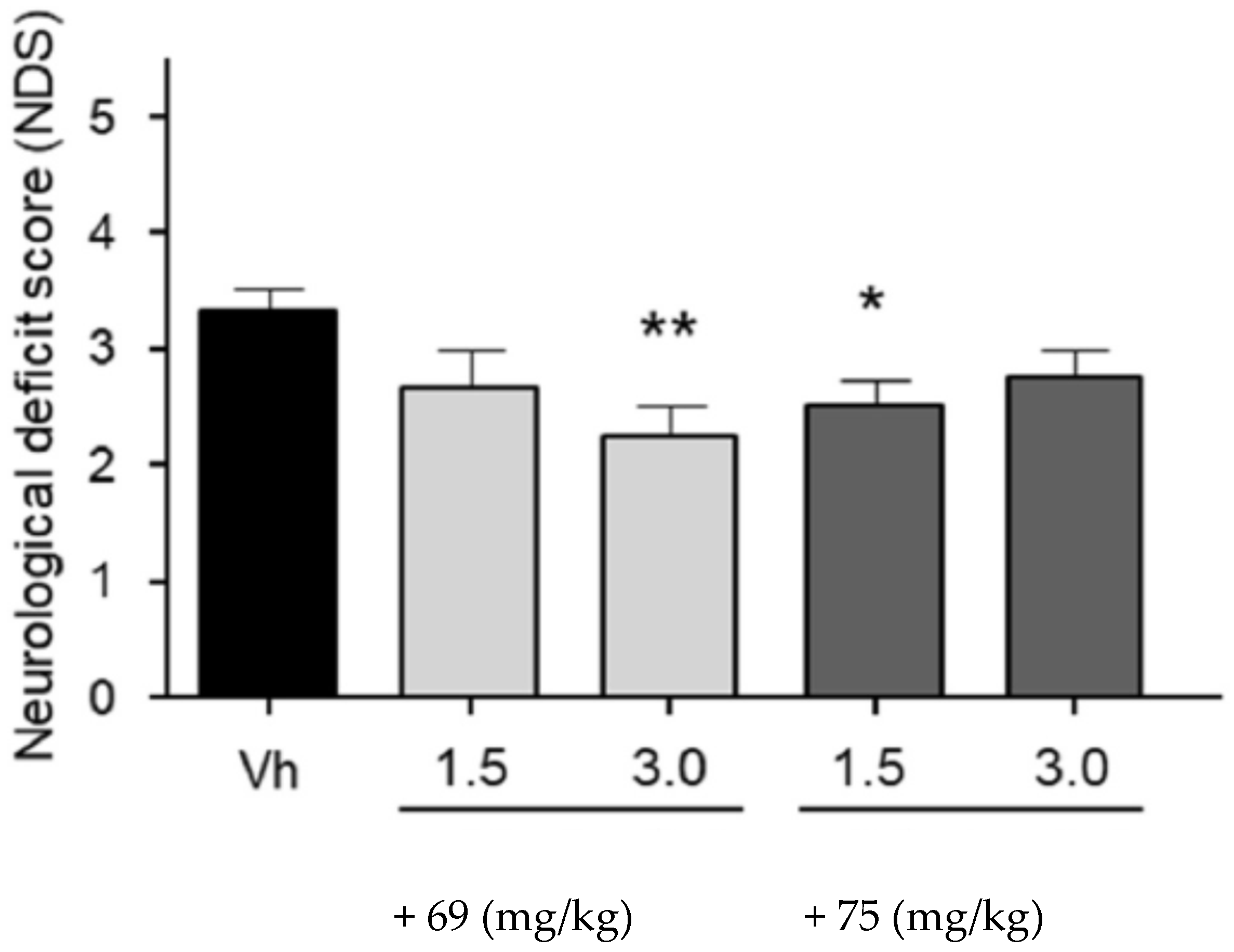
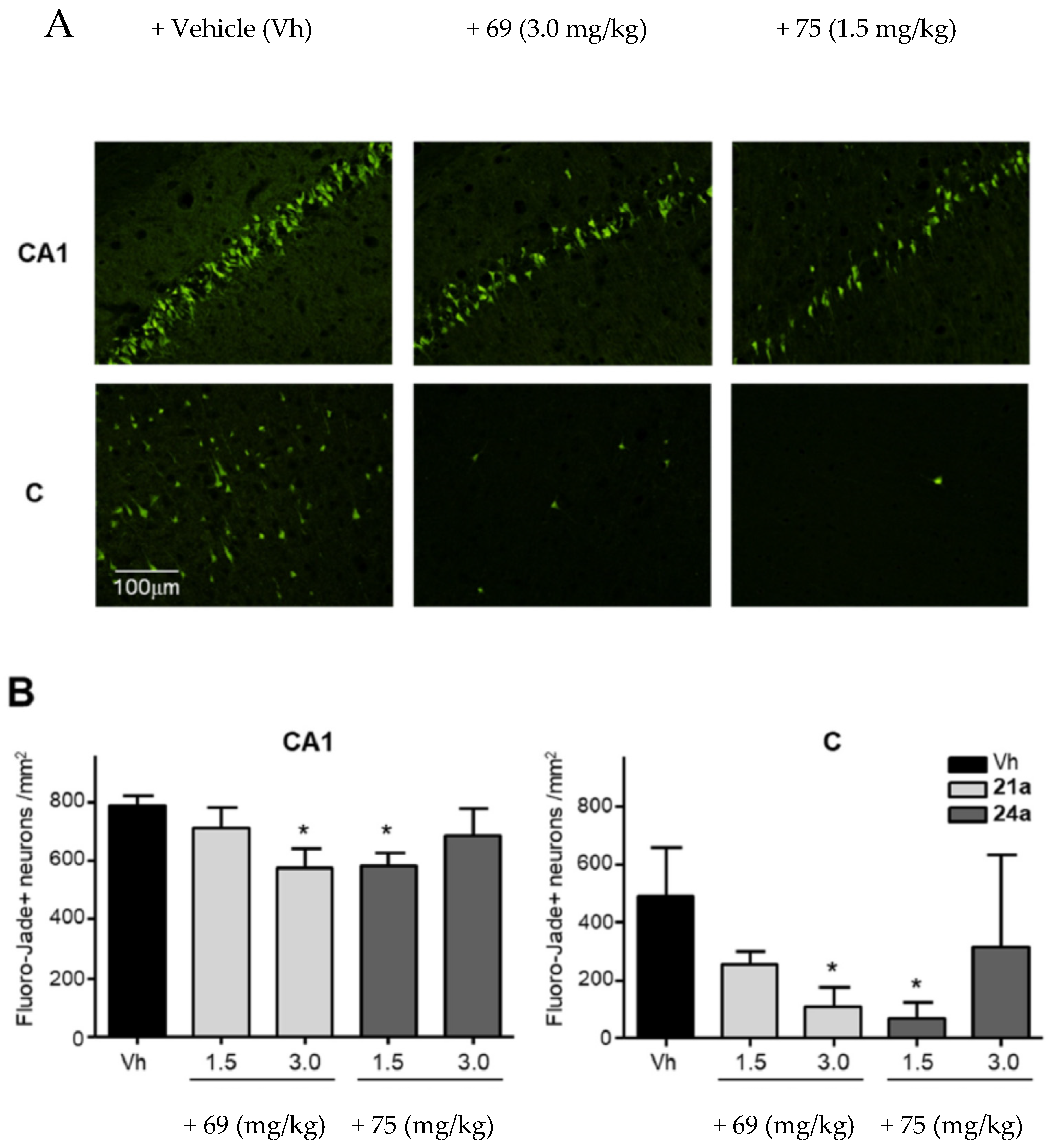
3.9. Neuroprotection of PBN-Based Nitrones 66–97
4. Summary and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Buccellato, F.R.; D’Anca, M.; Tartaglia, G.M.; Del Fabbro, M.; Scarpini, E.; Galimberti, D. Treatment of Alzheimer’s Disease: Beyond Symptomatic Therapies. Int. J. Mol. Sci. 2023, 24, 13900. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative Stress in Alzheimer’s Disease: Are We Connecting the Dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef]
- Olanow, C.W. An Introduction to the Free Radical Hypothesis in Parkinson’s Disease. Ann. Neurol. 1992, 32, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, H. Neuroprotection by Radical Avoidance: Search for Suitable agents. Molecules 2009, 14, 5054–5102. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Delanty, N. Oxidative Injury in Diseases of the Central Nervous System: Focus on Alzheimer’s Disease. Am. J. Med. 2000, 109, 577–585. [Google Scholar] [CrossRef]
- Cummings, J.L. Vascular Subcortical Dementias: Clinical Aspects. Dementia 1994, 5, 177–180. [Google Scholar] [CrossRef]
- Harman, D. Role of Free Radicals in Aging and Disease. Ann. N. Y. Acad. Sci. 1992, 673, 126–141. [Google Scholar] [CrossRef]
- Sack, C.A.; Socci, D.J.; Crandall, B.M.; Arendash, G.W. Antioxidant Treatment with Phenyl-α-tert-butyl nitrone (PBN) Improves the Cognitive Performance and Survival of Aging Rats. Neurosci. Lett. 1996, 205, 181–184. [Google Scholar] [CrossRef]
- Hammer, B.; Parker, W.D., Jr.; Bennett, J.P., Jr. NMDA Receptors Increase OH Radicals in vivo by Using Nitric Oxide Synthase and Protein Kinase C. Neuroreport 1993, 5, 72–74. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Culcasl, M.; Gaven, F.; Pietri, S.; Bockaert, J. Nitric Oxide, Superoxide and Peroxynitrite: Putative Mediators of NMDA-nduced Cell Death in Cerebellar Granule Cells. Neuropharmacology 1993, 32, 1259–1266. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative Stress, Glutamate, and Neurodegenerative Disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Henshaw, D.R.; Siwek, D.; Jenkins, B.G.; Ferrante, R.J.; Cipolloni, P.B.; Kowall, N.W.; Rosen, B.R.; Beal, M.F. Involvement of Free Radicals in Excitotoxicity in vivo. J. Neurochem. 1995, 64, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Boerma, T.; Fat, D.M. Global and Regional Causes of Death. Br. Med. Bull. 2009, 92, 7–32. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Olney, J.W. Glutamate and the Pathophysiology of Hypoxic—Ischemic Brain Damage. Ann. Neurol. 1986, 19, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Zipfel, G.J.; Choi, D.W. The Changing Landscape of Ischaemic Brain Injury Mechanisms. Nature 1999, 399, A7–A14. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kanno, I.; Ibaraki, M.; Hatazawa, J.; Miura, S. Changes in Human Cerebral Blood Flow and Cerebral Blood Volume during Hypercapnia and Hypocapnia Measured by Positron Emission Tomography. J. Cereb. Blood Flow Metab. 2003, 23, 665–670. [Google Scholar] [CrossRef]
- Choi, D.W. Ischemia-induced Neuronal Apoptosis. Curr. Opin. Neurobiol. 1996, 6, 667–672. [Google Scholar] [CrossRef]
- Hossmann, K.A. Viability Thresholds and the Penumbra of Focal Ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef]
- Kontos, H.A. Oxygen Radicals in CNS Damage. Chem. Biol. Interact. 1989, 72, 229–255. [Google Scholar] [CrossRef]
- Zivin, J.A.; Fisher, M.; Degirolami, U.; Hemenway, C.C.; Stashak, J.A. Tissue Plasminogen Activator Reduces Neurological Damage after Cerebral Embolism. Science 1985, 230, 1289–1292. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Tissue Plasminogen Activator (tPA) and Matrix Metalloproteinases in the Pathogenesis of Stroke: Therapeutic Strategies. CNS Neurol. Disord. Drug Targets 2008, 7, 243–253. [Google Scholar] [CrossRef]
- Paschen, W. Role of Calcium in Neuronal Cell Injury: Which Subcellular Compartment is Involved? Brain Res. Bull. 2000, 53, 409–413. [Google Scholar] [CrossRef]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and Apoptotic Neuronal Death Signaling Pathways in Cerebral Ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive Oxygen Radicals in Signaling and Damage in the Ischemic Brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Iadecola, C.; Alexander, M. Cerebral Ischemia and Inflammation. Curr. Opin. Neurol. 2001, 14, 89–94. [Google Scholar] [CrossRef]
- Lo, E.H.; Moskowitz, M.A.; Jacobs, T.P. Exciting, Radical, Suicidal: How Brain Cells Die after Stroke. Stroke 2005, 36, 189–192. [Google Scholar] [CrossRef]
- Bailly, C.; Hecquet, P.-E.; Kouach, M.; Thuru, X.; Goossens, J.-F. Chemical Reactivity and Uses of 1-Phenyl-3-methyl-5-pyrazolone (PMP), also known as Edaravone. Bioorg. Med. Chem. 2020, 28, 115463. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; Zhang, Y. From Seeds of Apium graveolens Linn. to a Cerebral Ischemia Medicine: The Long Journey of 3-n-Butylphthalide. J. Med. Chem. 2020, 63, 12485–12510. [Google Scholar]
- Villamena, F.A.; Das, A.; Nash, K.M. Potential Implication of the Chemical Properties and Bioactivity of Nitrone Spin Traps for Therapeutics. Future. Med. Chem. 2012, 4, 1171–1207. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J. Recent Advances on Nitrones Design for Stroke Treatment. J. Med. Chem. 2020, 63, 13413–13427. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Neto, H.C.C.F.; Zimmerman, G.A.; Hensley, K.; Towner, R.A. Nitrone-based Therapeutics for Neurodegenerative Diseases: Their Use alone or in Combination with Lanthionines. Free Radic. Biol. Med. 2013, 62, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Towner, R.A.; He, T.; Hensley, K.; Maples, K.R. Translational Research Involving Oxidative Stress and Diseases of Aging. Free Radic. Biol. Med. 2011, 51, 931–941. [Google Scholar] [CrossRef]
- Costa, D.S.S.; Martino, T.; Magalhães, F.C.; Justo, G.; Coelho, M.G.P.; Barcellos, J.C.F.; Moura, V.B.; Costa, P.R.R.; Sabino, K.C.C.; Dias, A.G. Synthesis of N-Methylarylnitrones Derived from Alkyloxybenzaldehydes and Antineoplastic Effect on Human Cancer Cell Lines. Bioorg. Med. Chem. 2015, 23, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- O’Collins, V.E.; Donnan, G.A.; Macleod, M.R.; Howells, D.W. Hypertension and Experimental Stroke Therapies. J. Cereb. Blood Flow Metab. 2013, 33, 1141–1147. [Google Scholar] [CrossRef]
- Datta, P.K.; Reddy, S.; Sharma, M.; Lianos, E.A. Differential nephron HO-1 expression Following Glomerular Epithelial Cell Injury. Nephron Exp. Nephrol. 2006, 103, e131–e138. [Google Scholar] [CrossRef]
- Mandal, M.N.; Moiseyev, G.P.; Elliott, M.H.; Kasus-Jacobi, A.; Li, X.; Chen, H.; Zheng, L.; Nikolaeva, O.; Floyd, R.A.; Ma, J.-X.; et al. Alpha-phenyl-N-tert-Butylnitrone (PBN) Prevents Light-induced Degeneration of the Retina by Inhibiting RPE65 Protein Isomerohydrolase Activity. J. Biol. Chem. 2011, 286, 32491–32501. [Google Scholar] [CrossRef]
- Ewert, D.; Hu, N.; Du, X.; Li, W.; West, M.B.; Choi, C.-H.; Floyd, R.; Kopke, R.D. HPN-07, a Free Radical Spin Trapping Agent, Protects against Functional, Cellular and Electrophysiological Changes in the Cochlea Induced by Acute Acoustic Trauma. PLoS ONE 2017, 12, e0183089. [Google Scholar] [CrossRef]
- Varela-Nieto, I.; Murillo, S.; Rodríguez-de la Rosa, L.; Oset Gasque, M.J.; Marco-Contelles, J. Use of Radical Oxygen Species Scavenger Nitrones to Treat Oxidative Stress-mediated hearing Loss: State of the Art and Challenges. Front. Cell. Neurosci. 2021, 15, 711269. [Google Scholar] [CrossRef]
- Rosselin, M.; Poeggeler, B.; Durand, G. Nitrone Derivatives as Therapeutics: From Chemical Modification to Specific-targeting. Curr. Top. Med. Chem. 2017, 17, 2006–2022. [Google Scholar] [CrossRef]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant Therapy: Current Status and Future Prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Oliveira, C.; Benfeito, S.; Fernandes, C.; Cagide, F.; Silva, T.; Borges, F. NO and HNO Donors, Nitrones, and Nitroxides: Past, Present, and Future. Med. Res. Rev. 2018, 38, 1159–1187. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Kopke, R.D.; Choi, C.-H.; Foster, S.B.; Doblas, S.; Towner, R.A. Nitrones as Therapeutics. Free Radic. Biol. Med. 2008, 45, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Croitoru, M.D.; Ibolya, F.; Pop, M.C.; Dergez, T.; Mitroi, B.; Dogaru, M.T.; Tőkes, B. Nitrones Are Able to Release Nitric Oxide in Aqueous Environment under Hydroxyl Free Radical Attack. Nitric Oxide 2011, 25, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, K.V.; Jorgensen, K.A. Asymmetric 1,3-Dipolar Cycloaddition Reactions. Chem. Rev. 1998, 98, 863–910. [Google Scholar] [CrossRef]
- Durand, G.; Choteau, F.; Pucci, B.; Villamena, F.A. Reactivity of Superoxide Radical Anion and Hydroperoxyl Radical with alpha-Phenyl-N-tert-butylnitrone (PBN) Derivatives. J. Phys. Chem. A 2008, 112, 12498–12509. [Google Scholar] [CrossRef]
- Cao, X.; Phillis, J.W. α-Phenyl-tert-butyl-nitrone Reduces Cortical Infarct and Edema in Rats Subjected to Focal Ischemia. Brain Res. 1994, 644, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Durand, G. Synthetic Antioxidants. In Molecular Basis of Oxidative Stress; Villamena, F.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 377–406. [Google Scholar]
- Lapchak, P.A.; Araujo, D.M. Spin Trap Agents: A New Approach to Stroke Therapy. Drug News Perspect. 2002, 15, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Zausinger, S.; Hungerhuber, E.; Baethmann, A.; Hanns-Jürgen Reulen, J.; Schmid-Elsaesser, R. Neurological Impairment in Rats after Transient Middle Cerebral Artery Occlusion: A Comparative Study under Various Treatment Paradigms. Brain Res. 2000, 863, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.P.; Angiolini, P.; Tani, R.; Consales, G.; Bordi, L. Phenyl-T-butyl-nitrone is Active against Traumatic Shock in Rats. Free Rad. Res. Commun. 1986, 1, 321–327. [Google Scholar] [CrossRef]
- Dehouck, M.-P.; Cecchelli, R.; Green, A.R.; Renftel, M.; Lundquist, S. In vitro Blood-brain Barrier Permeability and Cerebral Endothelial Cell Uptake of the Neuroprotective Nitrone Compound NXY-059 in Normoxic, Hypoxic and Ischemic Conditions. Brain Res. 2002, 955, 229–235. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, P.; Zhang, G.; Wang, L.; Zhong, H.; Zhai, Z.; Wang, L.; Wang, Y. Therapeutic Effects of Tetramethylpyrazine Nitrone in Rat Ischemic Stroke Models. J. Neurosci. Res. 2012, 90, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.F.; Hema, K.C.; Ting, H.; Rheal, T. Anti-cancer Activity of Nitrones and Observations on Mechanism of Action. Anti-Cancer Agents Med. Chem. 2011, 11, 373–379. [Google Scholar]
- Floyd, R.A.; Hensley, K.; Bing, G. Evidence for Enhanced Neuro-inflammatory Processes in Neurodegenerative Diseases and the Action of Nitrones as Potential Therapeutics. J. Neural Transm. Suppl. 2000, 60, 387–414. [Google Scholar]
- Doggrell, S.A. Nitrone Spin on Cerebral Ischemia. Curr. Opin. Investig. Drugs 2006, 7, 20–24. [Google Scholar]
- Kuroda, S.; Tsuchidate, R.; Smith, M.L.; Maples, K.R.; Siesjo, B.K. Neuroprotective effects of a Novel Nitrone, NXY-059, after Transient Focal Cerebral Ischemia in the Rat. J. Cereb. Blood Flow Metab. 1999, 19, 778–787. [Google Scholar] [CrossRef]
- Macleod, M.R.; van der Worp, H.B.; Sena, E.S.; Howells, D.W.; Dirnagl, U.; Donnan, G.A. Evidence for the Efficacy of NXY-059 in Experimental Focal Cerebral Ischaemia is Confounded by Study Quality. Stroke 2008, 39, 2824–2829. [Google Scholar] [CrossRef] [PubMed]
- Godínez-Rubí, M.; Rojas-Mayorquín, A.E.; Ortuño-Sahagún, D. Nitric Oxide Donors as Neuroprotective Agents after an Ischemic Stroke-related Inflammatory Reaction. Oxid. Med. Cell. Longev. 2013, 2013, 297357. [Google Scholar] [CrossRef]
- Matias, A.C.; Biazolla, G.; Cerchiaro, G.; Keppler, A.T. α-Aryl-N-aryl Nitrones: Synthesis and Screening of a New Scaffold for Cellular Protection against an Oxidative Toxic Stimulus. Bioorg. Med. Chem. 2016, 24, 232–239. [Google Scholar] [CrossRef]
- Minnerup, J.; Sutherland, B.A.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for Stroke: Current Status and Future Perspectives. Int. J. Mol. Sci. 2012, 13, 11753–11772. [Google Scholar] [CrossRef]
- Chen, G.; Griffin, M.; Poyer, J.L.; McCay, P.B. HPLC Procedure for the Pharmacokinetic Study of the Spin-trapping Agent, alpha-Phenyl-N-tert-butyl Nitrone (PBN). Free Radic. Biol. Med. 1990, 9, 93–98. [Google Scholar] [CrossRef]
- Saito, K.; Yoshioka, H.; Cutler, R.G. A Spin Trap, N-tert-Butyl-α-phenylnitrone Extends the Life Span of Mice. Biosci. Biotech. Biochem. 1998, 62, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yoshioka, H.; Kazama, S.; Cutler, R.G. Release of Nitric Oxide from a Spin Trap, N-tert-Butyl-α-phenylnitrone, under Various Oxidative Conditions. Biol. Pharm. Bull. 1998, 21, 401–404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inanami, O.; Kuwabara, M. α-Phenyl N-tert-butyl Nitrone (PBN) Increases the Cortical Cerebral Blood Flow by Inhibiting the Breakdown of Nitric Oxide in Anesthetized Rats. Free Rad. Res. 1995, 23, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Phillis, J.W.; Clough-Helfman, C. Protection from Cerebal Ischemic Injury in Gerbils with the Spin Trap Agent N-tert-Butyl-aphenylnitrone (PBN). Neurosci. Lett. 1990, 116, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Clough-Helfman, C.; Phillis, J.W. The Free Radical Trapping Agent N-tert-Butylalpha-phenylnitrone (PBN) Attenuates Cerebral Ischaemic Injury in Gerbils. Free Rad. Res. Commun. 1991, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Phillis, J.W. alpha-Phenyl-tert-butyl-nitrone (PBN) Attenuates Hydroxyl Radical Production During Ischemia-reperfusion Injury of Rat Brain: An EPR Study. Free Rad. Res. Commun. 1993, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.-L.; Gu, J.-L.; Lysko, P.G.; Cheng, H.-Y.; Barone, F.C.; Feuerstein, G. Neuroprotective Effects of Phenyl-t-butyl-nitrone in Gerbil Global Brain Ischemia and in Cultured Rat Cerebellar Neurons. Brain Res. 1992, 574, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Pahlmark, K.; Smith, M.L.; Siesjö, B.K. Delayed treatment with the Spin Trap alpha-Phenyl-N-tert-butyl Nitrone (PBN) Reduces Infarct Size Following Transient Middle Cerebral Artery Occlusion in Rats. Acta Physiol. Scand. 1994, 152, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Folbergrova, J.; Zhao, Q.; Katsura, K.; Siesjö, B.K. N-tert-Butyl-alphaphenylnitrone Improves Recovery of Brain Energy State in Rats Following Transient Focal Ischemia. Proc. Natl. Acad. Sci. USA 1995, 92, 5057–5061. [Google Scholar] [CrossRef]
- Pahlmark, K.; Siesjö, B.K. Effects of the Spin Trap α-Phenyl-N-tert-butylnitrone (PBN) in Transient Forebrain Ischemia in the Rat. Acta Physiol. Scand. 1996, 157, 41–51. [Google Scholar] [CrossRef]
- Floyd, R.A. Role of Oxygen Free Radicals in Carcinogenesis and Brain Ischemia. FASEB J. 1990, 4, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative Damage to Brain Proteins, Loss of Glutamine Synthetase Activity, and Production of Free Radicals during Ischemia/Reperfusion-induced Injury to Gerbil Brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Panahian, N.; Chen, Y.I.; Beal, M.F.; Moskowitz, M.A.; Rosen, B.R.; Jenkins, B.G. Facilitation of Postischemic Reperfusion with a-PBN: Assessment Using NMR and Doppler Flow Techniques. Am. J. Physiol. 1997, 272, H1986–H1995. [Google Scholar] [CrossRef] [PubMed]
- Aronowski, J.; Strong, R.; Grotta, J.C. Reperfusion Injury: Demonstration of Brain Damage Produced by Reperfusion after Transient Focal Ischemia in Rats. J. Cereb. Blood Flow Metab. 1997, 17, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Aronowski, J.; Grotta, J.; Waxham, N.M. Ischemia Induced Translocation of Ca2+/Calmodulin-dependent Protein Kinase II: Potential Roles in Neuronal Damage. J. Neurochem. 1992, 58, 1743–1753. [Google Scholar] [CrossRef]
- Aronowski, J.; Waxham, M.N.; Grotta, J.C. Neuronal Protection and Preservation of Calcium/calmodulin-dependent Protein Kinase II and Protein Kinase C Activity by Dextrorphan Treatment in Global Ischemia. J. Cereb. Blood Flow Metab. 1992, 13, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Hiestand, O.M.; Haley, B.E.; Kindy, M.S. Role of Calcium in Inactivation of Calcium/calmodulin Dependent Protein Kinase II after Cerebral Ischemia. J. Neurol. Sci. 1992, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Pazos, A.J.; Green, E.J.; Busto, R.; McCabe, P.M.; Baena, R.C.; Ginsberg, M.D.; Globus, M.Y.-T.; Schneiderman, N.; Dietrich, W.D. Effects of Combined Postischemic Hypothermia and Delayed N-tert-Butyl-α-phenylnitrone (PBN) Administration on Histopathological and Behavioral Deficits Associated with Transient Global Ischemia in Rats. Brain Res. 1999, 846, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Niwa, M.; Iwai, T.; Uematsu, T. Involvement of Free Radicals in Cerebral Vascular Reperfusion Injury Evaluated in a Transient Focal Cerebral Ischemia Model of Rat. Free Radic. Biol. Med. 1999, 26, 722–729. [Google Scholar] [CrossRef]
- Schmid-Elsaesser, R.; Hungerhuber, E.; Zausinger, S.; Baethmann, A.; Reulen, H.J. Neuroprotective Effects of the Novel Brain-penetrating Antioxidant U-101033E and the Spin-trapping Agent alpha-Phenyl-N-tert-butyl Nitrone (PBN). Exp. Brain Res. 2000, 130, 60–66. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Shuaib, A. Neuroprotection by 2-h Postischemia Administration of Two Free Radical Scavengers, α-Phenyl-N-tert-butyl-nitrone (PBN) and N-tert-Butyl-(2-sulfophenyl)-nitrone (S-PBN), in Rats Subjected to Focal Embolic Cerebral Ischemia. Exp. Neurol. 2000, 163, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; He, Q.P.; Nakamura, L.; Csiszar, K. Free Radical Spin Trap α-Phenyl-N-tert-butyl-nitrone Inhibits Caspase-3 Activation and Reduces Brain Damage Following a Severe Forebrain Ischemic Injury. Free Rad. Biol. Med. 2001, 31, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Chapman, D.F.; Zivin, J.A. Pharmacological Effects of the Spin Trap Agents N-t-Butyl-phenylnitrone (PBN) and 2,2,6,6-Tetramethylpiperidine-N-oxyl (TEMPO) in a Rabbit Thromboembolic Stroke Model: Combination Studies with the Thrombolytic Tissue Plasminogen Activator. Stroke 2001, 32, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.; Bruhn, T.; Diemer, N.H. The Free Radical Spin-trap α-PBN Attenuates Periinfarct Depolarizations Following Permanent Middle Cerebral Artery Occlusion in Rats without Reducing Infarct Volume. Brain Res. 2003, 990, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Goda, W.; Satoh, K.; Nakashima, M.; Hara, A.; Niwa, M. PBN Fails to Suppress in Delayed Neuronal Death of Hippocampal CA1 Injury Following Transient Forebrain Ischemia in Gerbils. Neurosci. Lett. 2012, 517, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Samadi, A.; Soriano, E.; Revuelta, J.; Valderas, C.; Chioua, M.; Garrido, I.; Bartolomè, B.; Tomassolli, I.; Ismaili, L.; Gonzàlez-Lafuente, L.; et al. Synthesis, Structure, Theoretical and Experimental in vitro Antioxidant/Pharmacological Properties of α-Aryl, N-Alkyl Nitrones, as Potential Agents for the Treatment of Cerebral Ischemia. Bioorg. Med. Chem. 2011, 19, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Chioua, M.; Sucunza, D.; Soriano, E.; Hadjipavlou-Litina, D.; Alcàzar, A.; Ayuso, I.; Oset-Gasque, M.J.; Gonzàlez, M.P.; Monjas, L.; Rodrìguez-Franco, M.I.; et al. α-Aryl-N-alkyl nitrones, as Potential Agents for Stroke Treatment: Synthesis, Theoretical Calculations, Antioxidant, Anti-inflammatory, Neuroprotective, and Brain-blood Barrier Permeability Properties. J. Med. Chem. 2012, 55, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Arce, C.; Díaz-Castroverde, S.; Canales, M.J.; Marco-Contelles, J.; Samadi, A.; Oset-Gasque, M.J.; González, M.P. Drugs for Stroke: Action of Nitrone (Z)-N-(2-Bromo-5-hydroxy-4-methoxybenzylidene)-2-methylpropan-2-amine Oxide on Rat Cortical Neurons in Culture Subjected to Oxygen-glucose-deprivation. Eur. J. Med. Chem. 2012, 55, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Almarza, A.; Diez-Iriepa, D.; Chioua, M.; Chamorro, B.; Iriepa, I.; Martínez-Murillo, R.; Hadjipavlou-Litina, D.; Oset-Gasque, M.J.; Marco-Contelles, J. Synthesis, Neuroprotective and Antioxidant Capacity of PBN-related Indanonitrones. Bioorg. Chem. 2019, 86, 445–451. [Google Scholar] [CrossRef]
- Chamorro, B.; Diez-Iriepa, D.; Merás-Sáiz, B.; Chioua, M.; García-Vieira, D.; Iriepa, I.; Hadjipavlou-Litina, D.; López-Muñoz, F.; Martínez-Murillo, R.; González-Nieto, D.; et al. Synthesis, Antioxidant Properties and Neuroprotection of α-Phenyl-tert-butylnitrone Derived Homobisnitrones in in vitro and in vivo Ischemia models. Sci. Rep. 2020, 10, 14150. [Google Scholar] [CrossRef]
- Diez-Iriepa, D.; Iriepa, I.; López-Muñoz, F.; Marco-Contelles, J.; Hadjipavlou-Litina, D. Synthesis and Antioxidant Properties of Heterobisnitrones Derived from Benzene Dicarbaldehydes. Antioxidants 2022, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Diez-Iriepa, D.; Chamorro, B.; Talaván, M.; Chioua, M.; Iriepa, I.; Hadjipavlou-Litina, D.; López-Muñoz, F.; Marco-Contelles, J.; Oset-Gasque, M.J. Homo-tris-nitrones Derived from α-Phenyl-N-tertbutylnitrone: Synthesis, Neuroprotection and Antioxidant Properties. Int. J. Mol. Sci. 2020, 21, 7949. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, B.; Garcia-Vieira, D.; Diez-Iriepa, D.; Garagarza, E.; Chioua, M.; Hadjipavlou-Litina, D.; López-Muñoz, F.; Marco-Contelles, J.; Oset-Gasque, M.J. Synthesis, Neuroprotection, and Antioxidant Activity of 1,1′-Biphenylnitrones as α-Phenyl-N-tert-butylnitroneanalogues in in vitro Ischemia Models. Molecules 2021, 26, 1127. [Google Scholar] [CrossRef] [PubMed]
- Diez-Iriepa, D.; Iriepa, I.; Knez, D.; Gobec, S.; de los Ríos, C.; Bravo, I.; López-Muñoz, F.; Marco-Contelles, J.; Hadjipavlou-Litina, D. Polyfunctionalized α-Phenyl-tert-butyl(benzyl)nitrones: Multifunctional Antioxidants for Stroke Treatment. Antioxidants 2022, 11, 1735. [Google Scholar] [CrossRef]
- Escobar-Peso, A.; Martínez-Alonso, E.; Hadjipavlou-Litina, D.; Alcázar, A.; Marco-Contelles, J. Synthesis, Antioxidant and Neuroprotective Analysis of Diversely Functionalized α-Aryl-N-alkyl Nitrones, as Potential Agents for Ischemic Stroke Therapy. Eur. J. Med. Chem. 2024, 266, 116133. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
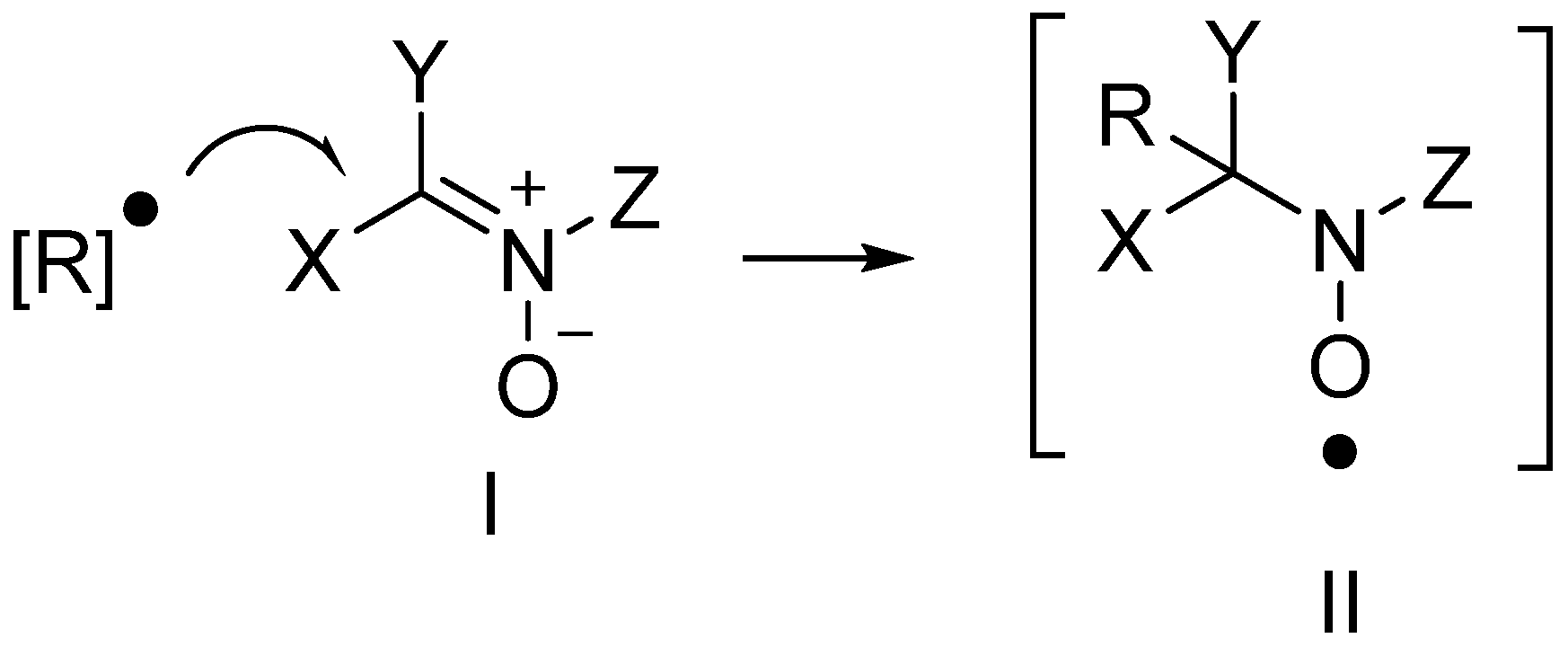{kind=link}
{kind=link}
| Nitrone | Structure | DPPH [RSA (%)] b | ORAC d | •OH [RSA (%)] e |
|---|---|---|---|---|
| 1 |  | 0.8 ± 0.7 | 0.0 ± 0.0 | 95.3 ± 0.8 |
| PBN |  | 1.4 ± 0.9 | 0.0 ± 0.0 | 94.9 ± 1.0 |
| 2 |  | 0.6 ± 0.3 | 0.01 ± 0.00 | 95.7 ± 0.5 |
| 3 |  | 31.1 ± 2.1 | 5.58 ± 0.33 | 96.3 ± 0.5 |
| 4 |  | 25.9 ± 1.8 | 7.36 ± 0.32 | 94.6 ± 0.9 |
| 5 |  | 0.8 ± 0.9 | 0.03 ± 0.00 | 96.3 ± 0.8 |
| 6 |  | 0.2 ± 0.2 | 0.03 ± 0.002 | 95.1 ± 1.3 |
| 7 |  | 87.6 ± 1.2 | 3.35 ± 0.07 | 96.5 ± 0.7 |
| 8 |  | 88.2 ± 1.0 | 2.93 ± 0.07 | 96.9 ± 0.7 |
| 9 |  | 40.7 ± 2.5 | 6.40 ± 0.35 | 95.7 ± 1.0 |
| 10 |  | 3.9 ± 3.8 | 0.01 ± 0.00 | 95.1 ± 1.1 |
| 11 |  | 1.6 ± 1.5 | 0.01 ± 0.00 | 94.9 ± 1.3 |
| 12 |  | 4.8 ± 1.8 | 7.98 ± 0.21 | nd c |
| 13 |  | 1.2 ± 1.1 | 3.69 ± 0.15 | nd c |
| Nitrone | LOX (%) d | AAPH (%) e (min) f | HO• g,h | O2• (%) i | NO (%) k,(m) | clogP |
|---|---|---|---|---|---|---|
 | 83 | No (0) | 90 | 15 | 2 (4.5) | 2.56 |
 | 22 | 5 (1) | 77 | 8 | No (No) | 1.32 |
 | No | 74 (25.2) | 97 | 24 | nd l (No) | 3.09 |
 | No | 67 (7.2) | 72 | 11 | 2 (3) | 2.83 |
 | No | 89 (84.6) | No | 6 | 2 (2) | 3.36 |
 | No | 47 (0.52) | 90 | 15 | 1 (2) | 3.15 |
 | 21 | No (53.7) | No | 16 | 1 (1) | 3.68 |
 | 59 | 90 (18) | No | 11 | 7 (8) | 0.5 |
 | No | 90 (88.2) | 52 | No | 9 (10) | 2.67 |
 | 24 | 87 (41.8) | 78 | 3 | 5 (6) | 3.36 |
 | 88 | 40 (8) | 93 | 10 | 4 (5.5) | 2.01 |
 | 59 | 37 (29.8) | 30 | 5 | 2 (4) | 1.86 |
 | 31 | 80 (43.8) | 79.5 | 5 | 3 (5) | 3.07 |
 | 42 | 57 (21.6) | 59 | 15 | 3 (4.5) | 2.57 |
| NDGA | 40 | |||||
| Caffeic acid | 15 | |||||
| Trolox | 63 (62.1) | 73 | ||||
| SNP | 54 (58) |
| Nitrone | LOX (%) a | AAPH (%) b (min) c | DPPH (RSA)(%) d | OH f | O2• (%) g | clogP |
|---|---|---|---|---|---|---|
| 14 | 65 | No (0) | 0.9 | 60 | 29 | 3.44 |
| 15 | No | 61 (83) | 81.9 | 77 | 8 | 5.87 |
| 16 | No | 9 (17.4) | 78.6 | No | No | 5.34 |
| 17 | 19 | No (0) | 0.6 | 98 | 19 | 3.04 |
| 18 | 21 | 64 (46) | nd h | 100 | No | 3.57 |
| 19 | 57 | 49 (47) | 0.4 | 54 | 2 | 3.29 |
| 20 | 30 | 81 (36.6) | Nd h | 98 | No | 3.83 |
| Caffeic acid | 15 | |||||
| Curcumin | 35.0 e | |||||
| Quercetin | 94.2 e | |||||
| Trolox | 63 (62.1) |
| Nitrone (Concentration) | Neuroprotection (%) |
|---|---|
| 14 (100 μM) | 101.3 ± 3.3 *** |
| 14 (250 μM) | 81.2 ± 5.0 *** |
| 20 (10 μM) | 77.6 ± 3.1 *** |
| 20 (100 μM) | 83.0 ± 2.7 *** |
| PBN (5 mM) | −13.4 ± 1.9 *** |
| Citicoline (100 μM) | 53.2 ± 1.8 |
| Nitrone | Concentration (μM) | Neuroprotection against Necrotic Cell Death (%) |
|---|---|---|
| 14 | 250 | 70.08 ± 7.52 * |
| 15 | 100 | 53.80 ± 6.01 |
| 15 | 250 | 56.95 ± 10.6 |
| 17 | 250 | 79.21 ± 19.55 |
| 18 | 1 | 47.48 ± 3.33 |
| 19 | 100 | 55.57 ± 12.51 |
| 20 | 10 | 45.37 ± 2.41 |
| PBN | 100 | 67.82 ± 4.70 ** |
| 250 | 73.20 ± 5.83 ** | |
| Citicoline | 100 | 61.26 ± 8.26 |
| R1 | R2 | Method | Time | Nitrone | Yield (%) |
|---|---|---|---|---|---|
| H | Bn | A (reflux) | 1 d | 66 | 68 |
| H | Me | A (MWI) | 20 min | 67 | 61 |
| H | Ph | B | 2 h | 68 | 57 |
| 2-OH | t-Bu | A (rt) | 2 d | 69 | 86 |
| 2-OH | Bn | A (reflux) | 5 d | 70 | 99 |
| 2-OH | Me | A (reflux) | 1 d | 71 | 73 |
| 2-OH | Ph | B | 4 d | 72 | 86 |
| 3-OH | t-Bu | A (MWI) | 9 h | 73 | 30 |
| 4-OH | t-Bu | A (MWI) | 5 h | 74 | 71 |
| 2-O-CH2-C≡CH | t-Bu | A (rt) | 4 d | 75 | 77 |
| 2-O-CH2-C≡CH | Bn | A (rt) | 7 d | 76 | 62 |
| 2-O-CH2-C≡CH | Me | A (rt) | 4 d | 77 | 60 |
| 3-O-CH2-C≡CH | t-Bu | A (rt) | 5 d | 78 | 30 |
| 4-O-CH2-C≡CH | t-Bu | B | 5 d | 79 | 28 |
| 2-OMe | t-Bu | A (reflux) | 1 d | 80 | 81 |
| 3-OMe | t-Bu | A (reflux) | 4 d | 81 | 72 |
| 4-OMe | t-Bu | A (reflux) | 3 d | 82 | 79 |
| 2-NO2 | t-Bu | A (MWI) | 6 h | 83 | 49 |
| 2-NO2 | Bn | A (MWI) | 50 min | 84 | 50 |
| 3-NO2 | t-Bu | A (MWI) | 9 h | 85 | 70 |
| 3-NO2 | Bn | A (MWI) | 2 h | 86 | 68 |
| 4-NO2 | t-Bu | A (reflux) | 4 d | 87 | 91 |
| 4-NO2 | Bn | A (MWI) | 1 h 15 min | 88 | 69 |
| 2-F | t-Bu | A (reflux) | 1 d | 89 | 66 |
| 3-F | t-Bu | A (MWI) | 1 h | 90 | 82 |
| 3-F | Bn | A (MWI) | 3.5 h | 91 | 89 |
| 4-F | t-Bu | A (reflux) | 2 d | 92 | 84 |
| 2-CF3 | t-Bu | A (reflux) | 2 d | 93 | 70 |
| 2-CF3 | Bn | A (MWI) | 1 h 45 min | 94 | 91 |
| 3-CF3 | t-Bu | A (MWI) | 7 h 30 min | 95 | 64 |
| 3-CF3 | Bn | A (MWI) | 1 h | 96 | 57 |
| 4-CF3 | t-Bu | A (reflux) | 1 d | 97 | 84 |
| Nitrone | Conc. | Neuroprotection (%) a |
|---|---|---|
 | 100 µM | 60.8 ± 1.6 |
| 250 µM | 69.0 ± 1.3 * | |
| 500 µM | 82.7 ± 3.2 ** | |
| 1 mM | 74.3 ± 4.3 * | |
 | 100 µM | 46.1 ± 1.3 |
| 250 µM | 72.2 ± 2.1 ** | |
| 500 µM | 48.6 ± 1.8 | |
| 1 mM | 67.9 ± 1.9 | |
| PBN | 5 mM | −59.4 ± 8.8 |
| NXY-059 | 250 µM | 56.8 ± 2.5 |
| Citicoline | 100 µM | 21.1 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marco-Contelles, J. α-Phenyl-N-tert-Butylnitrone and Analogous α-Aryl-N-alkylnitrones as Neuroprotective Antioxidant Agents for Stroke. Antioxidants 2024, 13, 440. https://doi.org/10.3390/antiox13040440
Marco-Contelles J. α-Phenyl-N-tert-Butylnitrone and Analogous α-Aryl-N-alkylnitrones as Neuroprotective Antioxidant Agents for Stroke. Antioxidants. 2024; 13(4):440. https://doi.org/10.3390/antiox13040440
Chicago/Turabian StyleMarco-Contelles, José. 2024. "α-Phenyl-N-tert-Butylnitrone and Analogous α-Aryl-N-alkylnitrones as Neuroprotective Antioxidant Agents for Stroke" Antioxidants 13, no. 4: 440. https://doi.org/10.3390/antiox13040440
APA StyleMarco-Contelles, J. (2024). α-Phenyl-N-tert-Butylnitrone and Analogous α-Aryl-N-alkylnitrones as Neuroprotective Antioxidant Agents for Stroke. Antioxidants, 13(4), 440. https://doi.org/10.3390/antiox13040440